

Abstract
Posttransplant lymphoproliferative disease (PTLD) is a potentially fatal complication after (solid organ) transplantation, which is highly associated with Epstein-Barr virus (EBV). The EBV-specific cytotoxic T cell response that is essential in controlling the virus in healthy individuals is suppressed in transplant recipients using immunosuppressive drugs. A primary EBV infection in EBV-seronegative patients receiving an EBV-seropositive donor organ or a reactivation in those who are already latently infected pretransplantation can lead to uninhibited growth of EBV-infected B cells and subsequently to PTLD. Effective preventive strategies, such as vaccines and antiviral agents, are lacking. Because not every transplant recipient with increasing EBV viral load develops PTLD, it is hard to decide how intensively these patients should be monitored and how and when a preemptive intervention should take place. There is a need for other tools to help predict the development of PTLD in patients at risk to make timing and strategy of preemptive intervention easier and more reliable.
The cornerstone of the treatment of patients with PTLD is restoring the host's immunity by reduction of immunosuppressive drug therapy. American and British guidelines recommend to add rituximab monotherapy or rituximab in combination with cyclophosphamide, doxorubicin, vincristine, and prednisolone, depending on histology and clinical characteristics. Although response to these therapies is good, toxicity is a problem, and PTLD still has a relatively high mortality rate. An evolving therapy, especially in PTLD occurring in allogeneic stem cell transplantation, is restoring the host's immune response with infusion of EBV-specific cytotoxic T cells. This may also play a role in the future in both prevention and treatment of PTLD in SOT.
Posttransplant lymphoproliferative disease (PTLD) is a life-threatening complication of solid organ transplantation. In the majority of cases, PTLD is associated with active replication of Epstein-Barr virus (EBV) after either primary infection or reactivation during treatment with immunosuppressive drugs. Although the incidence of PTLD is relatively high and its presence is associated with severe complications, there is no consensus on the optimal way to monitor for PTLD, nor are there uniform guidelines for intervention. Here, we provide an overview of the pathogenesis and clinical manifestations. Focusing on EBV-related PTLD, we discuss strategies for monitoring, diagnostic approach, and possibilities for (preemptive) treatment.
The EBV Life Cycle
Epstein-Barr virus is a gamma herpes virus, by which over 90% of the human population is infected.1,2 Primary infection usually occurs at early age. Children infected before the age of 10 are usually asymptomatic or present with upper respiratory tract infection. In adolescents and adults, primary EBV infection frequently presents as the classic syndrome of infectious mononucleosis (IM), characterized by the triad of fever, lymphadenopathy, and pharyngitis.1,3,4
Transmission usually occurs by contact with oral secretions, containing infectious virions.1,3,4 They enter the host through epithelial cells or naive B cells in the oropharynx.1,3-5 Moreover, the virus can be transmitted by blood or via transplanted allogeneic hematopoietic cells or solid donor organs.3
Epithelial cells may be infected directly or from virions at the surface of adjacent infected B cells. This leads to initiation of the lytic phase that is characterized by active replication, production of virions and lysis of the cell.6 The immediate-early lytic epitopes BZLF1 and BRLF1 are the first genes being transcribed and are the key products in initiating this lytic phase by activating viral and certain cellular promoters leading to a cascade of viral gene expression. In this cycle, the EBV genome is amplified more than 100-fold.2 Because this viral replication occurs in the oropharynx, large amounts of virus are shed into the saliva, spreading the virus to new hosts.1,7 The life cycle of EBV differs from those of other herpes viruses in that it has a less effective lytic replication system. The virus preferentially infects naive B cells in Waldeyer's ring, leading to initiation of the latent phase, eventually resulting in persistent viral infection. The lytic phase and latent phase occur at the same time. Additionally, by recirculating through Waldeyer's ring lytic replication can occur in B cells.1,2,6
The latent phase is divided in 3 successive gene transcription programs. Table 1 summarizes the characteristics of the different programs. After the virus has entered the naive B cell via binding of the EBV surface protein gp350 to its receptor CD21 and to HLA class II molecules at the surface membrane, the linear EBV genome of the virus changes into a circular DNA episome. This leads to the initiation of the first transcription program “the growth program” also known as “latency III” in the lymph node compartment, in which B cells are activated to become proliferating blasts1,4,8,9 (see Figure 1). In these lymphoblastoid cell lines, only a limited number of the nearly 100 viral genes that are expressed during the lytic phase appear at the cell surface. These cell surface proteins include 6 EBV nuclear antigens (EBNAs), 3 latent membrane proteins (LMPs), and 2 small EBV-encoded RNAs (EBERs).2,8 EBNA-2 is the master activator for latent gene transcription in this program. By upregulating the expression of LMP-1 and LMP-2, as well as many cellular proteins, EBNA-2 initiates a cascade that eventually leads to the growth and transformation of B cells. LMP-1 inhibits apoptosis and acts as an oncogene, leading to the development of B cell lymphomas when expressed in transgenic mice. It resembles in many aspects a constitutively active form of the B cell surface molecule CD40. By binding to several tumor necrosis factor receptor-associated factors, it activates the nuclear factor-κB pathway, which has a key role in the activation and differentiation of B cells. LMP-2 provides an important survival signal for B cells by mimicking the B cell receptor. However, it also blocks tyrosine kinase phosphorylation, leading to inhibition of the B cell receptor signaling. By this, it prevents unwanted antigen-triggered activation of latently infected B cells that would lead to entrance into the lytic cycle and cause the cell to die.1,4,10
TABLE 1.
Transcription programs in B cells used by EBV

FIGURE 1.
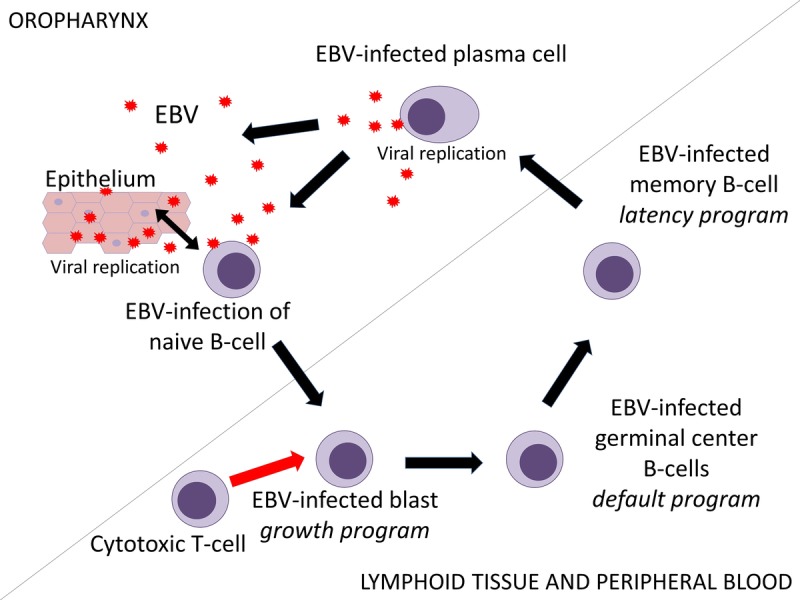
The EBV life cycle. In the oropharynx, epithelial cells and naive B cells can both be infected either directly by Epstein-Barr virions or by each other. Infection of epithelial cells leads to initiation of the lytic phase. At the same time, infection of naive B cells leads to initiation of the latent phase, which exists of 3 successive gene transcription programs: the growth program, the default program, and the latency program, resulting in latently infected memory B cells, in which the virus can persist asymptomatically for life. When these memory cells circulate through Waldeyer's ring, differentiating into plasma cells may lead to reactivation of the virus. The EBV-specific cytotoxic T cells are mostly directed against lymphoblasts expressing the growth program. Decrease in number and function of T cells due to immunosuppressive drugs might lead to uninhibited growth of these cells and eventually to PTLD.
