

Abstract
Pragmin is one of the few mammalian proteins containing the Glu‐Pro‐Ile‐Tyr‐Ala (EPIYA) tyrosine‐phosphorylation motif that was originally discovered in the Helicobacter pylori CagA oncoprotein. Following delivery into gastric epithelial cells by type IV secretion and subsequent tyrosine phosphorylation at the EPIYA motifs, CagA serves as an oncogenic scaffold/adaptor that promiscuously interacts with SH2 domain‐containing mammalian proteins such as the Src homology 2 (SH2) domain‐containing protein tyrosine phosphatase‐2 (SHP2) and the C‐terminal Src kinase (Csk), a negative regulator of Src family kinases. Like CagA, Pragmin also forms a physical complex with Csk. In the present study, we found that Pragmin directly binds to Csk by the tyrosine‐phosphorylated EPIYA motif. The complex formation potentiates kinase activity of Csk, which in turn phosphorylates Pragmin on tyrosine‐238 (Y238), Y343, and Y391. As Y391 of Pragmin comprises the EPIYA motif, Pragmin–Csk interaction creates a feed‐forward regulatory loop of Csk activation. Together with the finding that Pragmin and Csk are colocalized to focal adhesions, these observations indicate that the Pragmin–Csk interaction, triggered by Pragmin EPIYA phosphorylation, robustly stimulates the kinase activity of Csk at focal adhesions, which direct cell‐matrix adhesion that regulates cell morphology and cell motility. As a consequence, expression of Pragmin and/or Csk in epithelial cells induces an elongated cell shape with elevated cell scattering in a manner that is mutually dependent on Pragmin and Csk. Deregulation of the Pragmin–Csk axis may therefore induce aberrant cell migration that contributes to tumor invasion and metastasis.
Keywords: Cell motility, C‐terminal Src kinase (Csk), EPIYA motif, focal adhesion, Pragmin (Sgk223)
Pragmin, also known as Sgk223, was originally identified as a downstream effector of Rnd2, a member of the Rho family small GTPases, in neuronal cells.1 Pragmin associates with Rnd2 and thereby stimulates RhoA to induce cell contraction, which then inhibits neurite outgrowth by nerve growth factor. Pragmin also possesses a kinase domain in the C‐terminal region. However, the kinase domain does not seem to have Mg2+‐binding activity due to substitutions in key motifs that are characteristics of active kinases. Accordingly, Pragmin has been considered to be a pseudokinase.2, 3 Several studies have shown a functional link between Pragmin and oncogenesis. Increase in the level of Pragmin promotes invasion of advanced colon carcinoma cells.4 Pragmin is overexpressed in pancreatic ductal adenocarcinoma (PDAC) cells and ectopic expression of Pragmin in human pancreatic duct epithelial cells gives rise to an elongated mesenchymal‐like cell morphology, which is concomitantly associated with increased cell migration and invasion.5
Pragmin is one of the few mammalian (human) proteins that possess the Glu‐Pro‐Ile‐Tyr‐Ala (EPIYA) sequence motif.6 The EPIYA motif was originally discovered as a tyrosine phosphorylation motif present in variable numbers (from three to five) in the C‐terminal region of the Helicobacter pylori CagA oncoprotein.7, 8 Following delivery into gastric epithelial cells by the bacterial type IV secretion system, CagA undergoes tyrosine phosphorylation at the EPIYA motifs by Src family kinases (SFKs) or c‐Abl kinase.9 Once tyrosine‐phosphorylated, the CagA EPIYA motifs serve as docking sites for various Src homology 2 (SH2) domain‐containing host proteins such as SHP2 and the C‐terminal Src kinase (Csk).10, 11, 12 Aberrant activation of SHP2, a pro‐oncogenic tyrosine phosphatase, is associated with a variety of human malignancies.13, 14 The CagA–SHP2 interaction has also been considered to play a critical role in H. pylori‐associated gastric carcinogenesis.15 Csk is a protein kinase that specifically phosphorylates the C‐terminal tyrosine residue conserved among SFK members to inactivate their kinase activity.16 On its own, Csk is located predominantly in the cytoplasm as it does not contain the SH4 fatty‐acid acylation domain that mediates membrane anchoring. As all of the SFK members are membrane‐anchored, cytoplasmic Csk needs to be translocated to the plasma membrane in order to inhibit SFKs through phosphorylation. The membrane tethering of Csk is primarily mediated by binding to membrane‐associated proteins such as Csk binding protein (Cbp)/phosphoprotein associated with GEMS.17 Due to its inhibitory role on the pro‐oncogenic SFK members, Csk has been considered a tumor suppressor.16 However, recent studies suggest that Csk could also contribute to oncogenesis by phosphorylating non‐SFK substrates.18, 19, 20, 21, 22, 23 Following tyrosine phosphorylation by Csk, eukaryotic elongation factor 2 undergoes SUMOylation and subsequent cleavage, thereby causing chromosomal instability that stimulates neoplastic transformation of cells.22 Csk also mediates signals generated by the G protein‐coupled receptor, which induces actin stress fiber formation that regulates cell motility independently of SFKs.23 More recently, Src was found to act as a negative regulator of Ras, suggesting a stimulatory role of Csk in Ras signaling.24 Given these observations, it is likely that Csk exerts both pro‐oncogenic or anti‐oncogenic actions, depending on cell context.
In the present study, we investigated the biochemical and biological consequences of Pragmin–Csk complex formation. We found that Csk phosphorylates Pragmin on the EPIYA motif. We also found that Csk directly interacts with Pragmin through the tyrosine‐phosphorylated EPIYA motif and this interaction potentiates the kinase activity of Csk. The phospho‐EPIYA‐dependent Pragmin–Csk interaction creates a positive feedback loop of Csk activation at focal adhesions and thereby induces cell‐morphological transformation with elevated cell motility, deregulation of which provokes aberrant cell migration and invasion that contribute to oncogenesis.
Materials and Methods
Cell lines
AGS and MKN7 human gastric epithelial cells were cultured in RPMI‐1640 medium supplemented with 10% FBS. SYF cells, mouse embryonic fibroblast cells established from c‐src, yes, and fyn triple knockout mice25 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in DMEM with 10% FBS. AGS cells were transfected with expression vectors using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). MKN7 cells were treated with siRNA using Lipofectamine 3000 reagent (Invitrogen). SYF cells were treated with siRNA using Lipofectamine 2000 reagent (Invitrogen).
Expression vectors
Expression vectors used in this study are shown in Table S1. Recombinant lentiviruses that express Myc‐Pragmin‐WT, Myc‐Pragmin‐Y391F, and Csk‐WT‐Flag were generated using Lentivector Expression Systems (System Biosciences, Mountain View, CA, USA).
