

Supplemental digital content is available in the text.
Abstract
Background
Primary sclerosing cholangitis (PSC) is 1 of the leading causes of liver transplantation (LTX) in Scandinavia, and an increasing number of PSC patients have been transplanted in Norway during the last 2 decades. This trend is partly attributable to the recently established practice in Norway of offering LTX to PSC patients with cholangiocellular dysplasia. Based on the controversy associated with this practice, we herein aimed to report the main features and outcomes of our LTX program in PSC.
Methods
The primary indication for LTX (quality of life/end-stage liver disease or suspected neoplasia) was retrospectively determined for 222 patients undergoing LTX for PSC or other autoimmune liver diseases (primary biliary cirrhosis/autoimmune hepatitis) with at least 5 years of follow-up.
Results
In PSC patients impaired quality of life (43.5%) and end-stage liver disease (38.4%) were the most frequent indications for LTX, whereas suspected neoplasia accounted for 18.1%. The proportion of PSC patients with manifest encephalopathy, variceal bleeding, or ascites declined over time. In patients with suspected neoplasia as the primary indication for LTX (n = 25), neoplasia was confirmed in the explanted liver in 20 patients (80%). Five-year survival rates for PSC patients transplanted between 2001 and 2009 were 91.9% for patients receiving LTX due to impaired quality of life or end-stage liver disease and 83.3% for suspected neoplasia.
Conclusions
The PSC patients are increasingly listed for LTX at an earlier stage of their liver disease. In patients with suspected neoplasia before LTX, 5-year survival was acceptable, despite confirmation of neoplasia in 80% of the liver explants.
Primary sclerosing cholangitis (PSC) is a chronic inflammatory disease leading to concentric fibrosis of the intrahepatic and extrahepatic bile ducts.1 No medical therapy has so far proven to delay disease progression,2 and more than half of the patients will face an endpoint of liver transplantation (LTX) or death within 12 to 21 years after diagnosis.3,4 The high lifetime cumulative risk of developing cholangiocarcinoma (10-15%) in PSC further complicates the timing of LTX and accounts for death in almost half of the cases.5 The etiology of PSC is unknown,6 but like in primary biliary cirrhosis (PBC) and autoimmune hepatitis (AIH), an autoimmune component likely contributes to disease development.4,7 PSC is strongly associated with inflammatory bowel disease, predominantly in the form of ulcerative colitis affecting up to 70% to 80% of the PSC patients in Western countries.1,4 The rate of PSC progression is highly variable, and outcome prediction at the individual level is virtually impossible.2
In the Nordic countries, PSC is one of the leading indications for LTX, accounting for 15.2% of all liver transplants performed from 1982 to 2014 (www.scandiatransplant.org). Over the same period, the other autoimmune liver diseases, that is, PBC and AIH, were the primary indication in 9.0% and 4.2% of the recipients, respectively. In Norway, there has been a gradual increase in the annual number of patients transplanted due to PSC.8 Part of this is attributable to the recently established practice of offering LTX to PSC patients with confirmed cholangiocellular dysplasia in biliary brush cytology specimens.9-11 Median waiting time after listing for LTX in Norway is short (an overall median of 24 days for the period of 2005-2008),8 and the model of end-stage liver disease (MELD) score has so far not been used for organ allocation. According to the current Norwegian clinical practice, PSC patients are accepted for LTX either due to (1) significant impairment of quality of life in noncirrhotic patients (ie, severe recurrent bacterial cholangitis, asthenia, or debilitating pruritus), (2) end-stage liver disease (ie, cirrhosis, including overt encephalopathy, intractable ascites, and/or esophageal variceal bleeding), or (3) suspicion of bile duct neoplasia (ie, dysplasia as detected on brush cytology) without intrahepatic or extrahepatic malignancy according to relevant imaging modalities, and (4) selected cases of hilar cholangiocarcinoma (ie, according to the Mayo protocol, which was implemented at our clinic after the current study period).12
On the background of ongoing discussions as to the utility of a MELD-based allocation system for LTX in patients with PSC,13-16 as well the controversy related to the management of cholangiocellular dysplasia in the absence of suspected cholangiocarcinoma (ie, because up to one third of PSC patients may show such features17-19), we aimed to report on the main features of our practice on LTX in PSC and the observed outcomes.
