

Abstract
Nitric oxide, produced in pancreatic β cells in response to proinflammatory cytokines, plays a dual role in the regulation of β-cell fate. While nitric oxide induces cellular damage and impairs β-cell function, it also promotes β-cell survival through activation of protective pathways that promote β-cell recovery. In this study, we identify a novel mechanism in which nitric oxide prevents β-cell apoptosis by attenuating the DNA damage response (DDR). Nitric oxide suppresses activation of the DDR (as measured by γH2AX formation and the phosphorylation of KAP1 and p53) in response to multiple genotoxic agents, including camptothecin, H2O2, and nitric oxide itself, despite the presence of DNA damage. While camptothecin and H2O2 both induce DDR activation, nitric oxide suppresses only camptothecin-induced apoptosis and not H2O2-induced necrosis. The ability of nitric oxide to suppress the DDR appears to be selective for pancreatic β cells, as nitric oxide fails to inhibit DDR signaling in macrophages, hepatocytes, and fibroblasts, three additional cell types examined. While originally described as the damaging agent responsible for cytokine-induced β-cell death, these studies identify a novel role for nitric oxide as a protective molecule that promotes β-cell survival by suppressing DDR signaling and attenuating DNA damage-induced apoptosis.
INTRODUCTION
Type 1 (insulin-dependent) diabetes mellitus (T1D) is an autoimmune disease characterized by islet inflammation leading to selective destruction of insulin-secreting pancreatic β cells (1). Proinflammatory cytokines, such as interleukin-1 (IL-1), gamma interferon (IFN-γ), and tumor necrosis factor alpha (TNF-α), have been implicated as pathogenic factors that cause β-cell damage and destruction during the development of T1D (2–4). Nitric oxide, produced in micromolar quantities by β cells following cytokine-stimulated inducible nitric oxide synthase (iNOS) expression, mediates the inhibitory and destructive actions of cytokines on β cells (5–7). While inducing β-cell damage, nitric oxide also activates protective pathways that promote a temporally limited ability to recover from cytokine-mediated damage (8–10). Ultimately, cytokine-induced damage becomes irreversible and β cells undergo cell death by apoptosis (4, 10–12).
The DNA damage response (DDR) coordinates gene expression with DNA repair and cell cycle arrest (13). When DNA damage can no longer be repaired, the DDR promotes apoptosis (14). Ataxia telangiectasia mutated (ATM) is a primary transducer of the DDR that phosphorylates a number of substrates in response to DNA double-strand breaks (DSBs) (15). Nitric oxide induces single-strand breaks in β-cell DNA that, when sufficiently extensive, can lead to DSB formation (16–19). Recently, we have shown that the formation of DSBs in cytokine-treated β cells results in nitric oxide-dependent ATM activation and an ATM-dependent induction of β-cell apoptosis (18).
In this report, we make the novel observation that nitric oxide prevents DNA damage-induced apoptosis in response to camptothecin through inhibition of DDR signaling. Further DNA damage induced by nitric oxide can lead to DDR activation, but this occurs only when nitric oxide is no longer present or being produced at micromolar levels. When produced at micromolar levels, it suppresses phosphorylation of the DDR components H2AX (γH2AX formation), p53, and KRAB-associated protein 1 (KAP1) in response to various genotoxic agents, even in the presence of DSB. This effectively delays and uncouples DDR activation from DSB formation. Nitric oxide fails to suppress DDR activation in macrophages, fibroblasts, or hepatocytes, indicating that the inhibition of DDR activation may be selective for β cells. These findings identify a novel mechanism by which nitric oxide protects β cells from apoptosis by limiting DDR activation and DDR-mediated apoptosis in response to DNA-damaging agents, providing mechanistic insight into how nitric oxide regulates β-cell fate during cell stress.
MATERIALS AND METHODS
Materials and animals.
Male Sprague-Dawley rats (250 to 300 g) were purchased from Harlan (Indianapolis, IN). Rat insulinoma INS 832/13 cells were obtained from Chris Newgard (Duke University, Durham NC). RAW 264.7 cells were obtained from the Washington University Tissue Culture Support Center. HepG2 cells were obtained from the American Type Culture Collection (ATCC). Mouse embryo fibroblasts (MEFs) were obtained from Fumihiko Urano (Washington University, St. Louis, MO). Human insulinoma EndoC-βH1 cells were obtained from Raphael Scharfmann (Paris Descartes University, Paris, France) (20). RPMI 1640 medium, Dulbecco's modified Eagle medium (DMEM), minimum essential medium (MEM) alpha, Connaught Medical Research Laboratories (CMRL) 1066 medium, l-glutamine, sodium pyruvate, HEPES, penicillin, streptomycin, and β-mercaptoethanol were purchased from Invitrogen. Trypsin (0.05% in 0.53 mM EDTA) was purchased from Corning (Corning, NY). Human recombinant IL-1β and rat IFN-γ were purchased from PeproTech (Rocky Hill, NJ). The nitric oxide synthase inhibitor NG-monomethyl l-arginine (NMMA) was purchased from Axxora (San Diego, CA). The nitric oxide donors (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA/NO) (Enzo, Farmingdale, NY) and (Z)-1-[N-(3-aminopropyl)-N-(3-ammoniopropyl)amino]diazen-1-ium-1,2-diolate (DPTA/NO) (Cayman Chemical, Ann Arbor, MI) were dissolved in 10 mM NaOH prior to use. The nitric oxide scavenger 2-(4-carboxyphenyl)-4,5-dihydro-4,4,5,5-tetramethyl-1H-imidazolyl-1-oxy-3-oxide, monopotassium salt (cPTIO) was purchased from Cayman Chemical. H2O2, camptothecin, and menadione were purchased from Sigma-Aldrich (St. Louis, MO). The antibodies used and their sources were as follows: mouse anti-phospho-H2AX (Ser-139; γH2AX) (EMD Millipore, Billerica, MA); mouse anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) (Invitrogen); rabbit anti-cleaved caspase-3, rabbit anti-phospho-AMPK (where AMPK is AMP-activated protein kinase), rabbit anti-phospho-eIF2α (where eIF2α is eukaryotic initiation factor 2α) (Ser-51), and rabbit anti-phospho-p53 (Ser-15) (Cell Signaling Technology, Beverly, MA); rabbit anti-phospho-KAP1 (Ser-824) (Abcam, Cambridge, MA); rabbit anti-active mitogen-activated protein kinase (anti-MAPK) (extracellular signal-regulated kinase 1 and 2 [ERK1/2]) (Promega, Madison, WI); rabbit anti-poly(ADP-ribose) (anti-PAR) (Trevigen, Gaithersburg, MD); horseradish peroxidase (HRP)-conjugated donkey anti-rabbit and HRP-conjugated donkey anti-mouse antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA).
