

Abstract
The major mechanism of imatinib (IM) resistance of CML is the reactivation of ABL kinase either through BCR‐ABL gene amplification or mutation. We investigated the cytotoxicity of a pan‐ABL tyrosine kinase inhibitor, ponatinib, and a pan‐histone deacetylase inhibitor, panobinostat, against IM‐resistant CML cells in vitro. Two different IM‐resistant cell lines, K562/IM‐R1 and Ba/F3/T315I were evaluated in comparison with their respective, parental cell lines, K562 and Ba/F3. K562/IM‐R1 overexpressed BCR‐ABL due to gene amplification. Ba/F3/T315I was transfected with a BCR‐ABL gene encoding T315I‐mutated BCR‐ABL. Ponatinib inhibited the growth of both K562/IM‐R1 and Ba/F3/T315I as potently as it inhibited their parental cells with an IC 50 of 2–30 nM. Panobinostat also similarly inhibited the growth of all of the cell lines with an IC 50 of 40–51 nM. This was accompanied by reduced histone deacetylase activity, induced histone H3 acetylation, and an increased protein level of heat shock protein 70, which suggested disruption of heat shock protein 90 chaperone function for BCR‐ABL and its degradation. Importantly, the combination of ponatinib with panobinostat showed synergistic growth inhibition and induced a higher level of apoptosis than the sum of the apoptosis induced by each agent alone in all of the cell lines. Ponatinib inhibited phosphorylation not only of BCR‐ABL but also of downstream signal transducer and activator of transcription 5, protein kinase B, and ERK1/2 in both K562/IM‐R1 and Ba/F3/T315I, and the addition of panobinostat to ponatinib further inhibited these phosphorylations. In conclusion, panobinostat enhanced the cytotoxicity of ponatinib towards IM‐resistant CML cells including those with T315I‐mutated BCR‐ABL.
Keywords: Chronic myelogenous leukemia, combination therapy, imatinib‐resistant, panobinostat, ponatinib
Abbreviations
- AKT
protein kinase B
- CI
combination index
- HDAC
histone deacetylase
- HSP
heat shock protein
- IM
imatinib mesylate
- PACE
Ponatinib Ph‐positive acute lymphoblastic leukemia and CML Evaluation
- STAT5
signal transducer and activator of transcription 5
- TKI
tyrosine kinase inhibitor
Chronic myelogenous leukemia is a malignant transformation of hematopoietic stem cells. The clinical stages of CML consist of three different phases: a chronic phase that is characterized by excessive proliferation, but retains the capacity for differentiation; an accelerated phase that shows a rapid progression after 4–6 years of the chronic phase; and a blast crisis that results in a fatal acute leukemia.1 The genetic abnormality of CML is a reciprocal t(9,22)(q34;q11) chromosomal translocation, the so‐called Philadelphia chromosome. This abnormality generates a BCR‐ABL fusion gene, resulting in the expression of a leukemia‐specific oncoprotein, BCR‐ABL, which is a potent tyrosine kinase that plays a central role in CML pathogenesis.2, 3, 4, 5
Current first‐line treatment options for CML include the TKI IM, and the second‐generation agents, nilotinib and dasatinib. These TKIs all inhibit the BCR‐ABL tyrosine kinase and have dramatically improved the prognosis of CML patients.6, 7, 8, 9 Nevertheless, a small percentage of CML patients are primarily refractory or secondarily resistant to these TKIs.10, 11 Moreover, the prognosis of patients in blast crisis is still poor despite the use of these agents because of drug resistance. The major mechanism of drug resistance of CML is reactivation of the ABL kinase either through BCR‐ABL gene mutation or through gene amplification. Approximately 40% and 20% of the observed drug resistance is due to BCR‐ABL gene mutation and gene amplification, respectively.12, 13 Therefore, new agents that can overcome the reactivation of ABL kinase are needed.
Histone deacetylase inhibitors are emerging anticancer therapeutics. Histone deacetylase inhibitors promote the acetylation of histones in treated cells, which results in chromatin in an opened and transcriptionally permissive state, leading to apoptosis or the inhibition of proliferation. Recently, a pan‐HDAC inhibitor, panobinostat (formerly LBH589), has been reported to have promising anticancer activity.14 Panobinostat is a hydroxamate analog and clinical studies of this agent are currently underway for various hematological malignancies including Hodgkin's lymphoma, cutaneous T‐cell lymphoma, AML, myelodysplastic syndrome, and multiple myeloma.14, 15 Histone deacetylase inhibitors also induce the acetylation of non‐histone proteins such as HSP90, thereby inhibiting its chaperone function. If panobinostat has such a function, then panobinostat may suppress the association between HSP90 and its client protein, BCR‐ABL, leading to BCR‐ABL polyubiquitination and proteasomal degradation.13, 14, 16 Thus, HDAC inhibitors may overcome the cellular resistance of CML cells to TKIs.
The T315I mutation arises in the BCR‐ABL kinase domain from the beginning or during treatment with TKIs including IM, nilotinib, and dasatinib and this mutation has been identified in up to 20% of patients with TKI‐resistant CML.13, 17 This mutation confers CML resistance not only to IM but also to the second‐generation TKIs such as nilotinib and dasatinib.18, 19 The T315I residue is located in the gatekeeper region of the ATP‐binding site of BCR‐ABL, resulting in structural inhibition of the binding of IM, nilotinib, and dasatinib to this region.19, 20 A new pan‐ABL tyrosine kinase inhibitor, ponatinib, is structurally designed to accommodate T315I mutation through its carbon–carbon triple bond linkage.19 Ponatinib has been investigated in a phase II PACE clinical trial in patients who had CML or Philadelphia chromosome‐positive acute lymphoblastic leukemia with resistance or intolerance to nilotinib or dasatinib or with BCR‐ABL T315I mutation. By 12 months of treatment, 56% of 267 patients with chronic phase CML had achieved a major cytogenetic response.17 Thus, ponatinib is a promising therapeutic option in patients with all kinds of BCR‐ABL mutation, including T315I.