MATERIALS AND METHODS
We aimed to investigate all patients that had undergone LTX for PSC with a minimum of 5 years follow-up. Patients transplanted between 1984 and the end of 2009 were included. As a comparison group, we also included patients undergoing LTX for the other autoimmune liver diseases (PBC and AIH). Between 1984 and 2009, a total of 252 patients underwent LTX due to PSC, PBC, or AIH at Oslo University Hospital Rikshospitalet, Oslo, Norway. From this cohort, 222 patients with PSC (n = 138), PBC (n = 59), and AIH (n = 25) were included in a retrospective, observational study design. End of follow-up was December 31, 2014. Twenty-five patients were excluded due to lack of written consent, and 5 patients because of insufficient clinical information for the retrospective establishing of an unambiguous primary indication for LTX. Patient data were obtained from the hospital patient records and the Nordic Liver Transplantation Registry (www.scandiatransplant.org/members/nltr), and the study was approved by the regional committee for research ethics.
The primary indication for LTX was (1) significant impairment of quality of life in patients without end-stage liver disease, including recurrent bacterial cholangitis in patients with PSC (all having undergone relevant endoscopic treatment attempts); (2) end-stage liver disease; (3) suspected neoplasia. Considerations in patients in whom hepatobiliary neoplasia pre-LTX was suspected were made by multidisciplinary discussions of compound information, including biliary brush cytology investigations (when ERCP was otherwise indicated), serum carbohydrate antigen 19-9 measurements, and radiological imaging (including a relevant selection of ultrasound, magnetic resonance imaging, computed tomography, endoscopic retrograde cholangiopancreatography, or positron emission tomography using 2-[18F]fluoro-2-deoxy-d-glucose) as previously described.10 Unsuspected hepatobiliary neoplasia was defined as a hepatobiliary neoplasia described in the routine reports on the histological examination of the explanted liver from patients where preoperative investigation had raised no such suspicion. Confirmed cholangiocarcinoma was considered a contraindication for LTX.
The MELD score was calculated for patients undergoing LTX after 1999, when the international normalized ratio was implemented in clinical assessments at our center. On instances where the routine brush cytology reports indicated elements of more than 1 grade of dysplasia (ie, moderate to high grade dysplasia17), the highest reported grading was used in the statistical analysis. All patients undergoing LTX in Norway are followed up at the Oslo University Hospital Rikshospitalet. The social welfare system in Norway is well developed and robust, ensuring all patients the benefit of follow-up at regular intervals as well as reliable and largely free access to medications or hospitalization when needed. Mortality data in the Nordic Liver Transplantation Registry are updated annually and guarantees that 100% of all deaths are recorded. No patients were lost to follow-up.
Statistical Analysis
All statistical analyses were performed using IBM SPSS Statistics v21.0 (IBM, Chicago, IL). Comparisons of continuous variables were done using the Mann-Whitney U test. P values less than 0.05 were considered statistically significant. Survival rates were estimated by Kaplan-Meier analysis and restricted to patients transplanted between 2001 and 2009. This restriction was made to (a) allow all patients at least 5 years of follow-up and (b) to avoid bias related to changes in operative technique (eg, in 2000, the piggyback technique and temporary portocaval shunting was introduced).8
RESULTS
One hundred thirty-eight PSC patients who received a liver allograft in Norway between 1984 and 2009 were included in the final analysis, together with a comparison group of 84 consecutive patients transplanted at our center for PBC (n = 59) or AIH (n = 25) during the same period (Table S1, SDC, http://links.lww.com/TXD/A14). As of December 31, 2014, all 222 patients had at least 5 years of follow-up and a total of 72 events (death [n = 72] or retransplantation [n = 37]) had occurred. Twenty-two PSC patients, 9 PBC patients, and 6 AIH patients received at least 1 retransplantation before December 31, 2014. Median age at the time of listing for PSC patients was 43.0 years (range, 17-71) for PBC patients 55.2 (range, 24-69) and for AIH 42.6 years (range, 19-69), respectively (Table S1, SDC, http://links.lww.com/TXD/A14). This remained stable during the observed periods for all 3 groups. Complete clinical and biochemical data for calculation of MELD scores were available for 128 patients (91 patients with PSC, 24 with PBC, and 13 with AIH). All these patients were transplanted after 1999 and with a median MELD score of 15.0 (range, 6-40), 15.0 (range, 6-29), and 15.0 (range, 9-32) for PSC, PBC, and AIH, respectively. For PSC patients transplanted due to end-stage liver disease, the median MELD score (19.0 [range, 7-40]) was significantly higher than that for patients transplanted due to impaired quality of life (14.0 [range, 6-26], P < 0.001) or patients transplanted because of suspected neoplasia (10.0 [range, 6-27], P < 0.001). There was no significant difference in MELD score between PSC patients transplanted because of impaired quality of life and patients transplanted because of suspected neoplasia (P = 0.15).