Rodent islet isolation and cell culture.
Islets from male Sprague-Dawley rats were isolated and cultured as described previously (21). INS 832/13 cells, RAW 264.7 cells, MEFs, and EndoC-βH1 cells were cultured and plated as previously described (18, 22–24). HepG2 cells were maintained in MEM alpha containing 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and cultured at 37°C at 5% CO2 and 95% air. HepG2 cells were detached from culture plates using 0.05% trypsin and 0.53 mM EDTA and plated at a density of 250,000 cells/ml. All animal care and experimental procedures with rodents were approved by the Institutional Animal Care and Use Committees at the Medical College of Wisconsin (A3102-01).
Western blot analysis.
Cells/islets were lysed in Laemmli buffer, and Western blot analysis was conducted as previously described (25). The following dilutions of primary and secondary antibodies were used: γH2AX, 1:5,000; cleaved caspase-3, 1:1,000; GAPDH, 1:20,000; phospho-KAP1, 1:1,000; phospho-p53, 1:1,000; phospho-eIF2α, 1:1,000; phospho-AMPK, 1:1,000; phospho-ERK1/2, 1:2,000; PAR, 1:1,000; donkey anti-mouse antibody–horseradish peroxidase, 1:20,000; and donkey anti-rabbit antibody–horseradish peroxidase, 1:20,000. Bands were detected using chemiluminescence (26).
Nitrite determination.
Nitric oxide generation was assessed by measuring the accumulation of its stable metabolite nitrite in culture supernatants using the Griess assay (27).
Comet assay.
DNA damage was measured using the comet assay (28). DNA damage was quantified as the mean tail moment using the Comet Assay Software Project (CASP) program for 30 to 50 cells/condition.
Cell death assay.
Cell death was determined using the SYTOX Green nucleic acid stain (Invitrogen). After treatment, SYTOX Green was added to a final concentration of 5 μM, with duplicate wells receiving SYTOX Green and 120 μM digitonin. The cells were incubated at 37°C for 30 min, and fluorescence was determined at an excitation/emission of 504 nm/523 nm. Percent cell death was calculated by normalizing to the fluorescence in SYTOX Green plus digitonin duplicate wells for each condition (set as 100% cell death). Fluorescent and bright-field images of SYTOX-stained wells were captured using a Nikon TI-U inverted microscope.
Statistics.
Statistical analysis was performed using one-way analysis of variance with a Tukey-Kramer post hoc test. The minimum level of significance was a P value of <0.05, as defined in the figure legends.
RESULTS
Relationship between generation of nitric oxide and formation of γH2AX.
The formation of γH2AX is detectable within minutes of DSB induction (29). In pancreatic β cells, nitric oxide produced endogenously following cytokine treatment or added exogenously using donors stimulates the formation of γH2AX (18). Consistent with previous studies, the donor DEA/NO stimulates γH2AX formation in INS 832/13 cells following a 120-min treatment (Fig. 1A) (18). This temporal induction of γH2AX formation is surprising, because DEA/NO has a short half-life (3 min), leading to a rapid induction of DNA damage (increase in mean tail moment) that is detectable within 15 min of treatment, yet γH2AX formation does not occur until most of the nitric oxide is liberated from the donor (Fig. 1B). Cytokines also stimulate the time-dependent formation of γH2AX in islets, with a maximal 4-fold increase following 36 h of incubation (Fig. 1C and D). Cytokine-induced γH2AX formation also does not correlate with nitric oxide-dependent DNA damage. DNA damage can be detected as early as 24 h post-cytokine treatment (10), yet the maximal accumulation of γH2AX occurs following 36 h of incubation (Fig. 1C). We have previously shown that the rates of cytokine-stimulated nitric oxide production decrease 6-fold between 24 and 36 h (10). Like the actions of nitric oxide donor compounds, cytokine-induced γH2AX formation is a late event that occurs hours after the induction of DNA damage and under conditions of lower rates of nitric oxide production (Fig. 1).
FIG 1.

Relationship between the generation of nitric oxide and the formation of γH2AX. (A) INS 832/13 cells (200,000 cells/400 μl medium) were treated with 1 mM DEA/NO for the indicated times, and γH2AX formation was determined by Western blotting. (B) The red line represents the calculated steady-state levels of nitric oxide liberated from 1 mM DEA/NO, and the squares show the experimentally determined levels of DNA damage (mean tail moment) induced in INS 832/13 cells treated for the indicated times with 1 mM DEA/NO. (C and D) Rat islets (200 islets/400 μl CMRL 1066 culture medium) were treated for the indicated times with IL-1 (10 U/ml) and IFN-γ (150 U/ml), and γH2AX formation was assessed by Western blotting (C) and quantified by densitometry (D). GAPDH was used as a control for equal protein loading. The results are representative (A and C) or the average ± standard error of the mean (SEM) of three independent experiments (B and D) with statistically significant differences versus the control; *, P < 0.05.
Exogenous and endogenous sources of nitric oxide inhibit γH2AX formation.
The temporal dissociation of DSBs and the formation of γH2AX in the presence of nitric oxide suggest that nitric oxide, while stimulating γH2AX formation due to DNA damage, may also actively inhibit or attenuate γH2AX formation. To address this hypothesis, dispersed rat islets were treated with DEA/NO for 2 h or treated with DEA/NO and DPTA/NO, a second nitric oxide donor with a longer half-life of ∼3 h (30). The calculated steady-state levels of nitric oxide liberated from each donor over time are shown in Fig. 2A. DEA/NO stimulates γH2AX formation in dispersed rat islets following 2 h of incubation (Fig. 2B). Alone, DPTA/NO does not induce γH2AX formation; however, in a concentration-related manner, this long-half-life nitric oxide donor inhibits DEA/NO-induced γH2AX formation, with complete inhibition at 600 μM DPTA/NO (Fig. 2B). Nitric oxide also inhibits γH2AX formation induced by genotoxic stressors. γH2AX formation induced by nitric oxide (DEA/NO), the topoisomerase inhibitor camptothecin, and H2O2 is attenuated in a concentration-dependent manner by DPTA/NO in INS 832/13 insulinoma cells (Fig. 2C). Maximal inhibition of γH2AX formation in response to each genotoxic agent was observed at 400 to 600 μM DPTA/NO (Fig. 2D). Consistent with the attenuation in INS 832/13 cells, DPTA/NO attenuates H2O2-induced γH2AX formation in dispersed rat islet cells (Fig. 2E).