Expression of the different EBV gene products at the surface of the lymphoblasts makes these infected B cells recognizable for cytotoxic T cells, stimulating a strong immune response. During IM in immunocompetent adults, up to 40% or 50% of the circulating cytotoxic T cells are directed against EBV-infected B cells resulting in the classic atypical lymphocytosis.1,4,8 However, this cytotoxic T cell response is insufficient to kill all infected B cells.
In the infected B cells that survive, EBNA-2 expression is switched off, and the virus will switch to the second transcription program, the “default program” or “latency II,” in which only the LMPs are expressed at the cell surface (see Figure 1). LMP-1 and LMP-2 are essential in producing the signals necessary for the blasts to differentiate into memory cells in the germinal center. Next, in these memory cells, the last transcription program, the “latency program” or “latency I,” is initiated, in which the expression of viral proteins is shut down, making the EBV-infected memory cells indistinguishable from the host's other memory cells, rendering them invisible to the immune system, and enabling an asymptomatic existence alongside the host (see Figure 1). During cell division, only the poorly immunogenic EBNA-1 is expressed periodically at the surface. EBNA-1 tethers the EBV genome to the host's chromosomal DNA, ensuring that the viral DNA is also replicated.3,4,7,8
In the latency program, normally, about 1 to 50 per million circulating memory B cells are infected with EBV. In contrast, during the early stages of IM, this number may increase by more than 1000 times. The latently infected memory cells circulate between the peripheral blood and Waldeyer's ring, where they can differentiate into plasma cells, leading to replication of the virus and reinfection of new naive B cells and epithelial cells, thereby initiating the whole process of becoming resting B cells again.3,4,6-8
A delicate balance exists between the host's immune system and the virus. In immunocompetent persons, a significant fraction of the total CD8+ T cell population is directed against EBV-specific epitopes to control the infection. About 0.2% to 2% is directed against lytic epitopes and about 0.05% to 1% against latent epitopes.6 Although this percentage is quite stable over time, an increase in EBV-specific CD8+ T cell response up to 14% is seen in people older than 60 years.6 Much less is known about the CD4 response to EBV infection. In peripheral blood, EBV-specific CD4+ T cells are detectable at a much lower frequency than the EBV-specific CD8+ T cells during primary infection. In the latent phase, the number of EBV-specific CD4+ T cells falls rapidly.6
EBV Infection and PTLD
In solid organ transplantation, EBV-naive patients have a much higher risk of developing PTLD than latently infected patients who are EBV-seropositive at the time of transplantation.
Treatment with immunosuppressive drugs after transplantation in latently infected patients can lead to disruption of the balance between the immune system and the latent virus and subsequently to reactivation of EBV. In immunosuppressed patients, the number of virus-infected memory B cells can be as much as 50 times higher than that in the healthy population. When due to a decrease in number and function of T cells, the lymphoproliferative blasts can no longer be controlled by the EBV-specific cytotoxic T cells, uninhibited growth of these cells may eventually lead to the development of PTLD.4,8,11,12 However, PTLD does not develop in every EBV-seropositive transplant recipient. One explanation is that most infected lymphoblasts transit into resting memory cells, and only cells that are not able to make this transition will start to proliferate freely. This happens when in the presence of a high level of viremia cells other than naive B cells, such as memory B cells and germinal center B cells, incidentally get infected in the Waldeyer's ring. These so-called bystander B cells may express the growth program, but are not able to switch to the default program. Uncontrolled growth of these bystander cells might lead to PTLD and could explain the heterogeneity of the disease. Another potential mechanism of lymphomatous derailment is the growth of memory cells accidently triggered to express the growth program. The latter mechanism is thought to play a role in the development of Burkitt lymphoma. Although most PTLD tumor cells express the growth program, occasionally, also the viral gene expression patterns of the default program and the latency program are seen.4,8
Primary EBV infection increases the chance of developing PTLD 6 to 76 times. The high virion peak in the absence of an adequate immune response gives a massive infection of B cells and possibly bystander B cells, increasing the chance of development of PTLD. The route of transmission can be either via contact with virions in oral secretions or via a donor organ from an EBV-seropositive donor, containing latently infected donor B cells.11,13
Incidence and Risk Factors of EBV-Associated PTLD
PTLD is a relatively common malignant complication after solid organ transplantation. A variation in incidence rates is seen according to the type of organ transplanted, the pretransplant EBV-serostatus, and the age of the organ recipient.13-15
The differences in incidence between the type of donor organ reflect the different levels of immunosuppression required and the amount of lymphoid tissue in the allograft. The highest incidence is found in small intestine transplant recipients (up to 32%), recipients of pancreas, heart, lung, and liver have an intermediate risk (3-12%), and the lowest incidence is found in renal transplant recipients (1-2%).13-15 In general, the incidence in pediatric patients is higher than that in adults due to the higher incidence of pretransplant negative EBV-serostatus among children.13-16 In Europe and the United States over 80% of PTLD cases is EBV-associated.16 The risk of developing an EBV-associated PTLD is highest in the first year posttransplantation due to primary infection of EBV-naive transplant recipients by an EBV-positive transplanted organ and to the occurrence of graft PTLD.17 The majority of cases of PTLD is of recipient's origin; however, PTLD limited to the graft occurs, in general, early after transplantation and is predominantly of donor origin.14
Because of the multidrug immunosuppressive drug regimens in transplant recipients, it has been difficult to clarify the contribution of specific immunosuppressive drugs to the development of PTLD. Cyclosporine-containing regimens were long thought to increase the risk of developing PTLD, but it is now thought that it is the net state of immunosuppression, rather than any individual agent, that determines the incidence. This is an entity difficult to measure.12,17,18 Cytomegalovirus infection also seems to be a risk factor for PTLD, probably due to its immunosuppressive effect.11,12,14,18
Non-EBV–Associated PTLD
Approximately 20% of PTLD cases are not associated with EBV. EBV-negative PTLD typically occurs about 7 to 10 years after transplantation. Because of improved survival of transplant patients, the proportion of late and EBV-negative PTLD seems to increase.14-16,18 The pathogenesis is not well understood. These lymphomas may be coincidental as seen in the nontransplanted population. However, some do seem to respond to reduction of immunosuppression (RIS).16,19,20 Possibly, these lymphomas develop due to a long-lasting deficient immune system under the influence of other as yet unidentified transforming viral agents.20 It has also been hypothesized that initially EBV-driven lymphoproliferations can become EBV-negative when cellular mutations replace the function of the EBV-oncogenes.21
Diagnosis of EBV-Related PTLD
The burden of the disease at the time of diagnosis correlates closely with the prognosis. Therefore, early diagnosis is of crucial importance.22 The diagnosis is made on clinical and histological grounds.