RNA interference
Csk‐specific siRNAs (target sequences 5′‐AGTACCCAGCAAATGGGCA‐3′ [100% identical between human and mouse Csk] and 5′‐ACTCGCCTTCTTAGAGTTT‐3′ [unique to human Csk]) were used to knock down Csk expression.26, 27 Pragmin‐specific siRNAs (target sequences 5′‐GTCACAGGCCAAGATAGAA‐3′6 and 5′‐CTGTTTTCTTCTGTAATTATA‐3′ designed using siDirect version 2.0; http://sidirect2.rnai.jp) were used to knock down human Pragmin expression. Control siRNA Luciferase (GL2) was purchased from Cosmo Bio (Tokyo, Japan).
Antibodies
Anti‐Pragmin polyclonal antibody A302‐675A (Bethyl Laboratories, Montgomery, TX, USA) was used as a primary antibody for immunoprecipitation, immunoblotting, and immunostaining. Anti‐Myc mAb 9E10 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐DDDDK polyclonal antibody PM020 (Medical & Biological Laboratories, Aichi, Japan), anti‐vinculin mAb hVIN‐1 (Sigma‐Aldrich, St. Louis, MO, USA) and anti‐Csk polyclonal antibody C‐20 (Santa Cruz Biotechnology) were used as primary antibodies for immunoblotting and immunostaining. Anti‐FLAG mAb M2 (Sigma‐Aldrich), anti‐phosphotyrosine mAb 4G10 (Millipore, Temecula, CA, USA), anti‐GST mAb B‐14 (Santa Cruz Biotechnology), anti‐His mAb 6C4 (Medical & Biological Laboratories), anti‐His mAb OGHis (Medical & Biological Laboratories), anti‐actin polyclonal antibody C‐11 (Santa Cruz Biotechnology), and anti‐p‐c‐Src (Y530) polyclonal antibody (Santa Cruz Biotechnology) were used as primary antibodies for immunoblotting.
Pragmin purification
Escherichia coli Rosetta2 (DE3) was transformed with pGEX6P2‐Pragmin‐WT‐His or pGEX6P2‐Pragmin‐Y391F‐His and was cultured with LB medium. Protein expression was induced by addition of 0.1 mM isopropyl‐1‐thio‐β‐D‐galactopyranoside (IPTG) at 18°C for 16 h. GST‐fused Pragmin‐WT‐His or Pragmin‐Y391F‐His was purified using Ni Sepharose excel (GE Healthcare, Uppsala, Sweden). For tyrosine‐phosphorylated Pragmin purification, E. coli BL21 (DE3) was cotransformed with pGEX6P2‐Pragmin‐WT‐His or pGEX6P2‐Pragmin‐Y391F‐His and pACYCDuet1‐v‐Src.28 The subsequent procedure was the same as non‐phosphorylated Pragmin. The Ni‐binding buffer contained 0.2 mM Na3VO4.
C‐terminal Src kinase purification
E. coli BL21 (DE3) was transformed with pGEX6P2‐Csk‐WT‐His and was cultured with LB medium. Protein expression was induced by addition of 0.1 mM IPTG and additional culture at 25°C for 16 h. GST‐fused Csk‐His was purified using glutathione Sepharose 4B (GE Healthcare). The GST tag was excised by treating the GST‐Csk‐His protein with PreScission Protease (GE Healthcare).
Src‐tail purification
E. coli BL21 (DE3) was transformed with pGEX6P2‐Src‐tail and cultured with LB medium. Protein expression was induced by addition of 0.4 mM IPTG for 7 h at 37°C. The GST‐fused Src‐tail was purified using glutathione Sepharose 4B (GE Healthcare).
Glutathione S‐transferase pull‐down assay
The GST pull‐down assay was carried out as described previously.28 The mixtures were washed with GST pull‐down buffer (50 mM Tris‐HCl [pH 7.5], 150 mM NaCl, 10 mM β‐mercaptoethanol, and 0.01% Triton X‐100).
In vitro kinase assay
Recombinant proteins were mixed with indicated combinations in kinase buffer (50 mM HEPES–NaOH [pH 8.0], 100 mM NaCl, 10 mM MgCl2, 0.1 mM Na3VO4, and 20 mM ATP‐Na).11 The reaction mixtures were incubated at 30°C and were subjected to SDS‐PAGE, followed by immunoblotting.
Immunoprecipitation and immunoblotting
Cells were lysed in lysis buffer (50 mM Tris‐HCl [pH 7.5], 100 mM NaCl, 5 mM EDTA, 1% Brij‐35, 2 mM Na3VO4, 10 mM NaF, 10 mM β‐glycerophosphate, 10 μg/mL leupeptin, 10 μg/mL trypsin inhibitor, 10 μg/mL aprotinin, and 2 mM PMSF)10 or (50 mM Tris‐HCl [pH 7.5], 200 mM NaCl, 5 mM EDTA, 1% Triton X‐100, 10% glycerol, 2 mM Na3VO4, 10 mM NaF, 10 mM β‐glycerophosphate, 10 μg/mL leupeptin, 10 μg/mL trypsin inhibitor, 10 μg/mL aprotinin, and 2 mM PMSF). Immunoprecipitation, immunoblotting, and quantification of chemiluminescence on the immunoblotted membrane were carried out as described previously.10, 11
Immunostaining
Immunostaining was carried out by modifying the protocol described previously.10, 28 Briefly, cells were fixed with Mildform 10N (Wako, Osaka, Japan) for 10 min and permeabilized with 0.25% Triton X‐100 for 10 min. The cells were then treated with primary antibodies and were visualized with Alexa Fluor‐conjugated secondary antibodies (Invitrogen). The nuclei were stained with DAPI. Images were obtained using the FV1200 confocal microscope system (Olympus, Tokyo, Japan).
Analysis of cell morphology
MKN7 cells were infected with recombinant lentiviruses and were observed at 48 h after infection by light microscope (Nikon, Tokyo, Japan).
Cell scatter assay
MKN7 cells were seeded at 1 × 103 cells in 3.5‐cm dishes and were cultured for 96 h. Cells were then infected with recombinant lentiviruses and their morphology was analyzed as described above.
Statistical analysis
Statistical analyses were carried out by Student's t‐test.