Characteristics of Transplanted PSC Patients
The PSC patients transplanted without suspicion of malignancy/dysplasia, that is, on the indication of impaired quality of life (43.5%) or end-stage liver disease (38.4%), altogether accounted for 81.9% (n = 113) of the cases, whereas patients with suspected malignancy/dysplasia accounted for the remaining 18.1% (n = 25) (Table 1). Intractable itching was the major indication for LTX in 7 of the 60 PSC patients in the impaired quality of life group, and severe itching was present, but not the main indication for LTX in 19 additional patients. Recurrent cholangitis was the main indication for LTX in 24 of the patients in the impaired quality of life group.
TABLE 1.
Main indication for liver transplantation in patients with PSC in Norway (Stratified by period)

All PBC patients were transplanted on the basis of end-stage liver disease (n = 36) or impaired quality of life (n = 23). All patients with AIH (n = 25) were transplanted due to end-stage liver disease (Table S2, SDC, http://links.lww.com/TXD/A14). No patients with PBC or AIH were listed for LTX due to suspected malignancy.
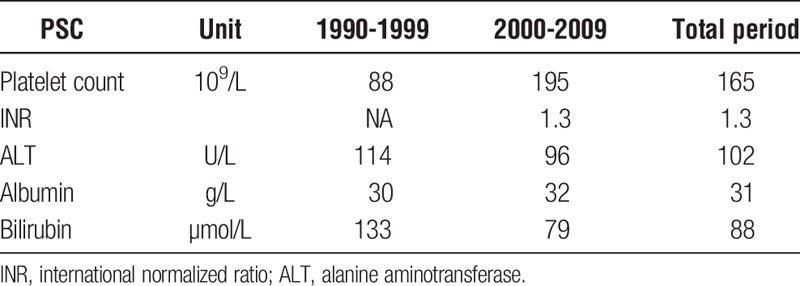
Encephalopathy was overall rare in the PSC group, and only 8.1% of the patients had encephalopathy grade 2 or higher at the time of acceptance for LTX (Table 2), whereas the corresponding figures for PBC and AIH patients were 25.0% and 47.6%, respectively (Table S3, SDC, http://links.lww.com/TXD/A14). We observed a decrease in PSC patients with encephalopathy grade 2 or higher, from 12.1% in the period 1990 to 1999 to 7.0% in the period 2000 to 2009 (P = 0.35) (Table 2). A history of variceal bleeding was recorded in 29.6% of the PSC patients during 1990 to 1999 compared with 18.0% during 2000 to 2009 (P = 0.18). In the AIH group, the tendency was similar, whereas, for PBC patients, there was a little difference between the periods (Table S3, SDC, http://links.lww.com/TXD/A14). Ascites was recorded in 46.7% of the PSC patients during 1990 to 1999 compared with 31.0% between 2000 and 2009 (P = 0.11). In accordance with these clinical observations, we also observed an increase in median platelet count (as a proxy for splenomegaly) and a decrease in median bilirubin at time of LTX (Table 3). Median albumin levels were below the normal range and stable throughout the different time periods (Table 3).
TABLE 2.
Clinical characteristics of patients with PSC at acceptance for liver transplantation (Stratified by period)

TABLE 3.