We hypothesized that the combination of panobinostat and ponatinib may overcome drug resistance and result in high therapeutic efficacy in CML through the combination of the different mechanisms of action of each agent. To test this hypothesis, the K562/IM‐R1 cell line and the Ba/F3/T315I cell line were used to evaluate the cytotoxicity of panobinostat and ponatinib. The K562/IM‐R1 cell line that was established in our previous study overexpresses BCR‐ABL due to BCR‐ABL gene amplification.21 The Ba/F3/T315I cell line is transfected with a BCR‐ABL gene encoding T315I‐mutated BCR‐ABL.22
Materials and Methods
Cell lines and reagents
Imatinib mesylate and panobinostat were kindly supplied by Novartis‐Pharma (Basel, Switzerland). Ponatinib was purchased from ARIAD Pharmaceuticals (Cambridge, MA, USA). Imatinib mesylate was dissolved in sterile water; panobinostat and ponatinib were dissolved in DMSO. The human CML cell line K562 and its IM‐resistant variant K562/IM‐R1, and the BCR‐ABL‐transfected cell line Ba/F3 and its IM‐resistant variant Ba/F3/T315I, were cultured in RPMI‐1640 media with 10% FBS and maintained in a 5% CO2‐humidified atmosphere at 37°C.
Proliferation assay
To evaluate the proliferative activity of each cell line, the XTT assay was carried out according to the manufacturer's instructions (Roche, Indianapolis, IN, USA) with slight modifications.21 Briefly, 1 mL cells (5 × 104/mL) was incubated for 24 h in a 24‐well plate, followed by the addition of a 10‐μL aliquot of different concentrations of IM, panobinostat, or ponatinib, alone or in combination. The cells were incubated for a further 72 h, and then a 100‐μL aliquot was transferred to a 96‐well microplate. The cells were mixed with 50 μL XTT and incubated for another 4 h at 37°C. Absorbance at 480 nm was analyzed using spectrophotometry with a fluorescent microplate reader (SpectraMax 250; Molecular Device Japan, Ashiya, Japan). The IC50 values were calculated from the growth inhibition curves generated for each treatment.
Calculation of CI
Combination index analysis provides quantitative information on drug interactions. The CI method was based on that described by Chou and Talalay,23 and the values were determined by using the IC50 values of their cell lines and the computer software CalcuSyn (version 2.0) (Biosoft, Great Shelford, UK). Combination index values less than, equal to, and more than 1 indicate synergism, additivity, and antagonism, respectively.
Quantitation of apoptotic cell death
To evaluate cytotoxicity, apoptotic cell death was determined as phosphatidylserine externalization by using the annexin V–FLUOS Staining kit (Roche). At 48 h after treatment, the cells were collected by centrifugation (253 g for 5 min) and washed in PBS and centrifuged again. The pellets were resuspended in 50 μL FITC‐conjugated annexin V mixed with 50 μL propidium iodide. Samples were added to 500 μL HEPES buffer and analyzed by FACS analysis using FACSCanto II (BD Bioscience, Franklin Lakes, NJ, USA). Annexin V‐positive cells were considered to be apoptotic.
Western blot analysis
Protein lysates were extracted from the cells (1 × 107 cells) after treatment for 24 h using the Qproteome Mammalian Protein Prep Kit (Qiagen, Hilden, Germany). The lysates were applied to 7.5% or 12% Mini‐PROTEAN TGX Precast Gels (Bio‐Rad, Hercules, CA, USA) and were electrophoresed and transferred onto Immobilon‐P membranes (Millipore, Billerica, MA, USA). The membranes were probed using standard techniques with the primary antibodies, and subsequently with the secondary antibodies. Amersham ECL Prime Western Blotting Detection Reagent and ImageQuant LAS4000mini (GE Healthcare Life Sciences, Little Chalfont, UK) were used to visualize and quantify protein signals.
Rabbit polyclonal anti‐BCR, anti‐phospho‐BCR (Tyr177), anti‐AKT, anti‐phospho AKT (Ser473), anti‐ERK1/2, anti‐phospho‐ERK1/2, anti‐histone H3 and anti‐HSP70, rabbit monoclonal anti‐phospho‐STAT5 (Tyr694) (all from Cell Signaling Technology, Beverly, MA, USA), mouse monoclonal anti‐STAT5 (Santa Cruz Biotechnology, Dallas, TX, USA), rabbit polyclonal anti‐acetylated histone H3 (Millipore), mouse monoclonal anti‐HSP90 (Enzo Life Science, Farmingdale, NY, USA), and anti‐actin (Sigma, St. Louis, MO, USA) antibodies were used as the primary antibodies. Blocking One and Blocking One‐P (Nacalai Tesque, Kyoto, Japan) were used for the antibody dilutions. Anti‐rabbit IgG–HRP‐linked antibody (Cell Signaling Technology) and anti‐mouse IgG–HRP ‐linked antibody (GE Healthcare Life Sciences) were used as the secondary antibodies.
Activity of HDAC
Activity of HDAC was determined using an HDAC assay kit (Active Motif, Carlsbad, CA, USA). Briefly, 30 μL nuclear extracts, which were extracted from cell samples treated for 12 h with the Qproteome Nuclear Protein kit (Qiagen), were incubated with 1 mM HDAC substrate in the assay buffer in a total volume of 50 μL for 1 h at 37°C, followed by termination of the reaction by addition of 50 μL HDAC developing solution. The fluorescence intensity was measured at a wavelength of 405 nm.
Statistical analyses
All statistical analyses were carried out using Microsoft Excel 2007 software (Microsoft, Redmond, WA, USA). All graphs were generated using GraphPad Prism software (version 5.0) (GraphPad Software, San Diego, CA, USA). Values of P < 0.05 were considered statistically significant.