FIG 2.

Nitric oxide inhibits γH2AX formation. (A) Calculated steady-state concentrations of nitric oxide liberated from either 1 mM DEA/NO or 400 μM DPTA/NO over the indicated times. (B to E) γH2AX formation was determined by Western blotting under the following conditions. (B) Dispersed rat islet cells (100 islets/condition) were treated with 1 mM DEA/NO with or without increasing concentrations of DPTA/NO for 2 h. (C) INS 832/13 cells were treated with 1 mM DEA/NO, 25 μM camptothecin, or 100 μM H2O2 with or without increasing concentrations of DPTA/NO for the indicated times. (E) Dispersed rat islets were treated with 100 μM H2O2 with or without 600 μM DPTA/NO for 30 min. DPTA/NO was added 1 h prior to H2O2 treatment, and γH2AX formation was determined by Western blotting. GAPDH was used as a control for equal protein loading. The results are representative (B, C, and E) or the average ± SEM (D) of 3 independent experiments. There was a statistically significant decrease in γH2AX formation in panel D; *, P < 0.05.
Pancreatic β cells exposed to the proinflammatory cytokines IL-1 and IFN-γ produce micromolar levels of nitric oxide (5–7, 31, 32). The role of endogenously produced nitric oxide in the regulation of DNA damage-induced γH2AX formation was examined using INS 832/13 cells (Fig. 3A) and dispersed rat islet cells (Fig. 3C). Following 24 h of incubation with IL-1, INS 832/13 cells were treated for 1 h with camptothecin, and γH2AX formation was examined. IL-1 pretreatment attenuates camptothecin-induced γH2AX, and the NOS inhibitor NMMA attenuates this effect. In a similar manner, H2O2-induced γH2AX formation is attenuated in rat islets pretreated for 24 h with IL-1 plus IFN-γ, and NMMA attenuates this effect (Fig. 3A to D). These findings show that the levels of nitric oxide produced endogenously in β cells treated with IL-1 or IL-1 plus IFN-γ are sufficient to attenuate γH2AX formation in response to genotoxic agents.
FIG 3.

Nitric oxide produced endogenously during cytokine treatment inhibits γH2AX formation. (A and B) INS 832/13 cells (A) or dispersed rat islets (B) were untreated or treated with 10 U/ml IL-1 (A) or 10 U/ml IL-1 plus 150 U/ml IFN-γ (B) for 24 h in the presence or absence of 2 mM NMMA. Following cytokine pretreatment, the INS 832/13 cells were treated with 25 μM camptothecin for 1 h (A), and the dispersed islets were treated with 100 μM H2O2 for 30 min (B). (C and D) The levels of γH2AX were determined by Western blotting and quantified by densitometry (INS 832/13 cells [C]; dispersed rat islets [D]). GAPDH levels are shown as a protein-loading control. Nitrite formation (INS 832/13 cells [C]; dispersed rat islets [D]) was determined on culture supernatants. The results are representative (A and B) or the average and SEM (C and D) of the results of three independent experiments. The inhibition of camptothecin-induced γH2AX formation by IL-1 (C) and of H2O2-induced γH2AX formation by IL-1 plus IFN-γ (D) (*, P < 0.05) and the attenuation of this inhibition of γH2AX formation by NMMA (#, P < 0.05) achieved statistical significance.
Scavengers attenuate nitric oxide-dependent inhibition of γH2AX.
The nitric oxide scavenger cPTIO attenuates the inhibitory actions of DPTA/NO on camptothecin-induced γH2AX formation in INS 832/13 cells (Fig. 4A and B). cPTIO also attenuates the inhibitory actions of DPTA/NO on H2O2-induced γH2AX formation in rat islets (Fig. 4C and D). Like cPTIO, superoxide scavenges nitric oxide at diffusion-controlled rates, forming the powerful oxidant peroxynitrite (33, 34), and we have shown that superoxide attenuates nitric oxide donor-mediated loss of cellular ATP content, mitochondrial oxidative metabolism, and β-cell viability (35). Consistent with our previous studies (35), the superoxide-generating redox cycler menadione attenuates the inhibitory actions of DPTA/NO on γH2AX formation (Fig. 4E and F). These actions are attributable to nitric oxide, as decomposed DPTA/NO fails to inhibit DNA damage-induced γH2AX (Fig. 4G). The ability of scavengers of nitric oxide (cPTIO and superoxide) to attenuate the inhibitory actions of nitric oxide on γH2AX formation in response to DNA damage suggests that nitric oxide, or reactive products of its metabolism other than peroxynitrite, mediates this effect.
FIG 4.

Scavengers attenuate nitric oxide-dependent inhibition of γH2AX. (A) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO and with or without cPTIO for 2 h. (B) γH2AX was assessed by Western blotting and quantified by densitometry. (C and D) Dispersed rat islets were treated with H2O2 (100 μM) with or without DPTA/NO and with or without cPTIO for 30 min. DPTA/NO was added 1 h prior to H2O2 treatment. γH2AX levels were determined by Western blotting (C) and quantified by densitometry (D). (E and F) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO and with or without menadione at the indicated concentrations for 2 h, and γH2AX levels were determined by Western blotting (E) and quantified by densitometry (F). GAPDH levels are shown as a protein-loading control. (G) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO or decomposed DPTA/NO at the indicated concentrations for 1 h. The donor was decomposed by incubation in INS medium at 37°C for 48 h prior to treatment. The levels of γH2AX were determined by Western blotting with GAPDH used as a control for protein loading. The results are representative (A, C, E, and G) or the average and SEM (B, D, and F) of the results of one (G) or three (A to F) independent experiments. There were statistically significant differences between camptothecin plus DPTA/NO (B and F) or H2O2 plus DPTA/NO (D) and the same conditions with the nitric oxide scavenger; *, P < 0.05.