Biochemistry tests (Median values) at time of acceptance for liver transplantation in patients with PSC (Stratified by period)
LTX and Neoplasia in PSC Patients
Suspected neoplasia was the primary indication for LTX in 25 (18.1%) of the PSC patients. One of these patients had confirmed hepatocellular carcinoma, and another was diagnosed with carcinoma of the cystic duct after cholecystectomy before transplantation. For the remaining patients, suspicion was based on findings on biliary brush cytology and included parallel considerations of serum carbohydrate antigen 19-9 levels and radiological imaging. According to the routine pathology reports, histopathological examination confirmed malignancy in 20 of the 25 liver explants (6 cholangiocarcinomas, 1 hepatocellular carcinoma, 1 carcinoma in situ, and 12 with dysplasia of the bile ducts (Figure 1). For an additional 13 PSC patients, suspicion of neoplasia was raised during the final stage of medical examinations, but where the primary indication for LTX was impaired quality of life (n = 8) or end-stage liver disease (n = 5). Histopathological examination of these 13 liver explants reported no malignancy in 8 patients, whereas 5 patients had various levels of neoplastic changes: 2 cholangiocarcinomas, 1 adenocarcinoma of the gall bladder, 1 hepatocellular carcinoma, and 1 carcinoma in situ of the bile duct epithelium. In 3 additional PSC patients, neoplastic changes were incidentally discovered on histological examination of the explanted liver without any pre-LTX suspicion recorded (1 gallbladder carcinoma, 1 carcinoma in situ of the bile duct, and 1 low-grade dysplasia).
FIGURE 1.
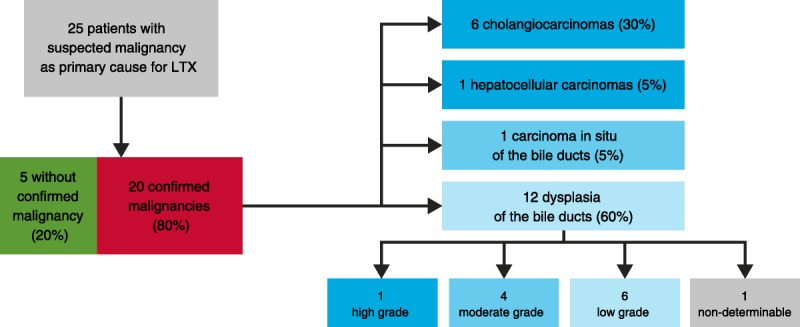
The relationship between pre- and post-pathological findings in patients with PSC undergoing LTX with a primary indication of suspected early stage neoplasia (n = 25).
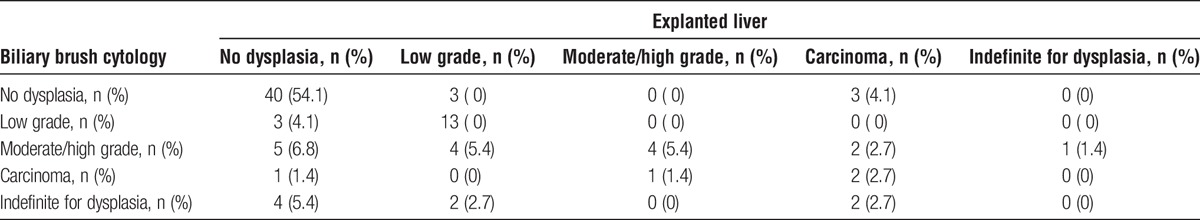
Pre-LTX routine biliary brush cytology reports were available for 74 of the 138 transplanted PSC patients in this series. Pre- and post-LTX findings of neoplasia on routine histopathological assessment in these instances are compared in Table 4. Sensitivity, specificity, positive, and negative predictive values (excluding cases indefinite for dysplasia) of brush cytology in diagnosis of neoplasia were 81.3%, 81.6%, 59.0%, and 93.0%, respectively. If considering only moderate-/high-grade dysplasia and carcinoma in situ as indicative for neoplasia, sensitivity and positive predictive value were 81.3% and 68.4%, respectively, whereas negative predictive value and specificity were 93.0% and 86.9%, respectively. If considering only high-grade dysplasia or carcinoma in situ as indicative of neoplasia, sensitivity, and positive predictive value were 72.7% and 80.0%, respectively, whereas negative predictive value and specificity were 93.0% and 95.2%, respectively.