Presenting Features
EBV infection after solid organ transplantation can present as IM with the classic features of fever, pharyngitis, cervical lymphadenopathy, hepatosplenomegaly, and atypical lymphocytosis. It can also manifest itself in a more organ-specific way by hepatitis, pneumonitis, gastrointestinal symptoms, or with hematological manifestations, such as leukopenia, thrombocytopenia, or hemolytic anemia.14
EBV-related PTLD is commonly localized extranodally (>90%), and patients therefore often present with symptoms related to the site of the disease and the type of organ(s) involved.5,14,16,23 However, lymphadenopathy and B-symptoms, such as (night) sweats, weight loss, and fever can also occur.16
Localization
Although PTLD can be localized in any organ, in heart and lung transplant recipients, the lymphoma is more often localized in the lung, and in liver transplant recipients, it is more often localized in the liver. In kidney transplant recipients, with the exception of graft PTLD, the transplanted organ is not the preferential localization, and lymphomas can occur at any site, of which the gastrointestinal tract is the most common (about 15%). Interestingly, in contrast to other solid organ transplant recipients, kidney transplant recipients also relatively often present with a central nervous system localization of the lymphoma.16,17
EBV-Specific Blood Tests
The EBV-serostatus of the recipient and the donor is routinely determined before transplantation by serological assays, detecting antibodies to EBV viral capsid antigen (VCA), early antigen, and EBNA-1.16 Once immunosuppressive therapy has been started, serological tests are no longer considered reliable due to delayed or absent humoral responses. Therefore, serology should not be used as a diagnostic tool for primary EBV infection or PTLD after transplantation.5,14
Quantification of EBV viral load by polymerase chain reaction may be useful for surveillance, diagnostic and disease monitoring of PTLD because an increase in viral load correlates with the development of EBV-related PTLD.14,16,24,25 However, there is a high interlaboratory variability in the qualitative and quantitative test results due to a wide array of commercially and in-house–developed assays.25-27 Changes in viral load in patients measured over time within the same institution may thus be more reliable.25-27 An international standard for calibration, created by the World Health Organization in 2011, aims to reduce the variation among assays.14
Another controversy is whether the viral load should be measured in plasma, in whole blood, or in lymphocytes. Whole blood or lymphocyte EBV viral load monitoring is more sensitive but less specific for the detection of early EBV reactivation as compared with plasma monitoring. The EBV plasma levels correlate better with the presence of EBV-positive PTLD, making the viral load measurement in plasma a more specific test for the detection of PTLD than the measurement in whole blood. However, the drawback of using plasma instead of whole blood is that in rare cases the diagnosis can be missed.14,24,28-30
A promising adjunctive laboratory test improving the specificity is measuring the EBV-specific T cell response. As previously noted, this response is decreased in the immunosuppressed organ transplant recipient. A low EBV-specific T cell response in combination with a high viral load has a high positive predictive value for the development of PTLD.14,31 However, the EBV-specific T cell enzyme-linked immunosorbent spot and tetramer assays to measure the T cell response are complex, costly, and difficult to implement in a routine diagnostic laboratory.14,16,32
Biopsy
Histological examination of tumor tissue is the gold standard for PTLD diagnosis. An excisional biopsy or resection of a whole lymph node is preferred over a core needle biopsy. Core needle biopsies should only be done when it is impossible to obtain an excisional biopsy.5,14-16
The World Health Organization classifies the disease in subgroups defined by whether they arise from a single cell (monoclonal) or multiple cells (polyclonal) (Table 2). The 4 major categories are early lesions, polymorphic PTLD, monomorphic PTLD, and classical Hodgkin lymphoma-type PTLD.33
TABLE 2.
WHO classification of PTLD
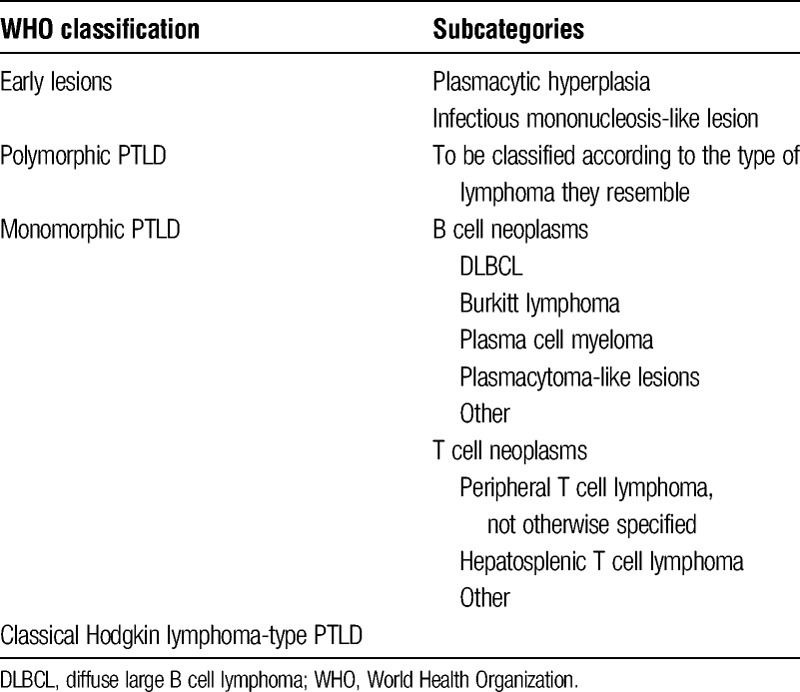
Early lesions are typically seen in the first year posttransplantation and are usually diagnosed in patients with primary EBV infection. This category is subdivided in plasmacytic hyperplasia and IM-like lesions, characterized by polyclonal B cell proliferation.
In the polymorphic category of PTLD, infiltrates are seen composed of a mixture of lymphocytes that may show malignant features, destroying the underlying tissue architecture.
Monomorphic PTLD is the most common category of PTLD in adult patients and is subdivided into 4 main types of PTLD, of which diffuse large B cell lymphoma is seen in the majority of cases. The other types are Burkitt lymphoma, plasma cell myeloma, and plasmacytoma-like lesions. The monomorphic T cell PTLD includes all the T cell and natural killer cell neoplasms. Any type of T cell lymphoma can be seen. T cell PTLD is predominantly EBV-negative. In practice, it may be hard to differentiate between early lesions, polymorphic, and monomorphic PTLD, because they represent a continuous spectrum of disease.
The last group, the classical Hodgkin lymphoma-type PTLD, resembles the histological pattern of a classic Hodgkin lymphoma, including the Reed-Sternberg cells. Both the monomorphic T cell and the classic Hodgkin lymphoma-type PTLD are typically late forms of the disease.16
Imaging
There are no large studies comparing different imaging techniques in PTLD. Contrast-enhanced computed tomography (CT) of the neck, chest, abdomen, and pelvis is helpful in making an initial diagnosis, identifying a site for biopsy, staging of the disease, and monitoring treatment response. Positron emission tomography (PET) has the advantage over CT that it can identify metabolically active sites of the disease that have not yet increased in size and is also better at detecting bone marrow involvement. It therefore increases the sensitivity.