The inhibition of γH2AX formation by nitric oxide is not due to changes in the levels of DNA damage. Camptothecin-induced γH2AX formation is completely inhibited by DPTA/NO, yet the nitric oxide donor does not change the level of DNA damage that accumulates in response to camptothecin (Fig. 5). These findings indicate that nitric oxide inhibits γH2AX formation in response to multiple genotoxic agents without modifying the extent of DNA damage.
FIG 5.

Nitric oxide-dependent inhibition of γH2AX occurs in the presence of DNA damage. (A) The levels of DNA damage in INS 832/13 cells were determined using the Comet assay following a 2-h treatment with 25 μM camptothecin with or without DPTA/NO. Representative comets are shown. (B) (Top) DNA damage was quantified and expressed as the mean tail moment. (Bottom) The formation of γH2AX and GAPDH levels (to confirm equal protein loading) were determined by Western blotting. The results are representative or the average and SEM of the results of three independent experiments. ns, not significant.
Nitric oxide prevents camptothecin-induced cell death.
Upon induction of DSBs, the principal DDR kinases ATM, ataxia telangiectasia and rad3-related protein (ATR), and DNA-dependent protein kinase (DNA-PK) activate multiple pathways that promote DNA repair, cell cycle arrest, and transcriptional processes in an attempt to repair the damage (13). When DNA damage becomes too extensive for repair, a proapoptotic signaling cascade is activated to remove the damaged cell from the population (14). Camptothecin is a classical inducer of DNA damage-induced apoptosis, doing so through inhibition of nuclear DNA topoisomerase I and the generation of DSBs (36). The effects of nitric oxide on camptothecin-induced activation of DDR components in addition to γH2AX were evaluated in INS 832/13 cells. Camptothecin stimulates rapid DDR activation, as evidenced by γH2AX formation, p53 (Ser15), and the ATM-dependent Ser824 phosphorylation of the transcriptional regulator KAP1 (37) within the first hour of incubation, and this action persists for up to 6 h (Fig. 6A). DDR activation is followed by caspase-3 activation (cleavage) that occurs following a 4- to 6-h camptothecin treatment (Fig. 6A). Similar to γH2AX formation, DPTA/NO prevents camptothecin-induced phosphorylation of p53 and KAP1 and the cleavage of caspase-3 (Fig. 6A). Under conditions where nitric oxide inhibits the phosphorylation of multiple signaling components of the DDR and the induction of cascades leading to apoptosis, it also activates pathways known to be responsive to oxidative stress. In the presence of camptothecin, nitric oxide stimulates the rapid and persistent phosphorylation of eIF2α at all time points examined. Nitric oxide also activates AMPK following 6 h of incubation and ERK1/2 following 1 h of incubation (Fig. 6A). These findings indicate that the inhibition of DDR activation by nitric oxide is not due to a loss of cell viability but is coupled to the activation of pathways that may afford protection from cellular stress (38).
FIG 6.

Nitric oxide prevents camptothecin-induced cell death. (A) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO (400 μM) for the indicated times or for 6 h with DPTA/NO. The cells were harvested, and cleaved caspase-3, phospho-KAP1 (S824), phospho-p53 (S15), γH2AX, phospho-eIF2α, phospho-AMPK, and phospho-ERK1/2 were analyzed by Western blotting, with GAPDH serving as a protein-loading control. (B) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO (300 μM) for 12 or 24 h, and cell death was assessed by SYTOX Green nucleic acid stain. (C) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO at the indicated concentrations for 24 h, and cell death was assessed by SYTOX Green nucleic acid stain. (D) Representative fluorescent and bright-field images (magnification, ×40) are shown for each condition in panel C. The morphology of SYTOX-positive cells in camptothecin with or without DPTA/NO (400 μM) is shown in fluorescent images (magnification, ×400) below. (E) INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO (300 μM) and with or without menadione at the indicated concentrations for 24 h, and cell death was measured by SYTOX staining. (F) Schematic depiction of the dual role of nitric oxide in the regulation of the DDR in β cells. The results are representative (A and D) or the average ± SEM (B, C, and E) of the results of three independent experiments with statistically significant differences between groups (*, P < 0.05) (B), differences in death compared to camptothecin alone (*, P < 0.05) (C), and differences from DPTA/NO alone (*, P < 0.05) or camptothecin plus DPTA/NO (#, P < 0.05) (E).
Since nitric oxide inhibits camptothecin-induced caspase-3 cleavage, we examined whether it also attenuates camptothecin-induced cell death. INS 832/13 cells were treated for 12 or 24 h with camptothecin in the presence or absence of DPTA/NO, and cell death was determined by SYTOX Green uptake (Fig. 6B). Alone, camptothecin kills ∼50% of INS 832/13 cells following a 12-h exposure and ∼80% after 24 h of incubation (Fig. 6B), and DPTA/NO attenuates camptothecin-induced killing of INS 832/13 cells to the levels observed in response to DPTA/NO alone (Fig. 6B). At DPTA/NO concentrations greater than 300 μM, nitric oxide toxicity increases in a concentration-dependent manner, although nitric oxide still reduces camptothecin-induced cell death to the levels induced by DPTA/NO alone (Fig. 6C and D). Consistent with the inhibition of apoptotic cell death, the classical morphology (condensed nuclei and formation of apoptotic bodies) in camptothecin-treated INS 832/13 cells is prevented in the presence of nitric oxide, where the cells do not take on a morphology consistent with apoptosis (Fig. 6D, bottom). Similar to the effects of superoxide observed on nitric oxide-dependent inhibition of the DDR (Fig. 4E), menadione prevents both the protective effects of DPTA/NO on camptothecin-induced cell death and the minor toxic effects of 300 μM DPTA/NO alone (Fig. 6E). These findings associate the suppression of DDR signaling by nitric oxide with protection of β cells from DNA damage-induced cell death, illustrating the dual role of nitric oxide in the regulation of the DDR in the β cell (Fig. 6F).
Nitric oxide does not protect β cells from H2O2-induced death.