TABLE 4.
Relationship between neoplastic changes found on routine histopathological investigations of biliary brush cytology specimens and the explanted liver for the 74 patients with primary sclerosing cholangitis for whom complete pretransplant and posttransplant pathology reports were available
Patient Outcomes After LTX During 2001 to 2009
Figure S1, SDC, http://links.lww.com/TXD/A14 shows the Kaplan-Meier plots for all 3 groups of PSC patients according to patient survival. One- and 5-year patient survival rates for PSC patients (n = 74) transplanted on the basis of impaired quality of life or end-stage liver disease were 94.6% and 91.9%, respectively. One- and 5-year patient survival rates were marginally better for patients transplanted on the basis of impaired quality of life (97.4% and 94.9%, respectively) than for end-stage liver disease (91.4% and 88.6%, respectively). One- and 5-year patient survival rates for the 24 PSC patients with pre-LTX suspicion of neoplasia transplanted between 2001 and 2009 were 95.8% and 83.3%, respectively (Figure S2, SDC, http://links.lww.com/TXD/A14). Seven deaths occurred in this group during the observation period; 3 from cholangiocarcinoma, 1 from pancreatic cancer, 1 from CNS infection, 1 from sepsis, and 1 from pulmonary infection.
One- and 5-year graft survival rates for patients with pre-LTX suspicion of neoplasia were 95.8% and 74.1%, respectively. When including only patients transplanted on indication of pre-LTX suspicion of neoplasia with confirmed malignancy (n = 18) in the survival analysis, 1- and 5-year patient survival rates was 94.4% and 77.8%.
One- and 5-year graft survival rates for patients transplanted on the basis of end-stage liver disease were 88.6% and 80% and for patients transplanted on the basis of impaired quality of life corresponding figures were 97.4% and 89.7%, respectively.
One- and 5-year patient survival rates in the PBC group (n = 24) were 95.8% and 91.7%, respectively. One- and 5-year patient survival rates for patients transplanted on the basis of AIH (n = 17) were 88.2% and 88.2% at both time points (Figure S3, SDC, http://links.lww.com/TXD/A14).
Retransplantations
The overall retransplantation rates for PSC, PBC, and AIH at the end of follow-up were 15.9%, 13.5%, and 24.0%, respectively. For PSC patients, the primary indications for retransplantation were recurrent disease (n = 10), biliary strictures without a firm diagnosis of recurrent disease or arterial insufficiency (n = 5), vascular complications (n = 6), and chronic allograft rejection (n = 1). For PBC patients, the primary indications for retransplantation were vascular complications (n = 4), biliary strictures without a firm diagnosis of arterial insufficiency (n = 5), ABO-incompatibility (n = 1), and chronic allograft rejection (n = 1). For AIH, the primary indications for retransplantations were vascular complications (n = 4) and biliary strictures without a firm diagnosis of arterial insufficiency (n = 2).
DISCUSSION
The purpose of the present report was to descriptively summarize the (real-world) experience on LTX in PSC in Norway. Over the years, PSC patients have been transplanted at an increasingly earlier disease stage, a trend not observed for patients with other autoimmune liver diseases (PBC and AIH). Generally, more than half of the PSC patients are now transplanted before end-stage liver disease, including LTX on the basis of biliary dysplasia and suspected malignancy (18.1% of the total LTX volume for PSC from 1984 to 2009).