Results
Direct interaction of Pragmin with Csk through tyrosine‐phosphorylated EPIYA motif
We previously reported that Pragmin forms a physical complex with Csk in mammalian cells and that the interaction is dependent on the EPIYA motif of Pragmin and the SH2 domain of Csk.6 To test whether the Pragmin–Csk interaction is direct or not, we undertook an in vitro binding assay using bacterially produced recombinant tyrosine‐phosphorylated Pragmin and Csk proteins. In E. coli, both GST‐Pragmin‐His and the EPIYA mutant (Y391F) were tyrosine‐phosphorylated in bacteria (Fig. S1a), indicating that tyrosine residues other than the EPIYA motif were also phosphorylated by v‐Src in E. coli. Next, recombinant Csk was purified (Fig. S1b). Using the purified GST‐Pragmin‐His and Csk‐His proteins, we undertook a GST pull‐down experiment and found that Csk specifically associates with tyrosine‐phosphorylated WT Pragmin but not with the Y391F Pragmin (Fig. S1c). From these observations, we concluded that Pragmin directly interacted with Csk in an EPIYA phosphorylation‐dependent manner.
C‐terminal Src kinase influences the level of Pragmin tyrosine phosphorylation in cells
Next, to investigate the impact of Pragmin on the kinase activity of Csk in cells, we transiently transfected a Flag‐tagged Csk vector together with a Myc‐tagged Pragmin vector into AGS human gastric epithelial cells. The cell lysates were then subjected to immunoblotting analysis with the use of an anti‐phosphotyrosine antibody. As a result, ectopic co‐expression of Pragmin and Csk increased the tyrosine phosphorylation levels of several cellular proteins including Pragmin (Fig. 1a). To determine whether the observed increase in Pragmin tyrosine phosphorylation was dependent on Csk kinase activity, we generated an expression vector for a mutant Flag‐tagged Csk (Csk‐K222R‐Flag) in which ATP‐binding K222 was replaced by arginine. When co‐expressed with Pragmin, the kinase‐dead Csk had little effect on the level of Pragmin tyrosine phosphorylation, whereas WT Csk greatly increased the level of phosphorylation (Fig. 1b). Pragmin and its one and only homolog Sgk269/pseudopodium‐enriched atypical kinase 1 (PEAK1) possess a pseudokinase domain that lacks the Mg2+‐binding motif.2 As Sgk269/PEAK1 was recently found to have kinase activity,29 it was possible that the weak increase in the tyrosine phosphorylation level of Pragmin in cells expressing the kinase‐dead Csk (Csk‐K222R‐Flag) was due to autophosphorylation of Pragmin through activation of its kinase activity after Csk binding. To test this idea, we generated an expression vector for a Pragmin mutant (Myc‐Pragmin‐K997R) in which K997, a putative ATP‐binding site, was replaced by arginine. Co‐expression of the K997 mutant together with the kinase‐dead Csk still gave rise to a weak increase in the level of Pragmin tyrosine phosphorylation (Fig. 1b), arguing against the idea that Pragmin possesses kinase activity. Although the mechanism by which kinase‐dead Csk slightly elevated the level of Pragmin tyrosine phosphorylation remained unknown, the kinase‐dead Csk might have competitively inhibited endogenous Csk–Cbp interaction and thereby attenuated Csk‐mediated SFK inhibition, which in turn induced a slight increase in the level of Pragmin tyrosine phosphorylation.
Figure 1.
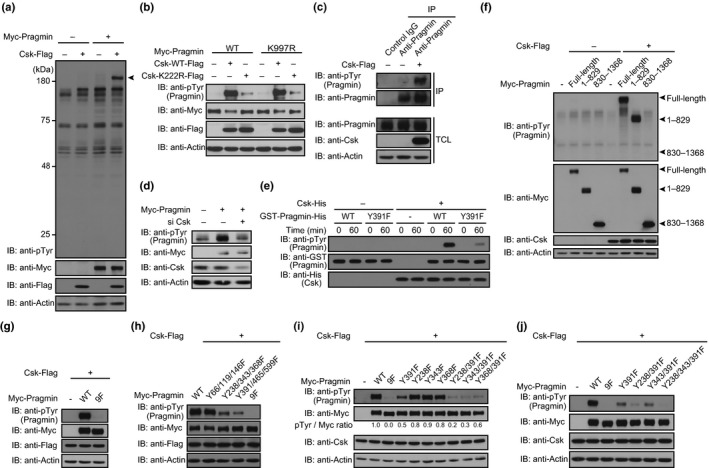
C‐terminal Src kinase (Csk) affects the level of Pragmin tyrosine phosphorylation (pTyr) in cells. (a) Immunoblot analysis of tyrosine‐phosphorylated proteins in AGS human gastric epithelial cells. Arrowhead indicates Myc‐Pragmin. (b) Immunoblot analysis of tyrosine‐phosphorylated Myc‐Pragmin in AGS cells. (c) Sequential immunoprecipitation (IP)–immunoblot (IB) analysis of tyrosine‐phosphorylated Pragmin in AGS cells. (d) Immunoblot analysis of SYF cell lysates. SYF cells treated with Csk‐specific siRNA were transfected with a Myc‐Pragmin vector. (e) Immunoblot analysis of Pragmin phosphorylation by Csk in vitro. (f–j) Immunoblot analysis of tyrosine‐phosphorylated Myc‐Pragmin in AGS cells.
We previously reported that SFKs are capable of Pragmin tyrosine phosphorylation.6 Given the above‐described observation, we next investigated the contribution of Csk to tyrosine phosphorylation of Pragmin. To do so, AGS cells were transfected with an expression vector for Csk‐Flag and the cell lysates were immunoprecipitated with an anti‐Pragmin antibody. The tyrosine phosphorylation level of endogenous Pragmin was increased following ectopic expression of Csk (Fig. 1c). Then, we examined the effects of Csk knockdown on the phosphorylation status of Pragmin. However, it is possible that Csk inhibition would induce strong SFK activation, resulting in hyperphosphorylation of Pragmin, which would hamper analysis of the contribution of Csk to Pragmin phosphorylation. To avoid this problem, we used SYF mouse embryonic fibroblast cells, which do not express SFKs.25 As there is currently no antibody that recognizes mouse Pragmin, SYF cells treated with Csk‐specific siRNA were also transfected with a Myc‐Pragmin vector to monitor the level of Pragmin tyrosine phosphorylation. The results of the experiment revealed that Csk knockdown reduced the level of Pragmin tyrosine phosphorylation (Fig. 1d). From these observations, we concluded that, in addition to SFKs, Csk is capable of tyrosine‐phosphorylating Pragmin.