H2O2 treatment leads to a rapid induction of DNA damage, overactivation of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP), and subsequent depletion of NAD+ and ATP due the addition and removal of PAR modifications (39, 40). Ultimately, these events lead to PARP-dependent β-cell death (40, 41). In contrast to camptothecin-induced cell death, nitric oxide does not prevent cell death induced by H2O2 (Fig. 7A). Consistent with this observation, H2O2-induced activation of PARP (PAR formation) is not inhibited in the presence of nitric oxide (Fig. 7B). These findings suggest that there is selectivity in the death pathways that are sensitive to inhibition by nitric oxide. Nitric oxide is protective in response to agents that stimulate ATM activation or DDR-associated apoptosis, but it does not prevent β-cell death in non-DDR-triggered cascades, such as PARP overactivation following H2O2 treatment.
FIG 7.
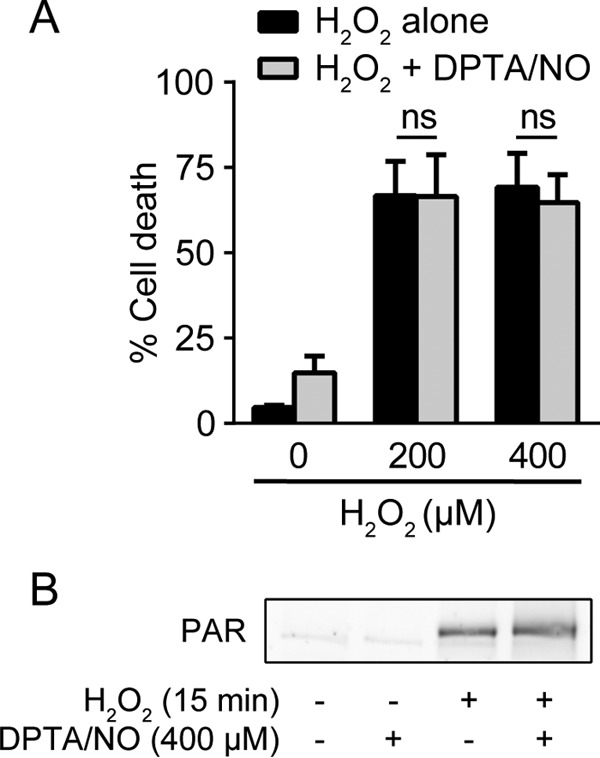
Nitric oxide does not protect against H2O2-induced cell death. (A) INS 832/13 cells were treated with H2O2 at the indicated concentrations for 6 h with or without DPTA/NO (300 μM), and cell death was assessed by SYTOX Green stain. (B) INS 832/13 cells were treated with H2O2 (100 μM) for 15 min with or without DPTA/NO (400 μM). DPTA/NO was added 1 h prior to H2O2 treatment. PAR formation was assessed by Western blotting. The results are the averages and SEM of three experiments (A) or representative of two independent experiments (B).
Cell type specificity of DDR inhibition by nitric oxide.
The inhibitory effects of nitric oxide on DDR activation were examined in a number of cell types to determine the cell type selectivity of the response. MEFs, RAW 264.7 macrophages, HepG2 hepatocytes, human insulinoma EndoC-βH1 cells, and INS 832/13 cells were treated for 2 h with camptothecin with or without increasing concentrations of DPTA/NO. While camptothecin stimulates γH2AX formation in each cell type, it is only in the human EndoC-βH1 and rat INS 832/13 cells that nitric oxide attenuates γH2AX formation (Fig. 8). In addition to γH2AX, nitric oxide also attenuates the stimulatory actions of camptothecin on the phosphorylation of the DDR substrates KAP1 and p53 in EndoC-βH1 and rat INS 832/13 cells. In contrast, nitric oxide does not inhibit camptothecin-induced KAP1 and p53 phosphorylation in RAW264.7 cells, MEFs, or HepG2 cells (Fig. 9). These findings suggest that the ability of nitric oxide to inhibit the DDR is not a general response of all cells and may be selective for terminally differentiated cells, such as β cells.
FIG 8.

Concentration-dependent effects of nitric oxide on γH2AX formation in multiple cell types. (A) MEFs, RAW 264.7 cells, HepG2 cells, EndoC-βH1 cells, and INS 832/13 cells were treated with camptothecin (25 μM) with or without DPTA/NO at the indicated concentrations for 2 h. γH2AX levels were assessed by Western blotting, with GAPDH shown as a control for protein loading. (B) Levels of γH2AX were quantified by densitometry and expressed as percentages of camptothecin-induced γH2AX for each cell type. The results are representative (A) or the averages ± SEM (B) of the results of three independent experiments. There were statistically significant decreases in γH2AX formation in EndoC-βH1 cells (*, P < 0.05) and in INS 832/13 cells (#, P < 0.05).
FIG 9.

Inhibition of the DDR by nitric oxide is selective for β cells. (A to D) RAW 264.7 cells (A), MEFs (B), HepG2 cells (C), and INS 832/13 cells (D) were treated with camptothecin (25 μM) with or without DPTA/NO (400 μM) for 2 h. Phospho-KAP1 (S824), phospho-p53 (S15), and γH2AX levels were determined by Western blotting. GAPDH is shown as a control for protein loading. (E) The phosphorylation of DDR substrates was quantified by densitometry, normalized to GAPDH, and expressed as percent inhibition of camptothecin-induced phosphorylation. The results are representative (A to D) or the average and SEM (E) of three independent experiments with statistically significant differences; *, P < 0.05.
DISCUSSION
In response to double-stranded-DNA strand breaks, the DDR is activated and functions to promote DNA repair. When DNA damage is too extensive, the DDR is also capable of removing the damaged cell by activating apoptosis. Through the induction of DNA strand breaks and DNA deamination, nitric oxide activates the DDR in multiple cell types (42–45). Nitric oxide is also an effective inhibitor of apoptotic cell death by inhibiting caspase activity via S-nitrosation of the active-site cysteine (53, 54). In this study, we identify a second mechanism by which nitric oxide attenuates apoptosis. We show that nitric oxide actively suppresses DDR activation and subsequent apoptosis in the presences of DNA damage. Specifically, nitric oxide attenuates the formation of γH2AX, the phosphorylation of KAP1 and p53, and the cleavage of caspase-3 in response to DNA damage. The inhibitory effects of nitric oxide on DDR activation are not limited to a particular form of genotoxic stress, as DDR activation in response to camptothecin, H2O2, and nitric oxide itself is suppressed when this free radical is present at micromolar levels. Associated with the inhibition of the DDR, nitric oxide also prevents β-cell apoptosis in response to camptothecin treatment but does not protect β cells from death induced by all types of DNA-damaging agents. Although activated by H2O2, the DDR does not contribute to β-cell death in response to the oxidant. The loss of β-cell viability in response to H2O2 is a consequence of PARP overactivation that results in the depletion of NAD and ATP (47). Consistent with this conclusion, nitric oxide does not inhibit PARP signaling or β-cell death mediated by this reactive species (Fig. 7). These findings suggest that the inhibitory actions of nitric oxide are restricted to apoptosis associated with DDR activation and provide a second mechanism by which nitric oxide protects β cells from apoptosis associated with DNA damage.