The PET should be combined with CT (PET-CT) to localize the exact site and size of the involved area. The disadvantage of PET is that no distinction can be made between malignancies and any other causes of increased isotope uptake, such as infection or inflammation. In addition, some T cell non-Hodgkin lymphomas and low grade B cell lymphomas are fluorodeoxyglucose (FDG)-negative.14,16,34,35
Depending on signs and symptoms, additional imaging of the central nervous system, gastrointestinal tract, or other organs should be performed. It has been argued that magnetic resonance imaging of the brain should be a part of the initial radiographic work-up.14,23
Prognostic Staging and Scoring of PTLD
No specific staging system exists for PTLD. The Ann Arbor staging system can be used to describe the sites of involvement and the presence of symptoms. Additionally, in all patients, the involvement of the transplanted organ should be evaluated.14,36 There is also no universally accepted scoring system for the prognosis of PTLD. The International Prognostic Index (IPI), used for non-Hodgkin lymphoma in the nontransplant setting, is based on disease stage, performance status, extranodal disease, lactate dehydrogenase (LDH), and age. Factors not included in this score that are associated with a poor prognosis in PTLD include tumor monoclonality, EBV-negative tumor, and graft involvement.22,37,38 Using the IPI-score to predict prognosis showed different outcomes in the few studies that have been done on this subject. The IPI-score alone seems inappropriate in PTLD, and a specific prognostic index should be developed.36,38
Reported survival rates for PTLD are quite variable and range from 40% to 60% at 5 years after diagnosis.17,18,22,23,37,39
Monitoring and Preemptive Intervention in PTLD
Because an increase in viral load generally precedes the development of PTLD, close monitoring of the EBV viral load in pretransplant EBV-seronegative patients with an EBV-seropositive donor organ, the so-called high-risk patients, gives the opportunity to intervene preemptively or at an early stage.14,16,25,40 The optimal frequency of monitoring is not certain. Because the doubling time of the viral load is about 1 to 8 days, weekly to twice weekly monitoring during the first year or at least during the first months after transplantation has been recommended. However, this may be a logistic challenge.14,16,41-43 Little is known about the natural course of an increase in EBV viral load, which makes it hard to determine when intervention should take place. Moreover, no clear cut-off point for a high or increasing EBV viral load can be given because of the high interlaboratory variability in test results, as described above.14,25 Data from the literature point to a positive predictive value of a high EBV load for the development of PTLD ranging from 50% to 65%, depending on the cut-off value used.14,29
The most frequently used preemptive intervention when the viral load becomes positive is RIS, which has been shown to decrease the incidence of PTLD.29 Rituximab, a human-mouse chimeric monoclonal anti-CD20 antibody that causes a depletion of circulating B cells including those infected with EBV, might be of additional value to this strategy. In a recent prospective study in 299 heart transplant recipients, a single dose of rituximab was given to patients with EBV reactivation and viral loads greater than 105 copies/mL, or to patients with primary infection, if they were not responding after 1 month of reduction in immunosuppression. This strategy turned out to be safe and effective.44 However, at present, we feel there is insufficient evidence to support the preemptive use of rituximab or other immuno- or chemotherapy. Considering the low predictive value of EBV load of about 50% to 65% for the development of PTLD, preemptive use of rituximab would lead to unnecessary exposure and thus B cell depletion and increased immunodeficiency in approximately half of the patients.14,29,40,45
Management of EBV Infection and EBV-Associated PTLD in the Solid Organ Transplant Recipient
Reduction of Immunosuppression
As previously noted, immunosuppressive drug therapy in transplant recipients decreases the T cell response necessary to control primary EBV infection or reactivation of a latent EBV infection, potentially resulting in the development of PTLD. Recovery of the host's immune system will allow EBV-specific cytotoxic T cells to proliferate and subsequently control the EBV-driven B cell proliferation. Reduction of immunosuppression has been used for several decades as a first-line approach in the management of PTLD.14,23,36 The response to RIS can be measured by assessing changes in EBV viral load, LDH, clinical symptoms, and tumor size.14,36
Reported response rates have been highly variable, due to both the heterogeneity of the disease and the differences in extent and duration of reduction in immunosuppressive drug therapy in the different studies. In some patients, RIS can lead to complete remission of the disease, and in others, additional treatment is indicated. Factors that are associated with not achieving a complete remission after RIS are: elevated LDH, organ dysfunction, multiorgan involvement, and older age.36,38,46
No consensus exists on which immunosuppressive agents to reduce or stop and which to continue. The American and British guidelines recommend in case of extensive disease to stop all antiproliferative agents, such as azathioprine and mycophenolate, and to continue with prednisolone only. In case of no critical illness, which is not well defined, they recommend to decrease the dose of calcineurin inhibitors (CNIs) by 50% and to maintain prednisolone in a dose of 7.5 to 10 mg per day.14,36 A limitation of this approach is the risk of allograft rejection, which is obviously especially threatening in lung and heart transplant recipients. Therefore, the organ function needs to be monitored weekly, and in some cases, RIS appears not to be possible at all.36 From 2 recently published retrospective French studies, it was concluded that maintenance of calcineurin inhibition in a reduced dose in kidney transplant recipients with PTLD might improve renal graft outcome. However, these trials were retrospective in nature, and the decision to either maintain or discontinue the CNIs was left to the physician. No information on critical illness was given. Therefore, a bias in patient selection cannot be ruled out.47,48
In kidney transplant recipients with cutaneous squamous cell carcinomas or with Kaposi sarcomas, a switch from CNIs to the inhibitor of mammalian target of rapamycin (mTOR) sirolimus was shown to have an antioncogenic effect.49,50 There is some evidence from experimental in vitro and in vivo studies and from some case reports that mTOR inhibition can also be effective in suppressing EBV-related malignancies.51-53 However, when used as maintenance therapy, several outcomes have been reported, including no difference, a decrease, or an increase in incidence of PTLD.54-56 So mTOR inhibitors might be beneficial in the treatment, but not in the prevention of PTLD.
Surgery and Radiotherapy
Surgery and radiotherapy can be used as an adjunctive to RIS in localized PTLD cases, such as PTLD limited to the graft, and can lead to long-term remission. Obviously, resection of the transplanted organ is not an option in tumors involving life-preserving organs, such as the heart.