Pragmin is a new substrate for Csk tyrosine kinase
These observations suggested that Pragmin is a new substrate of Csk for tyrosine phosphorylation. To directly test this idea, we expressed GST‐fused WT Pragmin or the Y391F Pragmin mutant in E. coli and affinity‐purified them (Fig. S2). Using the GST–Pragmin fusions, we carried out an in vitro kinase assay. As a result, Csk phosphorylated Pragmin, and the major site of Csk phosphorylation was the EPIYA motif, although non‐EPIYA sites were also tyrosine‐phosphorylated with less efficiency (Fig. 1e). Pragmin contains 21 tyrosine residues other than the tyrosine residue (Y391) in the EPIYA motif (Fig. S3). To determine actual tyrosine phosphorylation sites of Pragmin by Csk in cells, we used mammalian expression vectors for the Myc‐tagged N‐terminal Pragmin fragment (1–829), which contains nine tyrosine residues including the EPIYA motif, and the Myc‐tagged C‐terminal Pragmin fragment (830–1368), which contains 13 tyrosine residues.1 AGS cells were transiently transfected with the Csk vector together with the Myc‐Pragmin, Myc‐Pragmin (1–829), or Myc‐Pragmin (830–1368) vector. Immunoblotting analysis of the cell lysates revealed that Csk efficiently phosphorylated full‐length Pragmin and the N‐terminal Pragmin fragment but not the C‐terminal Pragmin fragment (Fig. 1f). Consistently, phenylalanine substitutions of the nine tyrosine residues present in the N‐terminal Pragmin fragment abolished Pragmin tyrosine phosphorylation by Csk (Fig. 1g). Given this, we next generated Y66/119/146F, Y238/343/368F, and Y391/465/599F mutants of Pragmin and expressed them together with Csk in AGS cells. Immunoblot analysis of the cell lysates showed that the level of Pragmin tyrosine phosphorylation was substantially reduced in the Y238/343/368F and Y391/465/599F mutants but not in the Y66/119/146F mutant (Fig. 1h). Thus, there is at least one tyrosine residue in Y238/343/368 and at least one tyrosine residue in Y391/465/599 that undergoes tyrosine phosphorylation by Csk. We next generated Y238F, Y343F, Y368F, Y238/391F, Y343/391F, and Y368/391F mutants of Pragmin, as Y391 constitutes the EPIYA motif and has been shown to undergo tyrosine phosphorylation. Expression of these Pragmin mutants together with Csk in AGS cells revealed that Y391 was the major site of Pragmin tyrosine phosphorylation, while Y238 and Y343 were also tyrosine phosphorylated (Fig. 1i). To consolidate the observation, we generated a Y238/343/391F mutant and found that the mutant did not undergo tyrosine phosphorylation by Csk in AGS cells (Fig. 1j). From these observations, we concluded that Csk phosphorylates Pragmin on Y238, Y343, and Y391; of these, Y391 is a primary site of phosphorylation.
Catalytic activation of Csk by complex formation with Pragmin
To determine whether the complex formation with Pragmin influences the kinase activity of Csk, we carried out an in vitro kinase assay of Csk using GST‐fused C‐terminal 13‐amino‐acid sequence of c‐Src (GST‐Src‐tail), which contains a well‐recognized tyrosine phosphorylation site of c‐Src by Csk. A bacterial expression vector for the GST‐fused Src‐tail was made and the fusion protein was expressed in E. coli. After affinity purification, GST‐Src‐tail was used as a substrate of Csk in an in vitro kinase assay. The results of the experiment convincingly indicated that the kinase activity of Csk was substantially potentiated in the presence of tyrosine phosphorylated WT Pragmin but not in the presence of Y391 Pragmin mutant (Fig. 2a,b). Although Csk has been reported to exist as a constitutively active form,16 the results of the experiment indicated that its enzymatic activity is further potentiated following complex formation with Pragmin. Interestingly, phosphorylation of GST‐Src‐tail by Csk was evident at 15 min after the onset of the reaction in the presence of tyrosine phosphorylated Pragmin. In contrast, there was little phosphorylation of GST‐Src‐tail after 1 h of incubation in the presence of non‐phosphorylated Pragmin (Fig. 2b). This difference was thought to be due to immediate complex formation between Csk and tyrosine phosphorylated Pragmin, which potentiates Csk kinase activity. In the case of non‐phosphorylated Pragmin, Csk needs to phosphorylate Pragmin before complex formation, which then potentiates Csk kinase activity (Fig. 2b). The results indicated the presence of a positive feedback regulatory loop of Csk activation, created by the complex formation between Csk and tyrosine phosphorylated Pragmin at the EPIYA motif.
Figure 2.
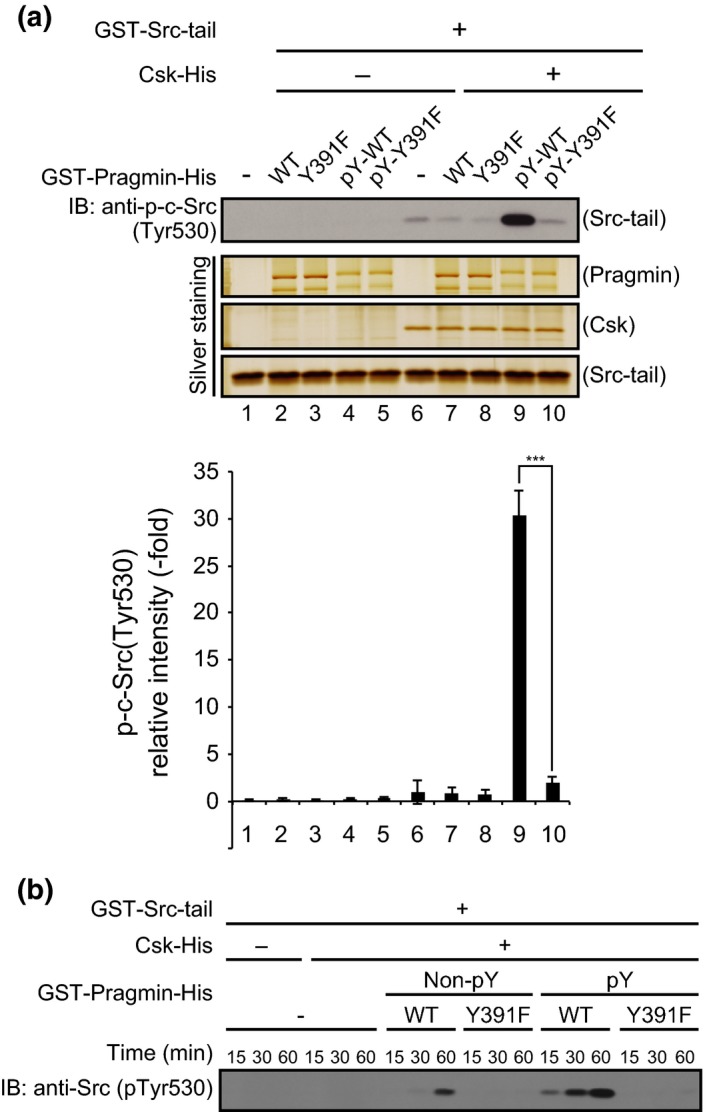
Activation of C‐terminal Src kinase (Csk) by complex formation with Pragmin. (a,b) In vitro kinase assay of Csk using GST‐Src‐tail as a substrate. (a) Column graph: Quantitative analysis of band intensities. Relative intensities were calculated by defining the intensity of lane 6 as 1. Error bars represent means ± SD (n = 3).