Whether supplied exogenously or produced endogenously following cytokine treatment, nitric oxide causes DNA base oxidation and deamination, resulting in single-strand breaks (16, 17, 19) When these breaks become sufficiently extensive, DSBs arise, and this results in DDR activation and γH2AX formation (18). The ability of nitric oxide to induce DNA damage and DDR activation has been shown in multiple cell types, including pancreatic β cells (18, 42, 43, 44). In addition to causing DNA damage, we now show that low micromolar levels of nitric oxide suppress DDR activation induced by camptothecin and H2O2 and by nitric oxide itself (Fig. 2 and 3). These findings suggest that nitric oxide plays a dual role in controlling the response of β cells to cytokines, acting as the inducer of DNA damage that leads to DDR activation while simultaneously suppressing this pathway when present at micromolar levels and effectively uncoupling DSB formation from DDR signaling.
Peroxynitrite, produced at diffusion-controlled rates during the reaction of nitric oxide with superoxide (34), is commonly viewed as the reactive species responsible for damage in cells making nitric oxide (19). We have shown that β cells lack the ability to generate superoxide and therefore peroxynitrite in response to cytokines (35, 49). When forced to generate peroxynitrite following treatment with the superoxide-generating redox cycler menadione and a chemical donor of nitric oxide, superoxide scavenges nitric oxide and protects β cells from nitric oxide-mediated decreases in cellular ATP, oxidative metabolism, and the induction of nitric oxide-dependent gene expression (35, 49). This protection from nitric oxide-mediated damage occurs under conditions where β cells generate peroxynitrite to levels that are comparable to the levels generated by activated macrophages (35). Using this model system, we show that the inhibitory actions of nitric oxide donors on γH2AX formation are prevented by cotreatment with menadione (Fig. 4E). Furthermore, menadione results in significant prevention of both the protective actions of nitric oxide on camptothecin-induced cell death and the toxic actions of nitric oxide alone (Fig. 6E). These findings are consistent with previous studies showing loss of nitric oxide-dependent effects when superoxide is present and suggest that DDR inhibition and protection from DNA damage-induced apoptosis in response to nitric oxide are not due to peroxynitrite formation but are dependent on nitric oxide or other reactive metabolites of nitric oxide metabolism. Consistent with this interpretation, the nitric oxide scavenger cPTIO attenuates the inhibitory actions of nitric oxide on DNA damage-induced γH2AX formation (Fig. 4A to D). Importantly, the inhibition of γH2AX formation by nitric oxide is not a consequence of reduced DNA damage, a finding that is not surprising given the DNA-damaging properties of nitric oxide itself (Fig. 5).
The extent of DNA damage in response to cytokine treatment may be one determinant of whether β cells recover from cytokine-mediated damage or undergo cell death by apoptosis (10, 18), and the DDR may control this response. Once activated following DNA damage, the DDR coordinates a response to repair the damage or, if DNA damage is too extensive, initiates a cell death program (13). ATM plays a central role in regulating these responses to DSBs during DDR activation (15). In cytokine-treated pancreatic β cells, ATM is activated in a nitric oxide-dependent manner but does not participate in the repair of damaged DNA (18). Alternatively, ATM appears to direct the induction of β-cell apoptosis (18). Cytokines have been shown to stimulate the induction of apoptotic cascades and caspase activation, leading to β-cell death (50), and this process is believed to contribute to the loss of β-cell mass during the development of autoimmune diabetes (12). Recently, ATM has been identified as one regulator of cytokine-induced apoptosis that couples DNA damage accumulation with the cleavage of caspase-3 (18). While cytokines can induce apoptosis, β cells have a temporally limited ability to recover from cytokine-mediated damage (8, 10, 11). Treatment of rat, mouse, or human islets with cytokines results in the nitric oxide-dependent inhibition of insulin secretion, oxidative metabolism, and protein synthesis and the induction of DNA damage (4). Not only does the inhibition of nitric oxide generation prevent these damaging actions of cytokines, the addition of NOS inhibitors to islets pretreated for 24 h with cytokines, followed by continued culture with a NOS inhibitor and cytokines for 8 h, results in complete restoration or repair of each of the damaging actions of cytokines (5–9, 11). This ability to recover from nitric oxide-mediated damage is temporally limited, as the inhibitory and destructive effects of cytokines become irreversible following 36 h of incubation (10, 11). The irreversible damage at 36 h correlates ATM (γH2AX formation) and caspase-3 activation with a 6-fold decrease in cytokine-induced nitric oxide production by β cells (10). We interpret these findings to suggest that nitric oxide has a dual role in the β-cell response to cytokines. Nitric oxide is responsible for mediating much of the damage associated with cytokine treatment (4). However, nitric oxide also activates protective pathways, such as AMPK, c-Jun N-terminal kinase, and the adaptive protective unfolded-protein response (UPR), that facilitate recovery from cytokine-induced damage (38, 51, 52), and as we show in this study, nitric oxide attenuates activation of ATM and the DDR and thereby prevents the induction of apoptosis. It is only when β cells no longer have the capacity to recover from cytokine-mediated damage (11) that apoptotic death is triggered, and this occurs when the β cell no longer has the capacity to produce micromolar levels of nitric oxide (10).