Furthermore, surgery is important in the management of local complications, such as gastrointestinal bleeding or perforation.14,36
Rituximab Monotherapy and Cytotoxic Chemotherapy
In patients with progressive PTLD despite RIS, additional therapy is necessary. However, the optimal treatment schedule has not yet been established.14,36
Rituximab monotherapy has a high response rate in B cell PTLD, which is superior to response rates reached in aggressive B cell lymphomas in the nontransplant population. The combined overall response rate in 3 prospective phase 2 trials with rituximab monotherapy was 55%. Additionally, no severe toxic effects were reported.14,57 Therefore, this could be used as first-line therapy. However, up to 57% of patients with an initial partial or complete response to rituximab monotherapy showed disease progression at 1 year after treatment. The median overall patient survival in the 3 trials was 14.9 to 42 months.57,58 Risk factors associated with a poor response to rituximab monotherapy are older than 60 years at diagnosis, poor performance status, elevated LDH, and longer time interval between transplantation and PTLD.58
Combination of rituximab and chemotherapy consisting of cyclophosphamide, doxorubicin, vincristine, and prednisolone (CHOP) has shown much better long-term disease control.58 However, the treatment-related mortality (TRM) of CHOP has been reported to be as high as 13% to 50% in this population, which is much higher than in the nontransplant setting.36 In a recent relatively large prospective phase 2 trial, sequential treatment with 4 cycles of rituximab followed by four 3-week cycles of rituximab in combination with CHOP (R-CHOP) led to an increase in response rate to 90%. The median overall survival was 6.6 years, which is longer than in the 3 prospective trials applying 4 to 8 cycles of rituximab monotherapy. Treatment-related mortality was 11%, being lower than previously reported TRM in first-line R-CHOP. This can be explained by the lower tumor mass and by an improved performance status due to the initial treatment with rituximab only.57 A low toxicity was also observed in a multicenter prospective study in a pediatric population, in which low-dose chemotherapy (cyclophosphamide and prednisone) was used with simultaneous rituximab in patients in whom first-line therapy failed. After 2 years, the event-free survival was 71%, and the overall survival was 83%. Of the 10 patients who died, 3 died due to infection while receiving therapy and 7 due to PTLD.59 However, these regimens are still considerably more toxic than rituximab monotherapy, and TRM is also considerably higher than TRM of R-CHOP in the nontransplant setting.
The British guidelines recommend to administer rituximab monotherapy to patients without any of the above mentioned risk factors who fail to respond to RIS, and R-CHOP in patients who fail to achieve an adequate remission despite previous RIS and rituximab monotherapy. They advise to consider first-line treatment with R-CHOP in addition to RIS in patients with high-risk or clinically aggressive lymphoma.36 Sequential therapy with rituximab and R-CHOP might then be a safe and effective option.
Other Strategies
A vaccine for EBV-seronegative transplant recipients would be of great value to prevent PTLD. However, several barriers have thus far hampered the development of a safe and effective vaccine. First, it is not clear which combination of the many viral proteins encoded by EBV is the best to use in a vaccine. In a phase 2 clinical trial including 181 EBV-seronegative healthy adults given either recombinant EBV subunit glycoprotein 350 (gp350) vaccine or placebo, there seemed to be a reduction in the development of IM in the vaccinated group, but not in the rate of EBV infection.60,61 The same was observed for a smaller HLA-B0801–positive EBV-seronegative group vaccinated with an HLA-B0801–restricted CD8 T cell peptide epitope vaccine corresponding to EBV latency protein EBNA-3A.60 Second, it is difficult to develop clinical trials to test the vaccine's effectiveness. There are no good markers to detect the development of EBV-related tumors, and it may take years between primary infection and the development of malignancy. In transplant recipients, the occurrence of tumors is generally faster, but they may be less responsive to immunization before transplantation due to organ disease interfering with the immune response. In a small phase 1 study with the EBV gp350 vaccine given to children with chronic kidney disease awaiting transplantation, immune responses to the vaccine declined rapidly. One of the subjects even developed PTLD 30 to 40 weeks posttransplantion.60,62 Third, animal models to test EBV vaccines are limited, because EBV only infects the human species.60
The role of treatment with antiviral agents, such as acyclovir and ganciclovir, in the prevention of PTLD is uncertain. These drugs can inhibit lytic EBV-DNA replication, but the cells proliferating in PTLD are predominantly latently infected. Still, inhibition of lytic replication may decrease the infection of B cells and subsequently prevent the growth of latently infected B cells and therefore decrease the risk of developing PTLD. Ganciclovir is in vitro more active against EBV than acyclovir and would be the drug of choice.9,14,36,43,63 A concomitant advantage of the use of ganciclovir is that it inhibits the development of CMV infection, which is a cofactor in the development of PTLD.14,43,64 More promising is the addition of pharmacological induction of EBV lytic infection, by, for example, arginine butyrate, making the EBV-infected cells more susceptible to antiviral treatment with ganciclovir.9 However, currently, there is insufficient evidence to support the use of prophylactic antiviral agents.
Another way of restoring the host's immune response to EBV is the administration of EBV-specific cytotoxic T lymphocytes (CTLs).14,36 Because T cell targeting is HLA-specific, the T cells should be HLA-matched to the recipient to be effective and to avoid graft-versus-host disease. Therefore, autologous EBV-CTLs or HLA-matched EBV-CTLs from EBV-seropositive blood donors can be used. This approach has been successfully used in the treatment of PTLD after allogenic stem cell transplantation, and also preliminary results in the solid organ transplant setting are promising. However, the generation and cloning of EBV-CTLs is time-consuming and expensive.65-68 Recently, T cells directed against viral antigens instead of viable viral components have been produced in a more simplified culture technique in 2 to 3 weeks with good results in a phase 1/2 study.69,70 An additional problem of the use of CTLs in solid organ transplant recipients is the need for ongoing immunosuppressive drug therapy limiting the persistence of the CTLs. To address this problem, EBV-CTLs resistant to CNI have recently been developed and appeared successful in mouse models.71,72 If these hurdles can be overcome, in the future, CTLs might also be used in the prevention of PTLD in patients with a high viral load at risk for developing PTLD. Exciting results have also recently been reported for the treatment of B cell malignancies, such as B-acute lymphoblastic lymphoma and B-NHL with chimeric antigen receptor T cells (CAR-T cells). The CAR-T cells are autologous or allogeneic T cells which are transduced with a lentiviral or retroviral CAR. The cells then express an antigen recognition part (originating from an antibody), a signalling part of a T cell and 1 or more costimulatory domains. Thus far, mostly targeting of the B cell antigen CD19 has been explored, but also other tumor- or lineage-specific antigens can be targeted.73 Recently, the use of the bisphosphonate pamidronate was tested in mouse models as a new therapeutic option. Pamidronate can induce the intracellular accumulation of isopentenyl pyrophosphate, leading to activation and expansion of a class of γδ T cells, the so-called Vγ9Vδ2-T cells. These cells have natural killer cell characteristics, with antiviral and antitumor activities in vitro and in a humanized mouse model in vivo, and can trigger a cell-death signalling pathway leading to lysis of EBV-transformed autologous human lymphoblastoid B cells. Pamidronate-expanded human Vγ9Vδ2-T cells prevented the development of EBV-related lymphoproliferative disease (EBV-LPD) in mice inoculated with these EBV-transformed lymphoblastoid B cells and induced regression in mice with established EBV-LPD. Pamidronate treatment inhibited the development of EBV-LPD in immunodeficient mice infused with human peripheral blood mononuclear cells. The drug is inexpensive and already in clinical use for the treatment of osteoporosis, which makes it an attractive new treatment option. Thus far, no studies in humans with PTLD and pamidronate have been performed.74,75
Future options will definitely also include more targeted therapy or precision medicine. Specific inhibitors of critical molecules in the signaling pathways that drive proliferation and survival of lymphoma cells, such as Bruton tyrosine kinase inhibitors and phosphatidylinositol-3 (PI3)-kinase inhibitors, are already available. Biologic research should focus on the identification of the relevant pathways in PTLD. Preliminary in vitro data show that activation of apoptosis or blocking of cell cycle-related genes by small molecules in EBV-transformed B cells can be effective against EBV-related lymphoma or PTLD.76,77 The PI3K-Akt-mTOR signaling pathway is a frequently activated pathway in human cancers, including PTLD, and has also been analyzed as a potential target. Herpesviruses, such as EBV, can activate this pathway and subsequently enhance their viral replication and latency. Selective mTOR inhibitors were the first agents developed to target this pathway.78,79 In vitro, dual therapy with a PI3Kδ inhibitor was superior in attenuating proliferation of EBV-related B cell lymphomas.79-81 These agents are now in clinical trials.