Biological consequence for complex formation between Pragmin and Csk
Ectopic expression of Pragmin in AGS cells has been reported to induce an elongated cell shape.6 We therefore investigated the effect of Csk on the Pragmin‐mediated morphogenetic change of cells. To do so, MKN7 gastric epithelial cells were co‐infected with a lentivirus transducing Csk and a lentivirus transducing WT or Y391F Pragmin, which lacks the EPIYA motif. At 48 h after infection, cell morphology was examined by light microscopy. Cell elongation was found in 10.8%, 4.4%, and 5.5% of cells singly expressing WT Pragmin, Pragmin Y391F, and Csk, respectively (Fig. 3a,b). Co‐expression of Csk and WT Pragmin, but not Y391F Pragmin, substantially augmented the morphological change (27.6%) (Fig. 3a,b). We also investigated whether the cell‐morphological changes induced by the Pragmin–Csk interaction was associated with altered cell motility. Whereas single expression of Csk or Pragmin did not substantially induce cell scattering, co‐expressing of Csk and Pragmin did markedly induce cell scattering in MKN7 cells (Fig. 3c), indicating that activation of Csk by Pragmin plays a key role in the cell‐scattering phenotype. The observation also indicated that the cell‐morphological changes induced by Csk or Pragmin is a prerequisite for induction of cell scattering, which required both Csk and Pragmin. Possibly, the threshold for induction of the cell‐morphological changes by Csk or Pragmin was lower than that required for induction of cell scattering by Csk or Pragmin, respectively. To test this idea, we investigated whether the morphological change induced by ectopic expression of Csk and Pragmin was dependent on endogenous Pragmin and Csk, respectively. To do so, we treated MKN7 cells with Csk‐specific siRNA and then were infected with a lentivirus transducing Pragmin. At 48 h after infection, cell morphology was examined by light microscopy. Cell elongation induced by overexpressing Pragmin was significantly reduced by Csk knockdown (Fig. 4a–c). In a reciprocal experiment, we treated MKN7 cells with Pragmin‐specific siRNA and then infected a lentivirus transducing Csk. Cell elongation induced by overexpressing Csk was again reduced by knockdown of Pragmin (Fig. 4d–f). Taken together, these results provided additional evidence for the presence of a positive feedback regulatory loop of Csk activation by forming a complex between Csk and Pragmin.
Figure 3.
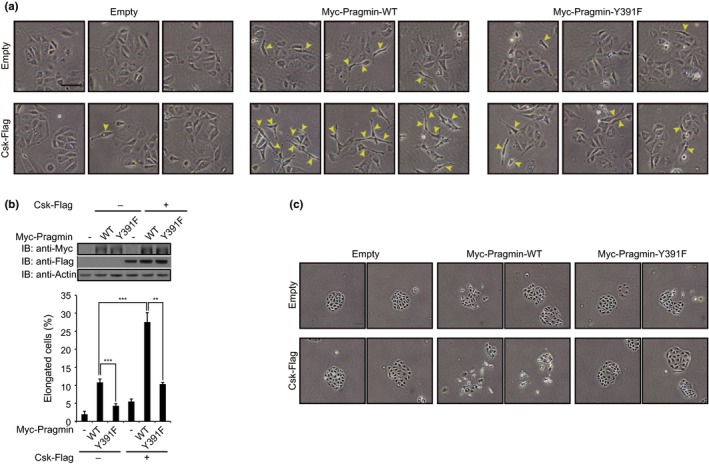
Effect of Pragmin and/or C‐terminal Src kinase (Csk) expression on cell morphology and cell motility. (a) Cell‐morphological analysis of MKN7 human gastric epithelial cells infected with recombinant lentiviruses. Yellow arrowheads indicate elongated cells. Scale bar = 100 μm. (b) Top, immunoblot analysis of total cell lysates prepared from lentivirus‐infected MKN7 cells shown in (a). Bottom, percentage of elongated cells shown in (a). Error bars represent means ± SD. **P < 0.01, ***P < 0.001 (Student's t‐test, n = 3). (c) Cell scatter assay of MKN7 cells infected with recombinant lentiviruses. Scale bar = 100 μm.
Figure 4.
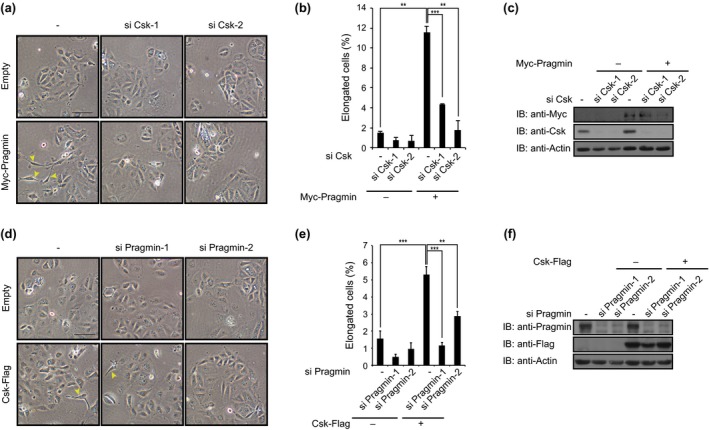
Downregulation of cell‐morphological changes by specific knockdown of Pragmin or C‐terminal Src kinase (Csk). (a,d) Cell‐morphological analysis of MKN7 human gastric epithelial cells transfected with siRNAs and infected with recombinant lentiviruses. Yellow arrowheads indicate elongated cells. Scale bar = 100 μm. (b,e) Percentage of elongated cells shown in (a,d). Error bars represent means ± SD. **P < 0.01, ***P < 0.001 (Student's t‐test, n = 3). (c,f) Immunoblot analysis of total cell lysates prepared from MKN7 cells shown in (a,d).