The Nordic countries have generally had a favorable organ donor situation with short waiting list; and in 2013, the median waiting time for first LTX was 41 days excluding urgent call listings.20 This is the reason why MELD scores are currently not used to prioritize patients for LTX in Norway and likely an important factor behind the excellent patient survival rates. Classification of patients on the waiting list according to MELD score leads to prioritization of patients with the most advanced liver disease for LTX. However, high MELD scores are also associated with increased perioperative and postoperative mortality, and a decline in survival after clinical implementation of the MELD criteria has been suggested by some authors.21 We observed favorable survival rates for patients transplanted on the basis of impaired quality of life compared with end stage liver disease, supporting notions that advanced cirrhosis is an independent risk factor for post-LTX mortality in patients with PSC.22 Survival after LTX for biliary dysplasia is acceptable; however, several types of studies are necessary to corroborate these results and justify general implementation (eg, definition of robust markers for dysplasia detection, detailed studies on the natural history of dysplasia in PSC, controlled studies evaluating the optimal timing for transplantation, and identification of which PSC patients with evidence of cholangiocellular dysplasia in whom LTX may not be justified). For both PBC and AIH patients, the survival outcomes compare favorably to a recent study based on data from the European Liver Transplant Registry.23
The variability of disease course in PSC, especially the high lifetime risk of cholangiocarcinoma, makes optimal timing of LTX difficult. Multiple prognostic models regarding PSC patients have been published.2,24-26 Although useful for patient stratification on the group level, clinical practice guidelines advise against use of prognostic models in individual patients.27 A strong relationship between bile duct dysplasia and the presence of cholangiocarcinoma has been suggested.17 In this study, no relationship between disease stage and dysplasia was observed, showing that prognostic models for cirrhosis development in PSC have little relevance in forecasting malignancies. The most commonly used method for early cholangiocarcinoma detection in PSC is assessment of brush cytology specimens, obtained at endoscopic retrograde cholangiography by brushing in a region of a suspicious stricture.11 No prospectively validated screening algorithm for early cholangiocarcinoma detection in PSC has so far been established.2,28 The problem with the method is the variable sensitivity2 and the lack of consensus of appropriate management (ranging from LTX to observation). Comparing real-life (routine pathology reports) brush cytology findings with the post-LTX histopathology reports yielded sensitivity and specificity for the brush cytology evaluation of 81.3% and 81.6%, respectively. When considering only high-grade dysplasia or carcinoma in situ as indicative of neoplasia, sensitivity and specificity were 72.7% and 95.2%, respectively.
In Norway, PSC patients are now accepted for LTX on definitive detection of dysplasia in brush cytology samples. Liver transplantation on the basis of biliary dysplasia has been routine practice at our center since 2000, and we are still one of the few transplant centers worldwide with this practice. Decisions to list patients are taken at weekly consensus meetings including surgical, medical, and radiological expertise. The considerations made for recommending transplantation are also thoroughly discussed with the patients. In the present study population, we found that this real-life practice included cases of compound assessments, and even 1 case with histological findings of carcinoma after cholecystectomy. The groups are too small to generate survival rates for different grades of dysplasia in this series, but the overall 5-year survival rate of 83.3% for patients with a primary indication of suspected neoplasia is good and exceeding results after LTX for manifest cholangiocarcinoma after neoadjuvant chemoirradiation and brachytherapy.29 Manifest cholangiocarcinoma is generally associated with poor post-LTX survival and a contraindication for LTX,30 also in Norway, and delineating this group of patients from those with early neoplasia warranting LTX remains a major challenge.
A weakness of the study is the relative low number of patients, with a total of 138 patients with PSC alongside 84 patients with AIH or PBC for comparison. The small study size precludes controlling for all potential covariables and confounders in the analysis. As an example, comprehensive information on ursodeoxycholic acid treatment in patients with PSC and PBC or the effect of treatment with prednisone and azathioprine in patients with AIH were not available. This is mainly because the initial assessment and follow-up, in the majority of the patients, was done by the local hospital. Furthermore, the primary indication for LTX was established through a retrospective investigation of patient records rather than prospectively, and is thus potentially subjected to observer bias. Finally and notably, it is impossible to know how many of the PSC patients transplanted on the basis of dysplastic changes on brush cytology that would eventually have developed cholangiocarcinoma and for whom the procedure was therefore justified. Although it is generally believed that dysplasia precedes cholangiocarcinoma in PSC patients,31,32 there is no published data to document the natural history of cholangiocellular dysplasia PSC patients. All weaknesses aside, we believe the data provide an impetus for further research into the optimal timing for LTX in PSC, particularly in the context of biliary dysplasia.