The use of other agents, such as IVIg and interferon α, has been considered in the treatment of PTLD.14,36 However, no large clinical trials have been performed.
Conclusions and Recommendations
In every solid organ transplant donor and recipient, EBV serostatus should be determined before transplantation.
There is insufficient evidence for the use of prophylactic antiviral therapies in EBV-seronegative recipients with EBV-seropositive donors.
In EBV-seronegative recipients of EBV-seropositive donor organs, the EBV-viral load should be closely monitored at least during the first year post transplantation. The optimal frequency is uncertain. We recommend measurements of the viral load weekly to twice weekly for the first 3 months, then monthly in the fourth, fifth, and sixth month and every 3 months for the rest of the first year. There are insufficient data to give recommendations about viral load monitoring in the years beyond the first transplant year.
There is no consensus on whether the viral load should be measured in plasma or in whole blood. We recommend the use of plasma because of the higher specificity for the detection of EBV-positive PTLD of an increase in EBV viral load in plasma compared with whole blood.
There is no clear cutoff point for a high or elevated EBV viral load. When an increase in EBV viral load occurs, a careful clinical and, depending on signs and symptoms, radiological evaluation should be performed. In case of anatomical abnormalities and suspicion of PTLD, histology should be obtained.
For patients with an increasing EBV viral load, immunosuppression should be reduced. Currently, there is insufficient evidence to support the preemptive use of rituximab.
In patients with histologically proven PTLD, we recommend RIS when no additional risk factors associated with poor response or clinically aggressive disease are present. Rituximab monotherapy should be added in patients not responding. Patients who fail to achieve a remission after rituximab monotherapy and patients with high-risk or clinically aggressive lymphomas should be treated with R-CHOP immunochemotherapy.
Footnotes
Published online 15 December 2015.
This work was supported by a grant from Lymph&Co.
The authors declare no conflicts of interest.
M.L.N. collected data and wrote the article. M.J.K., F.J.B., S.T.P., and I.J.M.t.B. participated in writing the article and reviewed the article.
REFERENCES
- 1. Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000; 343: 481– 492. [DOI] [PubMed] [Google Scholar]
- 2. Tsurumi T, Fujita M, Kudoh A. Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol. 2005; 15: 3– 15. [DOI] [PubMed] [Google Scholar]
- 3. Odumade OA, Hogquist KA, Balfour HH., Jr Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev. 2011; 24: 193– 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004; 350: 1328– 1337. [DOI] [PubMed] [Google Scholar]
- 5. Nourse JP, Jones K, Gandhi MK. Epstein-Barr virus-related post-transplant lymphoproliferative disorders: pathogenetic insights for targeted therapy. Am J Transplant. 2011; 11: 888– 895. [DOI] [PubMed] [Google Scholar]
- 6. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol. 2007; 25: 587– 617. [DOI] [PubMed] [Google Scholar]
- 7. Thorley-Lawson DA, Duca KA, Shapiro M. Epstein-Barr virus: a paradigm for persistent infection—for real and in virtual reality. Trends Immunol. 2008; 29: 195– 201. [DOI] [PubMed] [Google Scholar]
- 8. Snow AL, Martinez OM. Epstein-Barr virus: evasive maneuvers in the development of PTLD. Am J Transplant. 2007; 7: 271– 277. [DOI] [PubMed] [Google Scholar]
- 9. Ghosh SK, Perrine SP, Faller DV. Advances in virus-directed therapeutics against Epstein-Barr virus-associated malignancies. Adv Virol. 2012; 2012: 509296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Küppers R. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol. 2003; 3: 801– 812. [DOI] [PubMed] [Google Scholar]
- 11. Cockfield SM. Identifying the patient at risk for post-transplant lymphoproliferative disorder. Transpl Infect Dis. 2001; 3: 70– 78. [DOI] [PubMed] [Google Scholar]
- 12. Stojanova J, Caillard S, Rousseau A, Marquet P. Post-transplant lymphoproliferative disease (PTLD): pharmacological, virological and other determinants. Pharmacol Res. 2011; 63: 1– 7. [DOI] [PubMed] [Google Scholar]
- 13. Sampaio MS, Cho YW, Shah T, Bunnapradist S, Hutchinson IV. Impact of Epstein-Barr virus donor and recipient serostatus on the incidence of post-transplant lymphoproliferative disorder in kidney transplant recipients. Nephrol Dial Transplant. 2012; 27: 2971– 2979. [DOI] [PubMed] [Google Scholar]
- 14. Allen UD, Preiksaitis JK; AST Infectious Diseases Community of Practice. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. Am J Transplant. 2013; 13 (Suppl 4): 107– 120. [DOI] [PubMed] [Google Scholar]
- 15. Green M, Michaels MG. Epstein-Barr virus infection and posttransplant lymphoproliferative disorder. Am J Transplant. 2013; 13 (Suppl 3): 41– 54. [DOI] [PubMed] [Google Scholar]
- 16. Parker A, Bowles K, Bradley JA, et al. Diagnosis of post-transplant lymphoproliferative disorder in solid organ transplant recipients - BCSH and BTS Guidelines. Br J Haematol. 2010; 149: 675– 692. [DOI] [PubMed] [Google Scholar]
- 17. Opelz G, Döhler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant. 2004; 4: 222– 230. [DOI] [PubMed] [Google Scholar]
- 18. Caillard S, Lamy FX, Quelen C, et al. Epidemiology of posttransplant lymphoproliferative disorders in adult kidney and kidney pancreas recipients: report of the French registry and analysis of subgroups of lymphomas. Am J Transplant. 2012; 12: 682– 693. [DOI] [PubMed] [Google Scholar]
- 19. Nelson BP, Nalesnik MA, Bahler DW, Locker J, Fung JJ, Swerdlow SH. Epstein-Barr virus-negative post-transplant lymphoproliferative disorders: a distinct entity? Am J Surg Pathol. 2000; 24: 375– 385. [DOI] [PubMed] [Google Scholar]
- 20. Dotti G, Fiocchi R, Motta T, et al. Epstein-Barr virus-negative lymphoproliferate disorders in long-term survivors after heart, kidney, and liver transplant. Transplantation. 2000; 69: 827– 833. [DOI] [PubMed] [Google Scholar]
- 21. Vereide DT, Sugden B. Lymphomas differ in their dependence on Epstein-Barr virus. Blood. 2011; 117: 1977– 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghobrial IM, Habermann TM, Maurer MJ, et al. Prognostic analysis for survival in adult solid organ transplant recipients with post-transplantation lymphoproliferative disorders. J Clin Oncol. 2005; 23: 7574– 7582. [DOI] [PubMed] [Google Scholar]