To gain insights into mechanisms underlying enhanced cell motility by the Pragmin–Csk interaction, we examined the subcellular localization of Pragmin and Csk by immunostaining. To this end, Myc‐Pragmin and Csk‐Flag were co‐expressed in AGS cells by lentivirus‐mediated transduction. As previously reported, both proteins were primarily distributed to the cytoplasm.6 However, a fraction of Pragmin was also co‐localized with Csk at focal adhesion spots (Fig. 5a). More importantly, endogenous Pragmin was found to be primarily distributed to the focal adhesion spots as determined by co‐staining with an anti‐Pragmin antibody and an anti‐vinculin antibody (Fig. 5b). Given this, we tested whether Pragmin interacts with a focal adhesion component(s) by co‐precipitation experiment. Vinculin was co‐immunoprecipitated with Pragmin in AGS cells (Fig. 5c). Vinculin might therefore recruit the Pragmin and Pragmin‐associated Csk to focal adhesion spots, in which Pragmin‐activated Csk stimulates cell‐morphological transformation with elevated cell motility.
Figure 5.
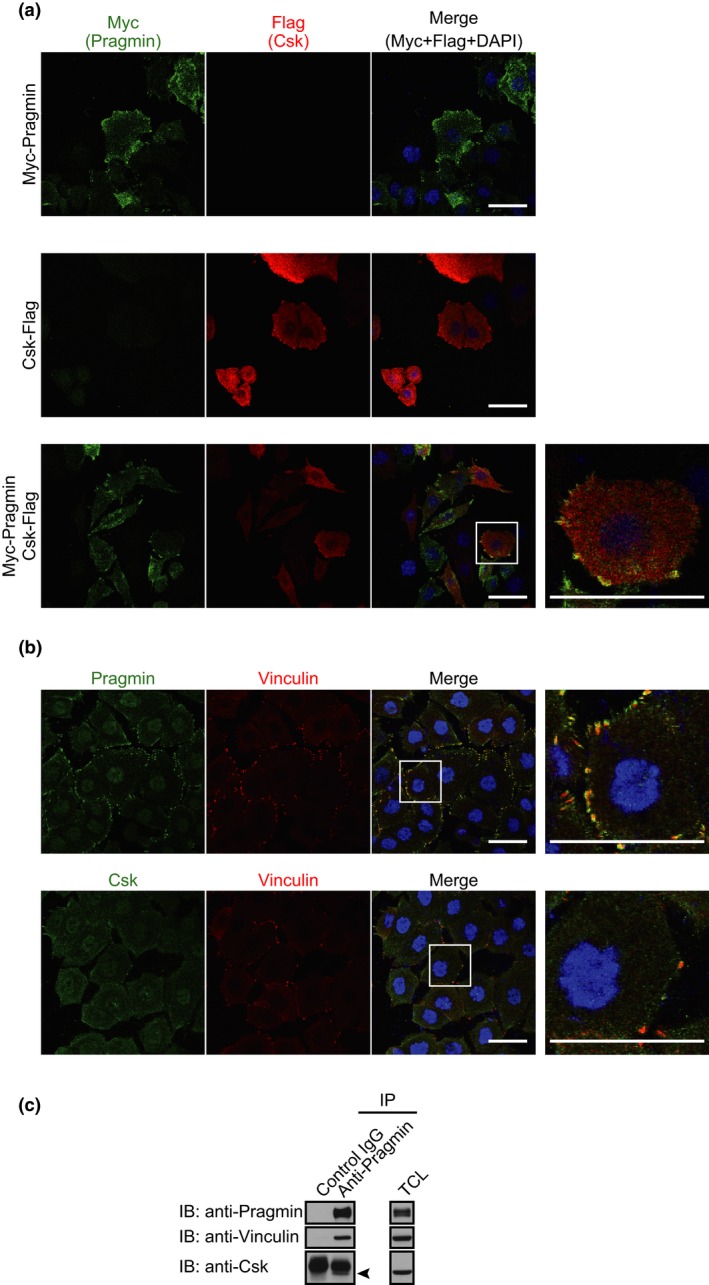
Distribution of Pragmin and C‐terminal Src kinase (Csk) to focal adhesions. (a,b) Confocal x‐y images are shown. Scale bar = 50 μm. (a) Immunostaining analysis of AGS human gastric epithelial cells infected with indicated lentiviruses. Anti‐Myc antibody (green), anti‐Flag antibody (red), DAPI (blue). (b) Immunostaining analysis of AGS cells. Upper, anti‐Pragmin antibody (green), anti‐vinculin antibody (red), DAPI (blue). Lower, anti‐Csk antibody (green), anti‐vinculin antibody (red), DAPI (blue). (c) Immunoprecipitation (IP)–immunoblot analysis of endogenous Pragmin in AGS cells. Arrowhead indicates the position of Csk. TCL, total cell lysates.
Discussion
In the present study, we showed that Pragmin directly interacts with Csk, a tyrosine kinase that negatively regulates SFK activity, through the tyrosine phosphorylated EPIYA motif. Following complex formation, Pragmin stimulates the kinase activity of Csk, which in turn phosphorylates Pragmin on Y238, Y343, and Y391. As Y391 constitutes the Pragmin EPIYA motif, the phosphorylation promotes Pragmin–Csk interaction, which further stimulates Csk kinase activity. The Pragmin–Csk interaction gives rise to morphological changes and induces scattering in epithelial cells, in which a fraction of Pragmin and Csk are co‐localized to focal adhesions, integrin‐containing multiprotein structures that govern cell–ECM interaction.30 The results of the present study therefore uncover a novel role of the Pragmin–Csk complex, which is dependent on tyrosine phosphorylation of the Pragmin EPIYA motif, in the regulation of cell shape and cell motility.
Although Csk shows a strong substrate tropism toward the C‐terminal inhibitory tyrosine residue that is highly conserved among the SFKs (Y530 in the case of human c‐Src), it also phosphorylates non‐SFK substrates.16 C‐terminal Src kinase phosphorylates the CD45 tyrosine phosphatase and thereby creates a binding site for Lck, a T cell‐specific SFK, while enhancing CD45 phosphatase activity.19 Following tyrosine phosphorylation by Csk, platelet endothelial cell adhesion molecule‐1 (PECAM‐1), an adhesion molecule expressed in hematopoietic and endothelial cells, acquires the ability to bind to SH2 domain‐containing phosphatases, SHP1 and SHP2.20 C‐terminal Src kinase also phosphorylates the P2X3 receptor, which is expressed in sensory neurons, and thereby attenuates the receptor function.21 Drosophila Csk phosphorylates large tumor suppressor (LATS) kinase, a key component of the tumor‐suppressive Hippo signaling pathway, indicating a role of Csk in organ/tissue size control.31 The present study adds Pragmin to the list of Csk substrates, while revealing a novel phosphorylation‐dependent functional interaction between Csk and Pragmin. Pragmin has been reported to undergo rapid tyrosine phosphorylation on the EPIYA motif following stimulation with growth factors such as epidermal growth factor.6, 32 C‐terminal Src kinase is considered to be constitutively active and its biological function is primarily determined by its subcellular localization through partner proteins.16 However, similar to Cbp,33 Csk kinase activity is substantially augmented following complex formation with Pragmin. As Csk phosphorylates Pragmin at the EPIYA motif to which Csk binds, Pragmin–Csk complex formation and subsequent enzymatic activation of Csk potentiate EPIYA phosphorylation of Pragmin, creating a positive feedback regulatory loop of Csk by Pragmin (Fig. 6). C‐terminal Src kinase therefore undergoes robust activation at sites where Pragmin–Csk interaction occurs.