Results
Synergistic cytotoxicity of panobinostat and ponatinib towards CML cell lines
Growth inhibitory effects of IM, or of panobinostat or ponatinib alone or in combination, on K562 cells, K562/IM‐R1 cells, Ba/F3 cells, and Ba/F3/T315I cells were determined using the XTT assay (Tables 1,2 and Figs 1,2). This assay indicated that K562/IM‐R1 cells and Ba/F3/T315I cells were 12‐fold and 13‐fold more IM‐resistant, respectively, than their parental counterpart (Table 1, Fig. 1). In contrast, panobinostat equally inhibited the growth of K562 cells, K562/IM‐R1 cells, Ba/F3 cells, and Ba/F3/T315I cells with IC50 values of between 40–51 nM (Table 2, Fig. 2). Ponatinib also inhibited the growth of both IM‐resistant K562/IM‐R1 cells and Ba/F3/T315I cells as potently as it inhibited the growth of their respective parental K562 and Ba/F3 cells (Table 2, Fig. 2). Importantly, the combination of panobinostat and ponatinib showed enhanced growth inhibitory effects on all of the cell lines compared to either agent alone (Table 2, Fig. 2). The CI that was calculated based on their IC50 values clearly showed the synergy between these agents in all of the cell lines (Fig. 3).
Table 1.
Sensitivity of CML cell lines to imatinib
| IC50 (μM) | ||||
|---|---|---|---|---|
| K562 | K562/IM‐R1 | Ba/F3 | Ba/F3/T315I | |
| Imatinib | 0.6 | 7.6 (12.6) | 0.6 | 7.8 (13.0) |
Cells were incubated with imatinib for 72 h, followed by XTT assay. IC50 values, which indicate the 50% growth inhibitory concentration, are shown. Numbers in parentheses indicate fold resistance relative to parental cells.
Table 2.
Sensitivity of CML cell lines to panobinostat and/or ponatinib
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| K562 | K562/IM‐R1 | Ba/F3 | Ba/F3/T315I | HL60 | |
| Panobinostat | 50.0 | 51.0 | 40.0 | 47.0 | 29.9 |
| Ponatinib | 2.0 | 3.8 | 5.0 | 30.0 | 796 |
| Combination | 0.7 | 1.3 | 3.7 | 10.0 | 32.1 |
Cells were incubated with the same concentration of panobinostat, ponatinib, or their combination for 72 h, followed by XTT assay. IC50 values, which indicate the 50% growth inhibitory concentration, are shown. HL60 cells that do not express the BCR‐ABL protein were used as a negative control.
Figure 1.
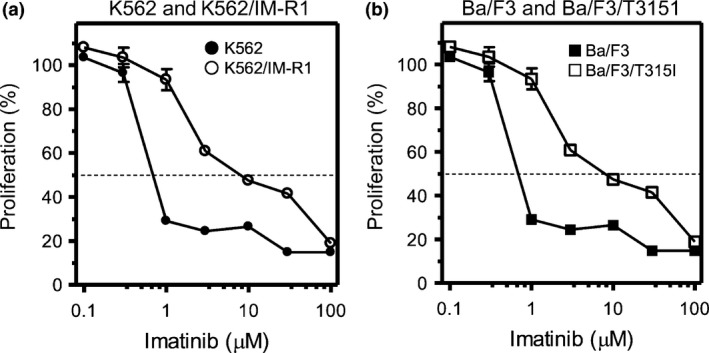
Cellular sensitivity to imatinib mesylate (IM). The CML cell lines and their respective IM‐resistant variants, K562 and K562/IM‐R1 cells (a), Ba/F3 and Ba/F3/T315I cells (b) were incubated for 72 h with IM at various concentrations, followed by the XTT assay for evaluation of cell proliferation. Proliferation was evaluated as a percentage of that in cells not treated with IM. The dotted line indicates 50% proliferation that was used to calculate the IC 50 values.
Figure 2.
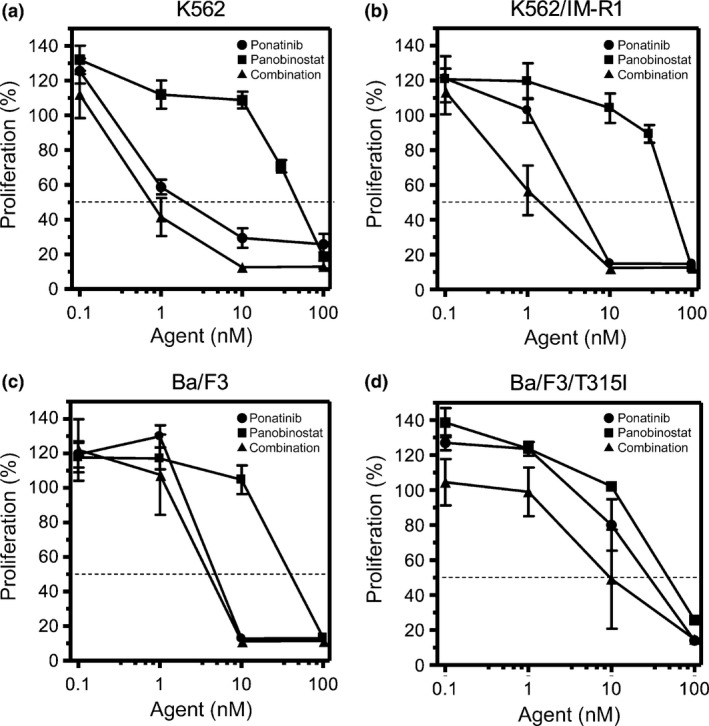
Cellular sensitivity to panobinostat and/or ponatinib. The CML cell lines and their respective imatinib mesylate (IM)‐resistant variants, K562 cells (a) K562/IM‐R1 cells (b), Ba/F3 cells (c), and Ba/F3/T315I cells (d) were incubated with ponatinib or panobinostat, alone or in combination, for 72 h at the indicated concentrations within the range of 0.1–100 nM, followed by XTT assays for evaluation of cell proliferation. Proliferation was evaluated as a percentage of that in cells not treated with ponatinib and/or panobinostat. The dotted line indicates 50% proliferation that was used to calculate the IC 50 values.
Figure 3.