In summary, by analyzing outcome for patients undergoing LTX for PSC in Norway in the period 1984 to 2009, we are able to demonstrate substantial changes in patterns of indications. Almost one fifth of the PSC patients are now being transplanted on the indication of cholangiocellular dysplasia. The relatively good long-term outcome after LTX for biliary dysplasia observed in the present retrospective series warrants further elaboration of the topic in prospective and controlled studies.
ACKNOWLEDGMENTS
The authors thank Stein Foss and the liver transplant coordinators in Oslo for valuable support in the continuous collection of data for the Nordic Liver Transplant registry. The authors are grateful to the Scandiatransplant team in Aarhus for technical maintenance of the system, Frank Pedersen, Ilse Duus Weinreich and Christian Mondrup in particular. Øystein Horgmo is acknowledged for kind assistance in preparing Figure 1.
Footnotes
The authors declare no conflicts of interest.
I.M.A., B.F., and T.H.K. participated in research design, writing of the article, performance of the research and participated in the data analysis. K.M.B., E.M., and E.S. participated in writing of the article and in the data analysis. O.P.F.C, P.J., P.D.L, and A.F. participated in the data analysis.
I.M.A. and B.F. contributed equally to this manuscript.
Supplemental digital content (SDC) is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal’s Web site (www.transplantationdirect.com).
REFERENCES
- 1. Hirschfield GM, Karlsen TH, Lindor KD, et al. Primary sclerosing cholangitis. Lancet. 2013; 382: 1587– 1599. [DOI] [PubMed] [Google Scholar]
- 2. Karlsen TH, Vesterhus M, Boberg KM. Review article: controversies in the management of primary biliary cirrhosis and primary sclerosing cholangitis. Aliment Pharmacol Ther. 2014; 39: 282– 301. [DOI] [PubMed] [Google Scholar]
- 3. Boonstra K, Weersma RK, van Erpecum KJ, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013; 58: 2045– 2055. [DOI] [PubMed] [Google Scholar]
- 4. Karlsen TH, Schrumpf E, Boberg KM. Update on primary sclerosing cholangitis. Dig Liver Dis. 2010; 42: 390– 400. [DOI] [PubMed] [Google Scholar]
- 5. Bergquist A, Ekbom A, Olsson R, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002; 36: 321– 327. [DOI] [PubMed] [Google Scholar]
- 6. Karlsen TH, Boberg KM. Update on primary sclerosing cholangitis. J Hepatol. 2013; 59: 571– 582. [DOI] [PubMed] [Google Scholar]
- 7. Mells GF, Kaser A, Karlsen TH. Novel insights into autoimmune liver diseases provided by genome-wide association studies. J Autoimmun. 2013; 46: 41– 54. [DOI] [PubMed] [Google Scholar]
- 8. Scholz T, Karlsen TH, Sanengen T, et al. [Liver transplantation in Norway through 25 years]. Tidsskr Nor Laegeforen. 2009; 129: 2587– 2592. [DOI] [PubMed] [Google Scholar]
- 9. Bjøro K, Brandsaeter B, Foss A, et al. Liver transplantation in primary sclerosing cholangitis. Semin Liver Dis. 2006; 26: 69– 79. [DOI] [PubMed] [Google Scholar]
- 10. Bjøro K, Schrumpf E. Liver transplantation for primary sclerosing cholangitis. J Hepatol. 2004; 40: 570– 577. [DOI] [PubMed] [Google Scholar]
- 11. Boberg KM, Jebsen P, Clausen OP, et al. Diagnostic benefit of biliary brush cytology in cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2006; 45: 568– 574. [DOI] [PubMed] [Google Scholar]
- 12. Darwish Murad S, Kim WR, Harnois DM, et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology. 2012; 143: 88– 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldberg DS, Camp A, Martinez-Camacho A, et al. Risk of waitlist mortality in patients with primary sclerosing cholangitis and bacterial cholangitis. Liver Transpl. 2013; 19: 250– 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goldberg DS, French B, Thomasson A, et al. Current trends in living donor liver transplantation for primary sclerosing cholangitis. Transplantation. 2011; 91: 1148– 1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goldberg D, French B, Thomasson A, et al. Waitlist survival of patients with primary sclerosing cholangitis in the model for end-stage liver disease era. Liver Transpl. 2011; 17: 1355– 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taniguchi M. Liver transplantation in the MELD era—analysis of the OPTN/UNOS registry. Clin Transpl. 2012; 41– 65. [PubMed] [Google Scholar]