Consistent with these protective roles, we show that nitric oxide attenuates the phosphorylation of the ATM substrates H2AX, KAP1, and p53 in β cells treated with genotoxic agents. ATM-dependent signaling promotes apoptosis following DNA damage (14), and the inhibition of this pathway by nitric oxide is associated with protection of β cells from caspase cleavage and cell death induced by camptothecin, an inducer of DNA damage-dependent apoptosis (Fig. 6). As ATM-dependent signaling is also involved in the initiation of apoptosis following cytokine-induced DNA damage (18), the inhibition of this pathway by nitric oxide may be a protective response affording β cells time to determine if the DNA damage can be repaired or if apoptosis should be initiated. Importantly, the inhibitory effects of nitric oxide on the DDR appear to be selective for β cells, as nitric oxide does not inhibit DDR activation in macrophages, fibroblasts, or hepatocytes (Fig. 8 and 9), even though it induces DNA damage in each cell type. It is interesting to speculate that attenuation of ATM activation by nitric oxide may be a protective response affording β cells time to determine if the DNA damage can be repaired or if apoptosis should be initiated. In the context of type 1 diabetes, induction of an apoptotic response would be predicted to limit further inflammation and may afford protection to the remaining β cells in the islet from additional inflammation. In summary, our findings are consistent with nitric oxide functioning as a mediator of cytokine-induced cellular damage and as a protective molecule that limits the activation of pathways leading to β-cell apoptosis.
ACKNOWLEDGMENTS
We thank Jennifer A. McGraw for technical assistance and Polly Hansen and Maria Feeney for helpful discussions related to this project.
This work was supported by the following grants: National Institutes of Health DK-052194 and AI-044458 (J.A.C), CA183593 (V.L.T.), and P30DK020595 (K.A.B.) and American Heart Association predoctoral fellowship 14PRE20380585 (B.J.O.).
B.J.O., K.A.B., A.N., N.H., V.L.T., and J.A.C. are responsible for the conception and design of the research; B.J.O. and K.A.B. performed the experiments; B.J.O., K.A.B., A.N., N.H., V.L.T., and J.A.C. analyzed the data; B.J.O., K.A.B., A.N., N.H., V.L.T., and J.A.C. interpreted the results of the experiments; B.J.O., K.A.B., A.N., and J.A.C. prepared the figures; B.J.O. and J.A.C. drafted the manuscript; B.J.O., K.A.B., A.N., N.H., V.L.T., and J.A.C. edited and revised the manuscript; B.J.O., K.A.B., A.N., N.H., V.L.T., and J.A.C. approved the final version of the manuscript.
We declare that we have no conflict of interest.
REFERENCES
- 1.Gepts W. 1965. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 2.Mandrup-Poulsen T, Bendtzen K, Nerup J, Dinarello CA, Svenson M, Nielsen JH. 1986. Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia 29:63–67. doi: 10.1007/BF02427283. [DOI] [PubMed] [Google Scholar]
- 3.Mandrup-Poulsen T, Bendtzen K, Nielsen JH, Bendixen G, Nerup J. 1985. Cytokines cause functional and structural damage to isolated islets of Langerhans. Allergy 40:424–429. doi: 10.1111/j.1398-9995.1985.tb02681.x. [DOI] [PubMed] [Google Scholar]
- 4.Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. 2013. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci 1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corbett JA, Lancaster JR Jr, Sweetland MA, McDaniel ML. 1991. Interleukin-1 beta-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 beta-induced inhibition of insulin secretion. J Biol Chem 266:21351–21354. [PubMed] [Google Scholar]
- 6.Southern C, Schulster D, Green IC. 1990. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an l-arginine-dependent nitric oxide generating mechanism. FEBS Lett 276:42–44. doi: 10.1016/0014-5793(90)80502-A. [DOI] [PubMed] [Google Scholar]
- 7.Welsh N, Eizirik DL, Bendtzen K, Sandler S. 1991. Interleukin-1 beta-induced nitric oxide production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology 129:3167–3173. doi: 10.1210/endo-129-6-3167. [DOI] [PubMed] [Google Scholar]
- 8.Corbett JA, McDaniel ML. 1994. Reversibility of interleukin-1 beta-induced islet destruction and dysfunction by the inhibition of nitric oxide synthase. Biochem J 299:719–724. doi: 10.1042/bj2990719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosales AL, Cunningham JM, Bone AJ, Green IC, Green MH. 2004. Repair of cytokine-induced DNA damage in cultured rat islets of Langerhans. Free Radic Res 38:665–674. doi: 10.1080/10715760410001697609. [DOI] [PubMed] [Google Scholar]
- 10.Hughes KJ, Chambers KT, Meares GP, Corbett JA. 2009. Nitric oxides mediates a shift from early necrosis to late apoptosis in cytokine-treated beta-cells that is associated with irreversible DNA damage. Am J Physiol Endocrinol Metab 297:E1187–E1196. doi: 10.1152/ajpendo.00214.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scarim AL, Heitmeier MR, Corbett JA. 1997. Irreversible inhibition of metabolic function and islet destruction after a 36-hour exposure to interleukin-1beta. Endocrinology 138:5301–5307. [DOI] [PubMed] [Google Scholar]
- 12.Eizirik DL, Mandrup-Poulsen T. 2001. A choice of death—the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44:2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 13.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos WP, Kaina B. 2013. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 16.Delaney CA, Green MH, Lowe JE, Green IC. 1993. Endogenous nitric oxide induced by interleukin-1 beta in rat islets of Langerhans and HIT-T15 cells causes significant DNA damage as measured by the ‘comet' assay. FEBS Lett 333:291–295. doi: 10.1016/0014-5793(93)80673-I. [DOI] [PubMed] [Google Scholar]
- 17.Fehsel K, Jalowy A, Qi S, Burkart V, Hartmann B, Kolb H. 1993. Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes 42:496–500. [DOI] [PubMed] [Google Scholar]
- 18.Oleson BJ, Broniowska KA, Schreiber KH, Tarakanova VL, Corbett JA. 2014. Nitric oxide induces ataxia telangiectasia mutated (ATM) protein-dependent gammaH2AX protein formation in pancreatic beta cells. J Biol Chem 289:11454–11464. doi: 10.1074/jbc.M113.531228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. 1999. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res 424:37–49. doi: 10.1016/S0027-5107(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 20.Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, Scharfmann R. 2011. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest 121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly CB, Blair LA, Corbett JA, Scarim AL. 2003. Isolation of islets of Langerhans from rodent pancreas. Methods Mol Med 83:3–14. [DOI] [PubMed] [Google Scholar]