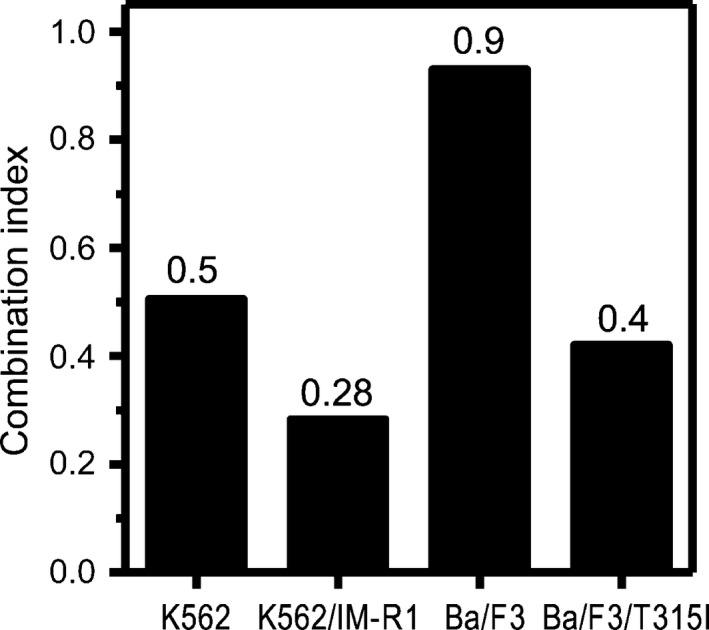
Combination index (CI) values evaluated for panobinostat and ponatinib. The CI values evaluated for panobinostat and ponatinib for the indicated cell lines were calculated using the computer software CalcuSyn (version 2.0). CI < 1, synergistic effect; Cl = 1, addictive effect; CI > 1, antagonistic effect.
We further evaluated drug‐induced apoptosis of these cells using flow cytometry. When the cells were treated for 48 h with panobinostat or ponatinib either alone or in combination, the combination of the two agents induced greater apoptotic cell death than the sum of apoptotic cell death induced by each agent alone in all of the cell lines (Table 3, Fig. 4a–d). Moreover, 30 nM ponatinib combined with 30 nM panobinostat was as cytotoxic as 50 nM ponatinib alone against Ba/F3/T315I cells (Fig. 4e).
Table 3.
Induction of apoptosis in CML cell lines by panobinostat and/or ponatinib
| Mean values (%) | ||||
|---|---|---|---|---|
| K562 | K562/IM‐R1 | Ba/F3 | Ba/F3/T315I | |
| Panobinostat | 20.4 | 17.8 | 5.4 | 12.4 |
| Ponatinib | 34.1 | 23.3 | 24.7 | 36.7 |
| Combination | 70.3 | 65.4 | 44.9 | 71.9 |
Cells were incubated with panobinostat, ponatinib, or their combination at a concentration of 10 nM (K562 cells, K562/IM‐R1 cells, Ba/F3 cells) or 30 nM (Ba/F3/T315I cells) per agent for 48 h, followed by flow cytometric analysis of apoptosis. Apoptotic cell death was evaluated by annexin V positivity. Mean values are shown.
Figure 4.

Effect of panobinostat and/or ponatinib on apoptosis of CML cell lines. The CML cell lines were treated for 48 h with the indicated concentrations of panobinostat or ponatinib, alone or in combination. Apoptotic cell death was evaluated by FACS analysis of annexin V positivity (a–d). (e) The pro‐apoptotic effect of treatment of Ba/F3/T315I cells with 50 nM ponatinib alone, with a combination of 30 nM ponatinib and 30 nM panobinostat, or with 30 nM panobinostat alone was also evaluated. N.S., not significant (P > 0.05).
Thus, panobinostat and ponatinib showed synergistic cytotoxicity towards IM‐resistant cell lines, whose resistance was caused by either BCR‐ABL gene amplification or by BCR‐ABL T315I mutation. The cytotoxicity was as potent as that shown towards IM‐sensitive cell lines.
Inhibition of BCR‐ABL autophosphorylation in CML cell lines by panobinostat and/or ponatinib
The protein expression levels of BCR‐ABL and autophosphorylated BCR‐ABL were determined by Western blotting of cell lines after treatment with panobinostat or ponatinib, alone or in combination. The constitutive expression level of BCR‐ABL was higher in K562/IM‐R1 cells than in K562 cells, indicating the overexpression of the ABL kinase by BCR‐ABL gene amplification in this cell line (Fig. 5a). This result was consistent with our previous findings.21
Figure 5.
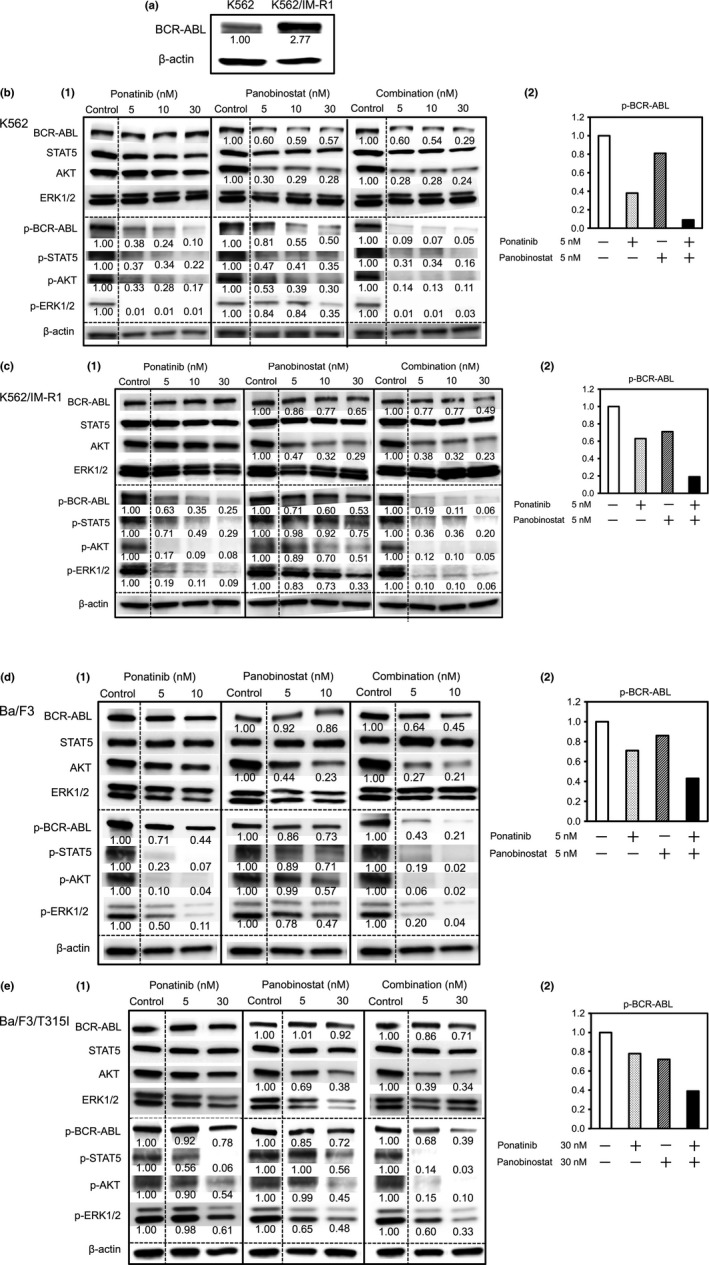
Effect of panobinostat and/or ponatinib on the expression of BCR‐ABL, phospho‐BCR‐ABL, and downstream signaling molecules in CML cell lines. All cell lines were treated for 24 h with panobinostat, ponatinib, or their combination at the indicated concentrations followed by Western blotting of BCR‐ABL expression (a). (b–e) (1) Western blotting of BCR‐ABL, STAT5, AKT, and ERK1/2 proteins (top) or their phosphorylated (p‐) forms (bottom). β‐Actin was assayed as a loading control. Band intensity was determined using densitometry and is indicated below each band. (b–e) (2) The combination of ponatinib with panobinostat at the indicated concentrations inhibited p‐BCR‐ABL expression more strongly than the sum of each agent alone.