- 23. Morgans AK, Reshef R, Tsai DE. Posttransplant lymphoproliferative disorder following kidney transplant. Am J Kidney Dis. 2010; 55: 168– 180. [DOI] [PubMed] [Google Scholar]
- 24. Tsai DE, Douglas L, Andreadis C, et al. EBV PCR in the diagnosis and monitoring of posttransplant lymphoproliferative disorder: results of a two-arm prospective trial. Am J Transplant. 2008; 8: 1016– 1024. [DOI] [PubMed] [Google Scholar]
- 25. Gulley ML, Tang W. Using Epstein-Barr viral load assays to diagnose, monitor, and prevent posttransplant lymphoproliferative disorder. Clin Microbiol Rev. 2010; 23: 350– 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hayden RT, Hokanson KM, Pounds SB, et al. Multicenter comparison of different real-time PCR assays for quantitative detection of Epstein-Barr virus. J Clin Microbiol. 2008; 46: 157– 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Preiksaitis JK, Pang XL, Fox JD, Fenton JM, Caliendo AM, Miller GG. Interlaboratory comparison of Epstein-Barr virus viral load assays. Am J Transplant. 2009; 9: 269– 279. [DOI] [PubMed] [Google Scholar]
- 28. Hakim H, Gibson C, Pan J, et al. Comparison of various blood compartments and reporting units for the detection and quantification of Epstein-Barr virus in peripheral blood. J Clin Microbiol. 2007; 45: 2151– 2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee TC, Savoldo B, Rooney CM, et al. Quantitative EBV viral loads and immunosuppression alterations can decrease PTLD incidence in pediatric liver transplant recipients. Am J Transplant. 2005; 5: 2222– 2228. [DOI] [PubMed] [Google Scholar]
- 30. Stevens SJ, Verschuuren EA, Verkuujlen SA, Van Den Brule AJ, Meijer CJ, Middeldorp JM. Role of Epstein-Barr virus DNA load monitoring in prevention and early detection of post-transplant lymphoproliferative disease. Leuk Lymphoma. 2002; 43: 831– 840. [DOI] [PubMed] [Google Scholar]
- 31. Smets F, Latinne D, Bazin H, et al. Ratio between Epstein-Barr viral load and anti–Epstein-Barr virus specific T-cell response as a predictive marker of posttransplant lymphoproliferative disease. Transplantation. 2002; 73: 1603– 1610. [DOI] [PubMed] [Google Scholar]
- 32. Macedo C, Donnenberg A, Popescu I, et al. EBV-specific memory CD8+ T cell phenotype and function in stable solid organ transplant patients. Transpl Immunol. 2005; 14: 109– 116. [DOI] [PubMed] [Google Scholar]
- 33. Swerdlow SH, Campo E, Harris NL, et al. In: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. International Agency for Research on Cancer; 2008. [Google Scholar]
- 34. Bianchi E, Pascual M, Nicod M, Delaloye AB, Duchosal MA. Clinical usefulness of FDG-PET/CT scan imaging in the management of posttransplant lymphoproliferative disease. Transplantation. 2008; 85: 707– 712. [DOI] [PubMed] [Google Scholar]
- 35. McCormack L, Hany TI, Hübner M, et al. How useful is PET/CT imaging in the management of post-transplant lymphoproliferative disease after liver transplantation? Am J Transplant. 2006; 6: 1731– 1736. [DOI] [PubMed] [Google Scholar]
- 36. Parker A, Bowles K, Bradley JA, et al. Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients - BCSH and BTS Guidelines. Br J Haematol. 2010; 149: 693– 705. [DOI] [PubMed] [Google Scholar]
- 37. Leblond V, Dhedin N, Mamzer Bruneel MF, et al. Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. J Clin Oncol. 2001; 19: 772– 778. [DOI] [PubMed] [Google Scholar]
- 38. Tsai DE, Hardy CL, Tomaszewski JE, et al. Reduction in immunosuppression as initial therapy for posttransplant lymphoproliferative disorder: analysis of prognostic variables and long-term follow-up of 42 adult patients. Transplantation. 2001; 71: 1076– 1088. [DOI] [PubMed] [Google Scholar]
- 39. Kremers WK, Devarbhavi HC, Wiesner RH, Krom RA, Macon WR, Habermann TM. Post-transplant lymphoproliferative disorders following liver transplantation: incidence, risk factors and survival. Am J Transplant. 2006; 6 (5 Pt 1): 1017– 1024. [DOI] [PubMed] [Google Scholar]
- 40. Wagner HJ, Cheng YC, Huls MH, et al. Prompt versus preemptive intervention for EBV lymphoproliferative disease. Blood. 2004; 103: 3979– 3981. [DOI] [PubMed] [Google Scholar]
- 41. Stevens SJ, Verschuuren EA, Pronk I, et al. Frequent monitoring of Epstein-Barr virus DNA load in unfractionated whole blood is essential for early detection of posttransplant lymphoproliferative disease in high-risk patients. Blood. 2001; 97: 1165– 1171. [DOI] [PubMed] [Google Scholar]
- 42. Funk GA, Gosert R, Hirsch HH. Viral dynamics in transplant patients: implications for disease. Lancet Infect Dis. 2007; 7: 460– 472. [DOI] [PubMed] [Google Scholar]
- 43. San-Juan R, Comoli P, Caillard S, Moulin B, Hirsch HH, Meylan P. Epstein-Barr virus-related post-transplant lymphoproliferative disorder in solid organ transplant recipients. Clin Microbiol Infect. 2014; 20 (Suppl 7): 109– 118. [DOI] [PubMed] [Google Scholar]
- 44. Choquet S, Varnous S, Deback C, Golmard JL, Leblond V. Adapted treatment of Epstein-Barr virus infection to prevent posttransplant lymphoproliferative disorder after heart transplantation. Am J Transplant. 2014; 14: 857– 866. [DOI] [PubMed] [Google Scholar]
- 45. Schubert S, Renner C, Hammer M, et al. Relationship of immunosuppression to Epstein-Barr viral load and lymphoproliferative disease in pediatric heart transplant patients. J Heart Lung Transplant. 2008; 27: 100– 105. [DOI] [PubMed] [Google Scholar]
- 46. Reshef R, Vardhanabhuti S, Luskin MR, et al. Reduction of immunosuppression as initial therapy for posttransplantation lymphoproliferative disorder(★). Am J Transplant. 2011; 11: 336– 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rabot N, Büchler M, Foucher Y, et al. CNI withdrawal for post-transplant lymphoproliferative disorders in kidney transplant is an independent risk factor for graft failure and mortality. Transpl Int. 2014; 27: 956– 965. [DOI] [PubMed] [Google Scholar]
- 48. Serre JE, Michonneau D, Bachy E, et al. Maintaining calcineurin inhibition after the diagnosis of post-transplant lymphoproliferative disorder improves renal graft survival. Kidney Int. 2014; 85: 182– 190. [DOI] [PubMed] [Google Scholar]
- 49. Euvrard S, Morelon E, Rostaing L, et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med. 2012; 367: 329– 339. [DOI] [PubMed] [Google Scholar]
- 50. Stallone G, Schena A, Infante B, et al. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med. 2005; 352: 1317– 1323. [DOI] [PubMed] [Google Scholar]