Figure 6.
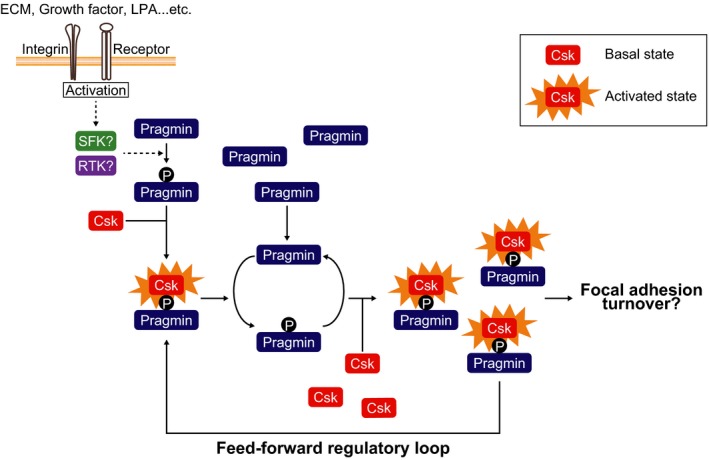
Proposed model for the feed‐forward activation of C‐terminal Src kinase (Csk) by EPIYA‐phosphorylated (p) Pragmin at focal adhesions. ECM, extracellular matrix; LPA, lysophosphatidic acid; RTK, receptor tyrosine kinase; SFK, Src family kinase.
Several studies have shown a connection between Pragmin and oncogenesis. In colon carcinoma cells, Pragmin stimulates the invasive phenotype in an EPIYA phosphorylation‐dependent manner.4 Pragmin is also overexpressed in PDAC cells, in which Pragmin is heavily tyrosine phosphorylated at the EPIYA motif.5 Ectopic expression of Pragmin in human pancreatic ductal epithelial cells to a level that was comparable to that in PDAC cells resulted in increased tyrosine phosphorylation of c‐Src at the C‐terminal inhibitory phosphorylation site (Y530),5 suggesting that the level of elevated Pragmin in PDAC cells is sufficient to activate Csk. In contrast, because of its well‐described role as a negative regulator of SFKs, Csk has been considered a tumor suppressor, despite lack of mutation in human cancers.16 In fact, downregulation of Cbp, which recruits Csk to the plasma membrane, provokes cytoplasmic retention of Csk, which maintains the membrane‐localized SFKs constitutively active.18 Nevertheless, the role of Csk in oncogenesis may be more complicated than expected. In particular, Src has recently been reported to negatively regulate the pro‐oncogenic Ras signaling pathway.24 In this instance, Src selectively binds to and phosphorylates the GTP‐loaded form of Ras on Y32, which inhibits Ras–Raf interaction while promoting Ras–GAP association and subsequent GTP hydrolysis. Thus, Csk‐mediated inactivation of SFKs potentiates the Ras action. C‐terminal Src kinase may therefore play both positive and negative roles in oncogenesis, depending on cell context.
The sequential assembly and disassembly of focal adhesions play a key role in cell migration.34 Loss of Csk in MEF cells impaired cell migration induced by the activation of G protein‐coupled receptor or receptor tyrosine kinase, which was concomitantly associated with a defect in focal adhesion turnover (assembly/disassembly).35 Ectopically expressed Csk was localized to focal adhesions and caused disruption of focal adhesion spots, redistribution of integrins, and inhibition of cell adhesion in HeLa cells.36 At focal adhesions, Csk may regulate integrin‐dependent cell–matrix interaction by phosphorylating both SFKs and non‐SFK substrates that constitute the focal adhesion machineries.30, 34 It is thus intriguing to speculate that spatiotemporal oscillation of the Csk activity underlies the assembly/disassembly of focal adhesions and that the phospho‐EPIYA‐dependent Pragmin–Csk interaction serves as a central player that ensures adequate turnover of cell adhesions that is crucial for cell motility. In this scenario, Pragmin is initially phosphorylated at the EPIYA motif by receptor tyrosine kinase and/or SFK, which are activated in response to mitogenic stimuli.6, 32 This initial Csk activation is then robustly amplified by the autocatalytic Pragmin–Csk interaction, which should be followed by the disruption of the Pragmin–Csk complex to terminate Csk activation. Although the mechanism underlying the complex disruption is currently unknown, it may include tyrosine dephosphorylation of the Pragmin EPIYA motif by phosphatases such as SHP2, which also localizes to focal adhesions.37 In turn, accelerated focal adhesion turnover through deregulation of the Pragmin–Csk axis may induce aberrant cell migration that contributes to tumor invasion and metastasis.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Data S1. Materials and methods.
Table S1. Expression vectors used in this study.
Fig. S1. In vitro interaction of Pragmin with C‐terminal Src kinase (Csk).
Fig. S2. Purification of recombinant Pragmin.
Fig. S3. Schematic representation of full‐length and truncated mutants of Pragmin.
Acknowledgments
We thank Dr. M. Negishi for providing Pragmin vectors. This work was supported by a Grant‐in‐Aid for Scientific Research (A) from the Ministry of Education, Culture, Sports, and Technology of Japan (Grant No. 25250016 to M.H.) and by a Grant‐in‐Aid for JSPS Fellows from Japan Society for the Promotion of Science (Grant No. 14J06406 to Y.S.).
Cancer Sci 107 (2016) 972–980
Funding Information
Japanese Ministry of Education, Culture, Sports, Science, and Technology (25250016); Japan Society for the Promotion of Science (14J06406).