Treatment with ponatinib inhibited the autophosphorylation of BCR‐ABL in all cell lines, indicating inhibition of ABL kinase activity. The expression level of BCR‐ABL was unchanged by this treatment. Treatment with panobinostat reduced the protein expression of BCR‐ABL in all cell lines, suggesting that it induced degradation of the BCR‐ABL protein (Fig. 5b–e). The detected autophosphorylation of BCR‐ABL was also reduced by panobinostat, which reflected the decreased expression of the BCR‐ABL protein in the presence of panobinostat. This degradation of BCR‐ABL might be attributable to the disruption of the chaperone function of HSP90 by panobinostat. It was noted that the combination of ponatinib with panobinostat augmented the inhibition of BCR‐ABL autophosphorylation in all of the cell lines (Fig. 5b–e).
Inhibition of BCR‐ABL downstream signal pathways in CML cell lines by panobinostat and/or ponatinib
BCR‐ABL‐dependent signal transduction is critical to the vigorous proliferation of CML cells. Signal transducer and activator of transcription 5, AKT, and ERK1/2 are signaling molecules that are located downstream in BCR‐ABL signal pathways.12 Ponatinib inhibited the phosphorylation of STAT5, AKT, and ERK1/2 in all of the cell lines including in the cell line with T315I mutated BCR‐ABL (Fig. 5b–e). Panobinostat also reduced the phosphorylation of these downstream signals in all of the cell lines in a concentration‐dependent manner (Fig. 5b–e). Moreover, the phosphorylation of downstream signals was more potently inhibited by the combination of ponatinib and panobinostat than by each single agent (Fig. 5b–e). The strong inhibition of phosphorylation of STAT5 in K562 and Ba/F3 cells, of AKT in K562/IM‐R1 and Ba/F3 cells, and of ERK1/2 in K562 and K562/IM‐R1 cells by 5 nM ponatinib alone, may have masked any additional effect of panobinostat. Interestingly, the expression of the AKT protein was reduced by panobinostat, suggesting the possibility that AKT is a client protein of HSP90. Thus, the inhibition of ABL‐kinase activity by ponatinib combined with the induced degradation of BCR‐ABL by panobinostat resulted in a synergistic decrease in the phosphorylation of BCR‐ABL and its downstream signaling pathways.
Inhibition of HDAC activity, induction of histone H3 acetylation, and augmentation of HSP70 protein expression in CML cell lines by panobinostat or panobinostat plus ponatinib
The major mechanism of action of panobinostat is inhibition of HDAC activity. The activity of HDAC was therefore measured in all of the cells after treatment with panobinostat or with panobinostat plus ponatinib. The activity of HDAC was significantly inhibited by panobinostat regardless of the presence of ponatinib in all of the cell lines (Fig. 6). Activity of HDAC was unchanged in these cell lines after treatment with IM or ponatinib alone (data not shown).
Figure 6.

Effect of panobinostat or panobinostat plus ponatinib on histone deacetylase (HDAC) activity in CML cell lines. All cell lines were treated for 12 h with the indicated concentration of panobinostat or panobinostat plus ponatinib. HDAC activity was then determined using an HDAC assay kit. Activity is indicated relative to that in the absence of treatment (100%). K562, *P = 0.026, **P = 0.023; K562/IM‐R1, *P = 0.047, **P = 0.013; Ba/F3, *P = 0.014, **P = 0.037; Ba/F3/T315I, *P = 0.025, **P = 0.041. N.S., not significant (P > 0.05).
Western blot analysis showed the acetylation of histone H3 in cells after treatment that included panobinostat (Fig. 7a). The level of the non‐histone protein, HSP90 was unchanged by treatment of K562 cells with panobinostat or with panobinostat plus ponatinib. Nevertheless, panobinostat increased the protein level of HSP70 in K562 cells (Fig. 7b), which is suggestive of inhibition of the chaperone function of HSP90.24
Figure 7.