- 17. Lewis JT, Talwalkar JA, Rosen CB, et al. Precancerous bile duct pathology in end-stage primary sclerosing cholangitis, with and without cholangiocarcinoma. Am J Surg Pathol. 2010; 34: 27– 34. [DOI] [PubMed] [Google Scholar]
- 18. Halme L, Arola J, Numminen K, et al. Biliary dysplasia in patients with primary sclerosing cholangitis: additional value of DNA ploidity. Liver Int. 2012; 32: 783– 789. [DOI] [PubMed] [Google Scholar]
- 19. Bangarulingam SY, Bjornsson E, Enders F, et al. Long-term outcomes of positive fluorescence in situ hybridization tests in primary sclerosing cholangitis. Hepatology. 2010; 51: 174– 180. [DOI] [PubMed] [Google Scholar]
- 20. Fosby B, Melum E, Bjøro K, et al. Liver transplantation in the Nordic countries—an intention to treat and post-transplant analysis from The Nordic Liver Transplant Registry 1982–2013. Scand J Gastroenterol. 2015; 50: 797– 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weismüller TJ, Fikatas P, Schmidt J, et al. Multicentric evaluation of model for end-stage liver disease-based allocation and survival after liver transplantation in Germany—limitations of the ‘sickest first’-concept. Transpl Int. 2011; 24: 91– 99. [DOI] [PubMed] [Google Scholar]
- 22. Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg. 1996; 2: 99– 108. [PubMed] [Google Scholar]
- 23. Schramm C, Bubenheim M, Adam R, et al. Primary liver transplantation for autoimmune hepatitis: a comparative analysis of the European Liver Transplant Registry. Liver Transpl. 2010; 16: 461– 469. [DOI] [PubMed] [Google Scholar]
- 24. Kim WR, Poterucha JJ, Wiesner RH, et al. The relative role of the Child-Pugh classification and the Mayo natural history model in the assessment of survival in patients with primary sclerosing cholangitis. Hepatology. 1999; 29: 1643– 1648. [DOI] [PubMed] [Google Scholar]
- 25. Ricci P, Therneau TM, Malinchoc M, et al. A prognostic model for the outcome of liver transplantation in patients with cholestatic liver disease. Hepatology. 1997; 25: 672– 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boberg KM, Rocca G, Egeland T, et al. Time-dependent Cox regression model is superior in prediction of prognosis in primary sclerosing cholangitis. Hepatology. 2002; 35: 652– 657. [DOI] [PubMed] [Google Scholar]
- 27. Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010; 51: 660– 678. [DOI] [PubMed] [Google Scholar]
- 28. Boberg KM, Lind GE. Primary sclerosing cholangitis and malignancy. Best Pract Res Clin Gastroenterol. 2011; 25: 753– 764. [DOI] [PubMed] [Google Scholar]
- 29. De Vreede I, Steers JL, Burch PA, et al. Prolonged disease-free survival after orthotopic liver transplantation plus adjuvant chemoirradiation for cholangiocarcinoma. Liver Transpl. 2000; 6: 309– 316. [DOI] [PubMed] [Google Scholar]
- 30. Brandsaeter B, Isoniemi H, Broomé U, et al. Liver transplantation for primary sclerosing cholangitis; predictors and consequences of hepatobiliary malignancy. J Hepatol. 2004; 40: 815– 822. [DOI] [PubMed] [Google Scholar]
- 31. Fleming KA, Boberg KM, Glaumann H, et al. Biliary dysplasia as a marker of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2001; 34: 360– 365. [DOI] [PubMed] [Google Scholar]
- 32. Bergquist A, Glaumann H, Stål P, et al. Biliary dysplasia, cell proliferation and nuclear DNA-fragmentation in primary sclerosing cholangitis with and without cholangiocarcinoma. J Intern Med. 2001; 249: 69– 75. [DOI] [PubMed] [Google Scholar]