References
- 1. Tanaka H, Katoh H, Negishi M. Pragmin, a novel effector of Rnd2 GTPase, stimulates RhoA activity. J Biol Chem 2006; 281: 10355–64. [DOI] [PubMed] [Google Scholar]
- 2. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298: 1912–34. [DOI] [PubMed] [Google Scholar]
- 3. Boudeau J, Miranda‐Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol 2006; 16: 443–52. [DOI] [PubMed] [Google Scholar]
- 4. Leroy C, Fialin C, Sirvent A et al Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Res 2009; 69: 2279–86. [DOI] [PubMed] [Google Scholar]
- 5. Tactacan CM, Phua YW, Liu L et al The pseudokinase SgK223 promotes invasion of pancreatic ductal epithelial cells through JAK1/Stat3 signaling. Mol Cancer 2015; 14: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Safari F, Murata‐Kamiya N, Saito Y, Hatakeyama M. Mammalian Pragmin regulates Src family kinases via the Glu‐Pro‐Ile‐Tyr‐Ala (EPIYA) motif that is exploited by bacterial effectors. Proc Natl Acad Sci USA 2011; 108: 14938–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Covacci A, Rappuoli R. Tyrosine‐phosphorylated bacterial proteins: Trojan horses for the host cell. J Exp Med 2000; 191: 587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Backert S, Selbach M. Tyrosine‐phosphorylated bacterial effector proteins: the enemies within. Trends Microbiol 2005; 13: 476–84. [DOI] [PubMed] [Google Scholar]
- 9. Mueller D, Tegtmeyer N, Brandt S et al c‐Src and c‐Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest 2012; 122: 1553–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higashi H, Tsutsumi R, Muto S et al SHP‐2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002; 295: 683–6. [DOI] [PubMed] [Google Scholar]
- 11. Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA x SHP‐2 signaling by interaction between CagA and C‐terminal Src kinase. J Biol Chem 2003; 278: 3664–70. [DOI] [PubMed] [Google Scholar]
- 12. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004; 4: 688–94. [DOI] [PubMed] [Google Scholar]
- 13. Chan RJ, Feng GS. PTPN11 is the first identified proto‐oncogene that encodes a tyrosine phosphatase. Blood 2007; 109: 862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev 2007; 17: 23–30. [DOI] [PubMed] [Google Scholar]
- 15. Ohnishi N, Yuasa H, Tanaka S et al Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA 2008; 105: 1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci 2012; 8: 1385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kawabuchi M, Satomi Y, Takao T et al Transmembrane phosphoprotein Cbp regulates the activities of Src‐family tyrosine kinases. Nature 2000; 404: 999–1003. [DOI] [PubMed] [Google Scholar]
- 18. Sirvent A, Benistant C, Pannequin J et al Src family tyrosine kinases‐driven colon cancer cell invasion is induced by Csk membrane delocalization. Oncogene 2010; 29: 1303–15. [DOI] [PubMed] [Google Scholar]
- 19. Autero M, Saharinen J, Pessa‐Morikawa T et al Tyrosine phosphorylation of CD45 phosphotyrosine phosphatase by p50csk kinase creates a binding site for p56lck tyrosine kinase and activates the phosphatase. Mol Cell Biol 1994; 14: 1308–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cao MY, Huber M, Beauchemin N, Famiglietti J, Albelda SM, Veillette A. Regulation of mouse PECAM‐1 tyrosine phosphorylation by the Src and Csk families of protein‐tyrosine kinases. J Biol Chem 1998; 273: 15765–72. [DOI] [PubMed] [Google Scholar]
- 21. D'Arco M, Giniatullin R, Leone V et al The C‐terminal Src inhibitory kinase (Csk)‐mediated tyrosine phosphorylation is a novel molecular mechanism to limit P2X3 receptor function in mouse sensory neurons. J Biol Chem 2009; 284: 21393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao Q, Liu BQ, Li H et al C‐terminal Src kinase (Csk)‐mediated phosphorylation of eukaryotic elongation factor 2 (eEF2) promotes proteolytic cleavage and nuclear translocation of eEF2. J Biol Chem 2014; 289: 12666–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lowry WE, Huang J, Ma YC et al Csk, a critical link of G protein signals to actin cytoskeletal reorganization. Dev Cell 2002; 2: 733–44. [DOI] [PubMed] [Google Scholar]
- 24. Bunda S, Heir P, Srikumar T et al Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc Natl Acad Sci USA 2014; 111: E3785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J 1999; 18: 2459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D. Association of Csk to VE‐cadherin and inhibition of cell proliferation. EMBO J 2005; 24: 1686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vang T, Abrahamsen H, Myklebust S et al Knockdown of C‐terminal Src kinase by siRNA‐mediated RNA interference augments T cell receptor signaling in mature T cells. Eur J Immunol 2004; 34: 2191–9. [DOI] [PubMed] [Google Scholar]
- 28. Nagase L, Hayashi T, Senda T, Hatakeyama M. Dramatic increase in SHP2 binding activity of Helicobacter pylori Western CagA by EPIYA‐C duplication: its implications in gastric carcinogenesis. Sci Rep 2015; 5: 15749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Kelber JA, Tran Cao HS et al Pseudopodium‐enriched atypical kinase 1 regulates the cytoskeleton and cancer progression. Proc Natl Acad Sci USA 2010; 107: 10920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wehrle‐Haller B. Assembly and disassembly of cell matrix adhesions. Curr Opin Cell Biol 2012; 24: 569–81. [DOI] [PubMed] [Google Scholar]
- 31. Stewart RA, Li DM, Huang H, Xu T. A genetic screen for modifiers of the lats tumor suppressor gene identifies C‐terminal Src kinase as a regulator of cell proliferation in Drosophila . Oncogene 2003; 22: 6436–44. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y, Wolf‐Yadlin A, Ross PL et al Time‐resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteomics 2005; 4: 1240–50. [DOI] [PubMed] [Google Scholar]
- 33. Wong L, Lieser SA, Miyashita O et al Coupled motions in the SH2 and kinase domains of Csk control Src phosphorylation. J Mol Biol 2005; 351: 131–43. [DOI] [PubMed] [Google Scholar]
- 34. Webb DJ, Parsons JT, Horwitz AF. Adhesion assembly, disassembly and turnover in migrating cells – over and over and over again. Nat Cell Biol 2002; 4: E97–100. [DOI] [PubMed] [Google Scholar]
- 35. McGarrigle D, Shan D, Yang S, Huang XY. Role of tyrosine kinase Csk in G protein‐coupled receptor‐ and receptor tyrosine kinase‐induced fibroblast cell migration. J Biol Chem 2006; 281: 10583–8. [DOI] [PubMed] [Google Scholar]
- 36. Bergman M, Joukov V, Virtanen I, Alitalo K. Overexpressed Csk tyrosine kinase is localized in focal adhesions, causes reorganization of αvβ5 integrin, and interferes with HeLa cell spreading. Mol Cell Biol 1995; 15: 711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein‐tyrosine phosphatase Shp‐2 regulates cell spreading, migration, and focal adhesion. J Biol Chem 1998; 273: 21125–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and methods.
Table S1. Expression vectors used in this study.
Fig. S1. In vitro interaction of Pragmin with C‐terminal Src kinase (Csk).
Fig. S2. Purification of recombinant Pragmin.
Fig. S3. Schematic representation of full‐length and truncated mutants of Pragmin.