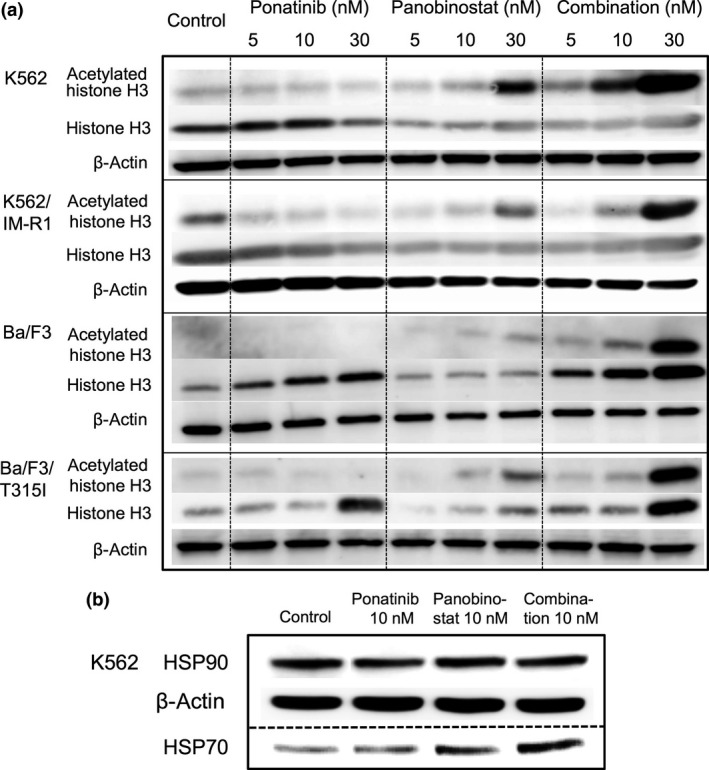
(a) Effect of panobinostat and/or ponatinib on the acetylation status of histone H3 and on the expression level of heat shock protein (HSP)70 in CML cell lines. All cell lines were treated for 24 h with panobinostat, ponatinib, or their combination at the indicated concentrations. Western blotting was then carried out to evaluate the protein expression levels of histone H3 and acetylated histone H3. β‐Actin was assayed as a loading control. (b) Western blotting was also used to evaluate the protein expression levels of HSP90 and HSP70 in K562 cells at 24 and 18 h, respectively, after treatment with 10 nM panobinostat, ponatinib, or their combination.
Discussion
Imatinib resistance in CML is attributed to a variety of mechanisms including reactivation of the BCR‐ABL kinase, increased efflux of the drug through P‐glycoprotein, and compensation for the loss of BCR‐ABL signaling by other kinase pathways such as Src‐family kinases.12, 25, 26, 27, 28, 29, 30, 31, 32 Therefore, to overcome IM resistance the use of new agents with anti‐CML activity that is mediated through different mechanisms of action is necessary. As the major reason for IM resistance is reactivation of the BCR‐ABL kinase, we investigated the cytotoxic effects of the new BCR‐ABL TKI ponatinib combined with the HDAC inhibitor panobinostat against IM‐resistant cell lines, including a cell line that expresses a BCR‐ABL kinase that is active due to T315I mutation.
The present study clearly shows that both ponatinib and panobinostat showed antiproliferative effects and cytotoxicity against the IM‐resistant variant K562/IM‐R1 cells with BCR‐ABL amplification and Ba/F3/T315I cells with BCR‐ABL with a T315I kinase mutation (Figs 2, 3, 4d). Moreover, the combination of panobinostat and ponatinib showed synergistic antiproliferative and cytotoxic effects on these cell lines (Fig. 3). Thus, ponatinib inhibited the constitutive ABL kinase activity in cells expressing gene‐amplified active ABL or in cells expressing ABL with a T315I mutation (Fig. 5b–e). This inhibition of ABL kinase activity was accompanied by inhibition of the phosphorylation of downstream signaling molecules. Panobinostat inhibited HDAC activity, resulting in acetylated histone H3, which might result in inhibition of the proliferation of CML cells.14 Moreover, HSP70 expression was augmented by panobinostat, suggesting that the chaperone function of HSP90 for BCR‐ABL was disrupted (Fig. 7b).24, 33 Therefore, synergistic cytotoxicity exerted by the combination of panobinostat and ponatinib was attributed to their different mechanisms of action: inhibition of the ABL kinase and inhibition of HDAC activity, respectively.
Fiskus et al.34 reported that panobinostat combined with nilotinib induced a greater loss of cell viability and apoptosis of IM‐resistant CML cells, including cells expressing BCR‐ABL with the T315I mutation, than panobinostat or nilotinib alone. In that study, it was not determined how panobinostat enhanced the activity of nilotinib, which structurally cannot contact the BCR‐ABL protein with the T315I mutation. For the present study, we therefore chose to study ponatinib, which can structurally contact the BCR‐ABL protein with the T315I mutation. Okabe et al.35 reported that the combination of vorinostat and ponatinib had synergistic effects against CML cells including cells expressing BCR‐ABL with the T315I mutation. In their study, co‐treatment with vorinostat and ponatinib was cytotoxic towards Ba/F3/T315I cells and ponatinib‐resistant Ba/F3 cells through reduction in the phosphorylation of CRKL, a signaling molecule downstream of BCR‐ABL, and through enhancement of caspase 3 and poly (ADP‐ribose) polymerase activity. Our present study showed that the combination of panobinostat and ponatinib also displayed synergistic antiproliferative and cytotoxic effects towards not only cells expressing BCR‐ABL with the T315I mutation but also towards cells with BCR‐ABL gene amplification (Figs 2, 3, 4d). Moreover, we showed that panobinostat and the combination of panobinostat with ponatinib inhibited HDAC activity (Fig. 6), induced histone H3 acetylation (Fig. 7a), and augmented HSP70 expression (Fig. 7b). These data suggest that the combination of panobinostat and ponatinib may hold promise for the treatment of IM‐resistant cells.