- 51. Cullis B, D'Souza R, McCullagh P, et al. Sirolimus-induced remission of posttransplantation lymphoproliferative disorder. Am J Kidney Dis. 2006; 47: e67– e72. [DOI] [PubMed] [Google Scholar]
- 52. Majewski M, Korecka M, Joergensen J, et al. Immunosuppressive TOR kinase inhibitor everolimus (RAD) suppresses growth of cells derived from posttransplant lymphoproliferative disorder at allograft-protecting doses. Transplantation. 2003; 75: 1710– 1717. [DOI] [PubMed] [Google Scholar]
- 53. Majewski M, Korecka M, Kossev P, et al. The immunosuppressive macrolide RAD inhibits growth of human Epstein-Barr virus-transformed B lymphocytes in vitro and in vivo: a potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc Natl Acad Sci U S A. 2000; 97: 4285– 4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kirk AD, Cherikh WS, Ring M, et al. Dissociation of depletional induction and posttransplant lymphoproliferative disease in kidney recipients treated with alemtuzumab. Am J Transplant. 2007; 7: 2619– 2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K. Posttransplant lymphoproliferative disorders after renal transplantation in the United States in era of modern immunosuppression. Transplantation. 2005; 80: 1233– 1243. [DOI] [PubMed] [Google Scholar]
- 56. Kauffman HM, Cherikh WS, Cheng Y, Hanto DW, Kahan BD. Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation. 2005; 80: 883– 889. [DOI] [PubMed] [Google Scholar]
- 57. Trappe R, Oertel S, Leblond V, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012; 13: 196– 206. [DOI] [PubMed] [Google Scholar]
- 58. Choquet S, Oertel S, Leblond V, et al. Rituximab in the management of post-transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol. 2007; 86: 599– 607. [DOI] [PubMed] [Google Scholar]
- 59. Gross TG, Orjuela MA, Perkins SL, et al. Low-dose chemotherapy and rituximab for posttransplant lymphoproliferative disease (PTLD): a Children's Oncology Group Report. Am J Transplant. 2012; 12: 3069– 3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cohen JI, Mocarski ES, Raab-Traub N, Corey L, Nabel GJ. The need and challenges for development of an Epstein-Barr virus vaccine. Vaccine. 2013; 31 (Suppl 2): B194– B196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sokal EM, Hoppenbrouwers K, Vandermeulen C, et al. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis. 2007; 196: 1749– 1753. [DOI] [PubMed] [Google Scholar]
- 62. Rees L, Tizard EJ, Morgan AJ, et al. A phase I trial of epstein-barr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. Transplantation. 2009; 88: 1025– 1029. [DOI] [PubMed] [Google Scholar]
- 63. Funch DP, Walker AM, Schneider G, Ziyadeh NJ, Pescovitz MD. Ganciclovir and acyclovir reduce the risk of post-transplant lymphoproliferative disorder in renal transplant recipients. Am J Transplant. 2005; 5: 2894– 2900. [DOI] [PubMed] [Google Scholar]
- 64. Opelz G, Daniel V, Naujokat C, Fickenscher H, Döhler B. Effect of cytomegalovirus prophylaxis with immunoglobulin or with antiviral drugs on post-transplant non-Hodgkin lymphoma: a multicentre retrospective analysis. Lancet Oncol. 2007; 8: 212– 218. [DOI] [PubMed] [Google Scholar]
- 65. Comoli P, Labirio M, Basso S, et al. Infusion of autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood. 2002; 99: 2592– 2598. [DOI] [PubMed] [Google Scholar]
- 66. Haque T, Wilkie GM, Jones MM, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007; 110: 1123– 1131. [DOI] [PubMed] [Google Scholar]
- 67. Haque T, McAulay KA, Kelly D, Crawford DH. Allogeneic T-cell therapy for Epstein-Barr virus-positive posttransplant lymphoproliferative disease: long-term follow-up. Transplantation. 2010; 90: 93– 94. [DOI] [PubMed] [Google Scholar]
- 68. Savoldo B, Goss JA, Hammer MM, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood. 2006; 108: 2942– 2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gerdemann U, Katari UL, Papadopoulou A, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther. 2013; 21: 2113– 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gerdemann U, Keirnan JM, Katari UL, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther. 2012; 20: 1622– 1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ricciardelli I, Blundell MP, Brewin J, Thrasher A, Pule M, Amrolia PJ. Towards gene therapy for EBV-associated posttransplant lymphoma with genetically modified EBV-specific cytotoxic T cells. Blood. 2014; 124: 2514– 2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ricciardelli I, Brewin J, Lugthart G, Albon SJ, Pule M, Amrolia PJ. Rapid generation of EBV-specific cytotoxic T lymphocytes resistant to calcineurin inhibitors for adoptive immunotherapy. Am J Transplant. 2013; 13: 3244– 3252. [DOI] [PubMed] [Google Scholar]
- 73. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371: 1507– 1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dharnidharka VR, Mohanakumar T. New approaches to treating B-cell cancers induced by Epstein-Barr virus. N Engl J Med. 2015; 372: 569– 571. [DOI] [PubMed] [Google Scholar]
- 75. Xiang Z, Liu Y, Zheng J, et al. Targeted activation of human Vγ9Vδ2-T cells controls Epstein-Barr virus-induced B cell lymphoproliferative disease. Cancer Cell. 2014; 26: 565– 576. [DOI] [PubMed] [Google Scholar]
- 76. Bernasconi M, Ueda S, Krukowski P, et al. Early gene expression changes by Epstein-Barr virus infection of B-cells indicate CDKs and survivin as therapeutic targets for post-transplant lymphoproliferative diseases. Int J Cancer. 2013; 133: 2341– 2350. [DOI] [PubMed] [Google Scholar]
- 77. Morscio J, Dierickx D, Tousseyn T. Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: what do we know so far? Clin Dev Immunol. 2013; 2013: 150835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015; 146: 53– 60. [DOI] [PubMed] [Google Scholar]
- 79. Liu X, Cohen JI. The role of PI3K/Akt in human herpesvirus infection: from the bench to the bedside. Virology. 2015; 479–480: 568– 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Furukawa S, Wei L, Krams SM, Esquivel CO, Martinez OM. PI3Kδ inhibition augments the efficacy of rapamycin in suppressing proliferation of Epstein-Barr virus (EBV)+ B cell lymphomas. Am J Transplant. 2013; 13: 2035– 2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Blachly JS, Baiocchi RA. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br J Haematol. 2014; 167: 19– 32. [DOI] [PubMed] [Google Scholar]