- 22.Freudenburg W, Moran JM, Lents NH, Baldassare JJ, Buller RM, Corbett JA. 2010. Phosphatidylinositol 3-kinase regulates macrophage responses to double-stranded RNA and encephalomyocarditis virus. J Innate Immun 2:77–86. doi: 10.1159/000243785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meares GP, Hughes KJ, Naatz A, Papa FR, Urano F, Hansen PA, Benveniste EN, Corbett JA. 2011. IRE1-dependent activation of AMPK in response to nitric oxide. Mol Cell Biol 31:4286–4297. doi: 10.1128/MCB.05668-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oleson BJ, McGraw JA, Broniowska KA, Annamalai M, Chen J, Bushkofsky JR, Davis DB, Corbett JA, Mathews CE. 2015. Distinct differences in the responses of the human pancreatic beta-cell line EndoC-betaH1 and human islets to proinflammatory cytokines. Am J Physiol Regul Integr Comp Physiol 309:R525–R534. doi: 10.1152/ajpregu.00544.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes KJ, Meares GP, Hansen PA, Corbett JA. 2011. FoxO1 and SIRT1 regulate beta-cell responses to nitric oxide. J Biol Chem 286:8338–8348. doi: 10.1074/jbc.M110.204768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khan P, Idrees D, Moxley MA, Corbett JA, Ahmad F, von Figura G, Sly WS, Waheed A, Hassan MI. 2014. Luminol-based chemiluminescent signals: clinical and non-clinical application and future uses. Appl Biochem Biotechnol 173:333–355. doi: 10.1007/s12010-014-0850-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. 1982. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–138. doi: 10.1016/0003-2697(82)90118-X. [DOI] [PubMed] [Google Scholar]
- 28.Olive PL, Banath JP. 2006. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 29.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 30.Keefer LK, Nims RW, Davies KM, Wink DA. 1996. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol 268:281–293. doi: 10.1016/S0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 31.Cetkovic-Cvrlje M, Eizirik DL. 1994. TNF-alpha and IFN-gamma potentiate the deleterious effects of IL-1 beta on mouse pancreatic islets mainly via generation of nitric oxide. Cytokine 6:399–406. doi: 10.1016/1043-4666(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 32.Heitmeier MR, Scarim AL, Corbett JA. 1997. Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J Biol Chem 272:13697–13704. doi: 10.1074/jbc.272.21.13697. [DOI] [PubMed] [Google Scholar]
- 33.Hughes MN. 2008. Chemistry of nitric oxide and related species. Methods Enzymol 436:3–19. doi: 10.1016/S0076-6879(08)36001-7. [DOI] [PubMed] [Google Scholar]
- 34.Szabo C, Ischiropoulos H, Radi R. 2007. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 35.Broniowska KA, Mathews CE, Corbett JA. 2013. Do beta-cells generate peroxynitrite in response to cytokine treatment? J Biol Chem 288:36567–36578. doi: 10.1074/jbc.M113.522243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pommier Y. 2006. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 37.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. 2006. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol 8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 38.Meares GP, Hughes KJ, Jaimes KF, Salvatori AS, Rhodes CJ, Corbett JA. 2010. AMP-activated protein kinase attenuates nitric oxide-induced beta-cell death. J Biol Chem 285:3191–3200. doi: 10.1074/jbc.M109.047365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo X, Kraus WL. 2012. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ha HC, Snyder SH. 1999. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meares GP, Fontanilla D, Broniowska KA, Andreone T, Lancaster JR Jr, Corbett JA. 2013. Differential responses of pancreatic beta-cells to ROS and RNS. Am J Physiol Endocrinol Metab 304:E614–E622. doi: 10.1152/ajpendo.00424.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clemons NJ, McColl KE, Fitzgerald RC. 2007. Nitric oxide and acid induce double-strand DNA breaks in Barrett's esophagus carcinogenesis via distinct mechanisms. Gastroenterology 133:1198–1209. doi: 10.1053/j.gastro.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 43.Murata M, Thanan R, Ma N, Kawanishi S. 2012. Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J Biomed Biotechnol 2012:623019. doi: 10.1155/2012/623019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka T, Kurose A, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. 2006. Nitrogen oxide-releasing aspirin induces histone H2AX phosphorylation, ATM activation and apoptosis preferentially in S-phase cells: involvement of reactive oxygen species. Cell Cycle 5:1669–1674. doi: 10.4161/cc.5.15.3100. [DOI] [PubMed] [Google Scholar]
- 45.Yang YC, Chou HY, Shen TL, Chang WJ, Tai PH, Li TK. 2013. Topoisomerase II-mediated DNA cleavage and mutagenesis activated by nitric oxide underlie the inflammation-associated tumorigenesis. Antioxid Redox Signal 18:1129–1140. doi: 10.1089/ars.2012.4620. [DOI] [PubMed] [Google Scholar]
- 46.Reference deleted.
- 47.Zong WX, Thompson CB. 2006. Necrotic death as a cell fate. Genes Dev 20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 48.Reference deleted.
- 49.Broniowska KA, Oleson BJ, McGraw J, Naatz A, Mathews CE, Corbett JA. 2015. How the location of superoxide generation influences the beta-cell response to nitric oxide. J Biol Chem 290:7952–7960. doi: 10.1074/jbc.M114.627869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurzov EN, Eizirik DL. 2011. Bcl-2 proteins in diabetes: mitochondrial pathways of beta-cell death and dysfunction. Trends Cell Biol 21:424–431. doi: 10.1016/j.tcb.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Chambers KT, Unverferth JA, Weber SM, Wek RC, Urano F, Corbett JA. 2008. The role of nitric oxide and the unfolded protein response in cytokine-induced beta-cell death. Diabetes 57:124–132. doi: 10.2337/db07-0944. [DOI] [PubMed] [Google Scholar]
- 52.Scarim AL, Nishimoto SY, Weber SM, Corbett JA. 2003. Role for c-Jun N-terminal kinase in beta-cell recovery from nitric oxide-mediated damage. Endocrinology 144:3415–3422. doi: 10.1210/en.2002-0112. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Billiar TR, Talanian RV, Kim YM. 1997. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun 240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 54.Mohr S, Zech B, Lapetina EG, Brune B. 1997. Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem Biophys Res Commun 238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]