The BCR‐ABL protein activates several molecular mechanisms to inhibit apoptosis.33 Thus, BCR‐ABL increases the phosphorylation and the activity of STAT5, resulting in increased expression of the anti‐apoptotic BCL‐XL protein. BCR‐ABL also induces activation of the phosphoinositide 3‐kinase pathway, resulting in activated AKT kinase that leads to the phosphorylation and inactivation of downstream signaling proteins such as Bcl‐2‐associated death promoter and caspase 9 that regulate apoptosis. Moreover, BCR‐ABL activates Ras/Raf/ERK1/2 and nuclear factor‐kB activities, which together inhibit apoptosis through multiple mechanisms.33, 36 In the present study, ponatinib inhibited both the autophosphorylation of BCR‐ABL and phosphorylation of downstream signaling molecules, resulting in the induction of apoptosis. It has been reported that panobinostat also decreases the levels of generic oncogenic targets, including not only BCR‐ABL but also Fms‐related tyrosine kinase 3, c‐Raf, and AKT through attenuation of the chaperone function of HSP90.14, 34, 37 Heat shock protein 90 is a non‐histone protein that is associated with HDAC6 and that functions as a chaperone of the BCR‐ABL protein, protecting it from degradation. This protection of BCR‐ABL supports activation of the aforementioned downstream pathways, cell cycle proliferation, and apoptosis.14 Accordingly, HSP90 has also been previously suggested to be an important therapeutic target of CML.34, 35
In the present study, panobinostat reduced BCR‐ABL and AKT protein expression (Fig. 5b–e), and increased HSP70 levels (Fig. 7b). These data are suggestive of a decreased chaperone function of HSP90 that would lead to the degradation of BCR‐ABL in both wild‐type and IM‐resistant cells.14, 33, 34, 38 Moreover, the induction of histone H3 acetylation by panobinostat (Fig. 7a) was consistent with a previous report that panobinostat did not exert antiproliferative effects without induction of acetylated histone H3.14 Importantly it was observed that the autophosphorylation of BCR‐ABL following treatment with the combination of ponatinib and panobinostat was lower than that following treatment with either agent alone. These results suggested that inhibition of the phosphorylation of BCR‐ABL by ponatinib combined with the degradation of BCR‐ABL and AKT induced by panobinostat would exert synergistic inhibitory effects on BCR‐ABL signaling.
In the HDAC assay, panobinostat alone or the combination of panobinostat with ponatinib reduced HDAC activity (Fig. 6). The reduction in HDAC activity tended to be greater when the cells were treated with ponatinib together with panobinostat compared to treatment with panobinostat alone (P = 0.33 for K562 cells, P = 0.16 for K562/IM‐R1 cells, P = 0.16 for Ba/F3 cells, and P = 0.18 for Ba/F3/T315I cells; panobinostat versus panobinostat plus ponatinib). Gonzalez‐Zuñiga et al.39 reported that, in Alzheimer's disease, c‐abl stabilized HDAC2, resulting in the repression of neuronal gene expression. In that study, a transgenic mouse model of Alzheimer's disease was treated with IM, which resulted in decreased HDAC2 levels, suggesting that the BCR‐ABL protein was also involved in stabilizing HDAC activity. Therefore, ponatinib might also be associated with the regulation of HDAC through unknown epigenetic mechanisms.
Adverse clinical effects of ponatinib include serious vascular occlusion events such as myocardial infarction or stroke.40 In the PACE trial, patients usually take ponatinib at a dose of 45 mg/day (as a starting dose).17 It was reported that the trough level of ponatinib reaches 40 nM when patients take ponatinib at a dose of 30 mg/day.41, 42 In our study, even in IM‐resistant cells that display active BCR‐ABL, including BCR‐ABL with the T315I mutation, combination therapy of panobinostat and ponatinib at a dosage of 30 nM per agent exerted synergistic cytotoxic effects (Fig. 4d). Flow cytometric analysis of apoptosis indicated that the combination of these two agents at a concentration of 30 nM each agent had similar cytotoxic effects towards Ba/F3/T315I cells as 50 nM ponatinib alone (Fig. 4e). O'Hare et al.19 reported that ponatinib suppressed the emergence of any single mutation of BCR‐ABL at a concentration of 40 nM. Our study suggested that, when panobinostat is combined with ponatinib, low concentrations of these agents might be sufficient to exert strong cytotoxic effects towards CML cells, including towards IM‐resistant cells. Therefore, combining a low concentration of ponatinib with a low concentration of panobinostat might be a possible solution to the problem of adverse effects of higher concentrations of ponatinib.
In conclusion, this is the first report on the combination of ponatinib and panobinostat. We have clearly shown that treatment with a combination of panobinostat and ponatinib synergistically overcame IM resistance that was mediated either though BCR‐ABL gene amplification or through BCR‐ABL T315I mutation. This synergistic effect might be attributable to inhibition of ABL kinase activity combined with degradation of the BCR‐ABL protein. The combination of these two agents might offer a new therapeutic strategy with excellent anti‐CML activity and reduced toxicity.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by a grant from Sugita Genpaku Memorial Obama Municipal Hospital (J110000964).
Cancer Sci 107 (2016) 1029–1038
Funding Information
Sugita Genpaku Memorial Obama Municipal Hospital (J110000964).
References
- 1. Kantarjian HM, Talpaz M. Definition of the accelerated phase of chronic myelogenous leukemia. J Clin Oncol 1988; 6: 180–2. [DOI] [PubMed] [Google Scholar]
- 2. Bartram CR, de Klein A, Hagemeijer A et al Translocation of c‐ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1983; 306: 277–80. [DOI] [PubMed] [Google Scholar]
- 3. Ren R. Mechanisms of BCR‐ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5: 172–83. [DOI] [PubMed] [Google Scholar]
- 4. Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984; 36: 93–9. [DOI] [PubMed] [Google Scholar]
- 5. Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr‐abl oncogene products. Science 1990; 247: 1079–82. [DOI] [PubMed] [Google Scholar]
- 6. Hochhaus A, O'Brien SG, Guilhot F et al Six‐year follow‐up of patients receiving imatinib for the first‐line treatment of chronic myeloid leukemia. Leukemia 2009; 23: 1054–61. [DOI] [PubMed] [Google Scholar]
- 7. Larson RA, Hochhaus A, Hughes TP et al Nilotinib versus imatinib in patients with newly diagnosed Philadelphia chromosome‐positive chronic myeloid leukemia in chronic phase: ENESTnd 3‐year follow‐up. Leukemia 2012; 26: 2197–203. [DOI] [PubMed] [Google Scholar]
- 8. Jabbour E, Kantarjian HM, Saglio G et al Early response with dasatinib or imatinib in chronic myeloid leukemia: 3‐year follow‐up from a randomized phase 3 trial (DASISION). Blood 2014; 123: 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kantarjian HM, O'Brien S, Jabbour E et al Impact of treatment end point definitions on perceived differences in long‐term outcome with tyrosine kinase inhibitor therapy in chronic myeloid leukemia. J Clin Oncol 2011; 29: 3173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corbin AS, Buchdunger E, Pascal F, Druker BJ. Analysis of the structural basis of specificity of inhibition of the Abl kinase by STI571. J Biol Chem 2002; 277: 32214–19. [DOI] [PubMed] [Google Scholar]
- 11. Hochhaus A, Saglio G, Larson RA et al Nilotinib is associated with a reduced incidence of BCR‐ABL mutations versus imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Blood 2013; 121: 3703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kantarjian HM, Giles F, Quintas‐Cardama A, Cortes J. Important therapeutic targets in chronic myelogenous leukemia. Clin Cancer Res 2007; 13: 1089–97. [DOI] [PubMed] [Google Scholar]
- 13. Soverini S, Iacobucci I, Baccarani M, Martinelli G. Targeted therapy and the T315I mutation in Philadelphia‐positive leukemias. Haematologica 2007; 92: 437–9. [DOI] [PubMed] [Google Scholar]
- 14. Atadja P. Development of the pan‐DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett 2009; 280: 233–41. [DOI] [PubMed] [Google Scholar]
- 15. Tan P, Wei A, Mithraprabhu S et al Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high‐risk myelodysplastic syndrome. Blood Cancer J 2014; 4: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics 2013; 7: 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cortes JE, Kim DW, Pinilla‐Ibarz J et al A phase 2 trial of ponatinib in Philadelphia chromosome‐positive leukemias. N Engl J Med 2013; 369: 1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jabbour E, Branford S, Saglio G, Jones D, Cortes JE, Kantarjian HM. Practical advice for determining the role of BCR‐ABL mutations in guiding tyrosine kinase inhibitor therapy in patients with chronic myeloid leukemia. Cancer 2011; 117: 1800–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Hare T, Shakespeare WC, Zhu X et al AP24534, a pan‐BCR‐ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation‐based resistance. Cancer Cell 2009; 16: 401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jabbour E, Kantarjian HM, Jones D et al Characteristics and outcomes of patients with chronic myeloid leukemia and T315I mutation following failure of imatinib mesylate therapy. Blood 2008; 112: 53–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morinaga K, Yamauchi T, Kimura S, Maekawa T, Ueda T. Overcoming imatinib resistance using Src inhibitor CGP76030, Abl inhibitor nilotinib and Abl/Lyn inhibitor INNO‐406 in newly established K562 variants with BCR‐ABL gene amplification. Int J Cancer 2008; 122: 2621–7. [DOI] [PubMed] [Google Scholar]
- 22. Kimura S, Naito H, Segawa H et al NS‐187, a potent and selective dual Bcr‐Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib‐resistant leukemia. Blood 2005; 106: 3948–54. [DOI] [PubMed] [Google Scholar]
- 23. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enz Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 24. Wu L, Yu J, Chen R et al Dual inhibition of Bcr‐Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib‐resistant CML cells. Clin Cancer Res 2015; 21: 833–43. [DOI] [PubMed] [Google Scholar]
- 25. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005; 105: 2640–53. [DOI] [PubMed] [Google Scholar]
- 26. Hochhaus A, Kreil S, Corbin AS et al Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002; 16: 2190–6. [DOI] [PubMed] [Google Scholar]
- 27. Gambacorti‐Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia‐chromosome‐positive leukaemias. Lancet Oncol 2003; 4: 75–85. [DOI] [PubMed] [Google Scholar]
- 28. Walz C, Sattler M. Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML). Crit Rev Oncol Hematol 2006; 57: 145–64. [DOI] [PubMed] [Google Scholar]
- 29. Gorre ME, Mohammed M, Ellwood K et al Clinical resistance to STI‐571 cancer therapy caused by BCR‐ABL gene mutation or amplification. Science 2001; 293: 876–80. [DOI] [PubMed] [Google Scholar]
- 30. Graham SM, Jørgensen HG, Allan E et al Primitive, quiescent, Philadelphia‐positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro . Blood 2002; 99: 319–25. [DOI] [PubMed] [Google Scholar]
- 31. Kumano K, Arai S, Hosoi M et al Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood 2012; 119: 6234–42. [DOI] [PubMed] [Google Scholar]
- 32. Sen R, Natarajan K, Bhullar J et al The novel BCR‐ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR‐associated ATP‐binding cassette transporter ABCG2. Mol Cancer Ther 2012; 11: 2033–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. George P, Bali P, Annavarapu S et al Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17‐AAG is highly active against human CML‐BC cells and AML cells with activating mutation of FLT‐3. Blood 2005; 105: 1768–76. [DOI] [PubMed] [Google Scholar]
- 34. Fiskus W, Pranpat M, Bali P et al Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr‐Abl‐expressing human leukemia cells. Blood 2006; 108: 645–52. [DOI] [PubMed] [Google Scholar]
- 35. Okabe S, Tauchi T, Kimura S et al Combining the ABL1 kinase inhibitor ponatinib and the histone deacetylase inhibitor vorinostat: a potential treatment for BCR‐ABL‐positive leukemia. PLoS ONE 2014; 9: e89080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cilloni D, Saglio G. CML: a model for targeted therapy. Best Pract Res Clin Haematol 2009; 22: 285–94. [DOI] [PubMed] [Google Scholar]
- 37. Fiskus W, Ren Y, Mohapatra A et al Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor‐{alpha} levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res 2007; 13: 4882–90. [DOI] [PubMed] [Google Scholar]
- 38. Bali P, Pranpat M, Bradner J et al Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005; 280: 26729–34. [DOI] [PubMed] [Google Scholar]
- 39. Gonzalez‐Zuñiga M, Contreras PS, Estrada LD et al c‐Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer's disease. Mol Cell 2014; 56: 163–73. [DOI] [PubMed] [Google Scholar]
- 40. FDA Drug Safety Communication . FDA requires multiple new safety measures for leukemia drug Iclusig; company expected to resume marketing. [Cited 12‐20‐2013] Available from URL: http://www.fda.gov/downloads/Drugs/DrugSafety/UCM380607.pdf
- 41. Goldman JM. Ponatinib for chronic myeloid leukemia. N Engl J Med 2012; 367: 2148–9. [DOI] [PubMed] [Google Scholar]
- 42. Cortes JE, Kantarjian H, Shah NP et al Ponatinib in refractory Philadelphia chromosome‐positive leukemias. N Engl J Med 2012; 367: 2075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]