

Abstract
In relation to recent advances in nanobiotechnologies, cancer‐targeted therapy using nano‐scaled drug carriers (nanocarriers) has been attracting enormous attention with success in clinical studies. Polymeric micelles, core–shell‐type nanoparticles formed through the self‐assembly of block copolymers, are one of the most promising nanocarrier, because their critical features such as size, stability, and drug incorporation efficiency and release rate can be modulated by designing the constituent block copolymers. The utilities of polymeric micelles have been reported not only in experimental tumor models in mice but also in clinical studies. In this article, we aim to explain the rationale of designing polymeric micelles for targeting intractable cancers such as pancreatic cancer, glioblastoma, and metastases. Also, we review recent progress in clinical studies on polymeric micelles incorporating anticancer drugs. In addition, we introduce the next generation of polymeric micelles as the platform integrated with smart functionalities such as targetability, environmental sensitivity, and imaging properties. Thus, polymeric micelles can realize safe and effective cancer therapy, and offer tailor‐made medicines for individual patients.
Keywords: Clinical trial, drug targeting, nanomedicine, polymeric micelles, theranostics
Polymeric micelles, core–shell‐type nanoparticles formed through the self‐assembly of block copolymers, have been widely recognized as a promising nanocarrier in cancer targeted therapy (Fig. 1). 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 Various molecular interactions, such as hydrophobic interactions, hydrogen bonding, electrostatic interaction, and metal complex formation in the core‐forming segments, can be a driving force of the formation of polymeric micelles.5, 6, 7, 8, 9 Accordingly, a wide range of therapeutic molecules including hydrophobic substances, charged compounds, and metal complexes can be stably and efficiently incorporated into the micellar core, and their release can be controlled in a sustained or environment‐sensitive manner.5, 6, 7, 8, 9 Compared with surfactant micelles, polymeric micelles show excellent stability characterized by low critical micelle concentration, glass state (solid) core, and kinetic stability.12, 13 Polymeric micelles have a narrowly distributed size controllable in the range of 10–100 nm,14 which should be in contrast with other clinically approved nanocarrier formulations (e.g., Doxil (Janssen Pharmaceutical Co., Titusville, NJ, USA), Abraxane (Celgene Co., Summit, NJ, USA)) with the size of 100 nm. Importantly, these properties, which might critically affect the performance as a drug carrier, can be optimized by fine‐tuning chemical structures and compositions of the micelle‐forming block copolymers.5, 6, 7, 8, 9, 15 In addition, installation of functional molecules such as environment‐responsive cleavable linkages and targetable ligands on the block copolymers allows construction of polymeric micelles with smart functionalities.5, 6, 7, 8, 9, 16, 17, 18, 19 Owing to the above‐mentioned prominent advantages beyond other drug vehicles, several micellar formulations of anticancer drugs are currently under evaluation in preclinical and clinical studies.8, 10 In this article, we review design rationales and recent advances of polymeric micelles for the delivery of anticancer drugs.
Figure 1.
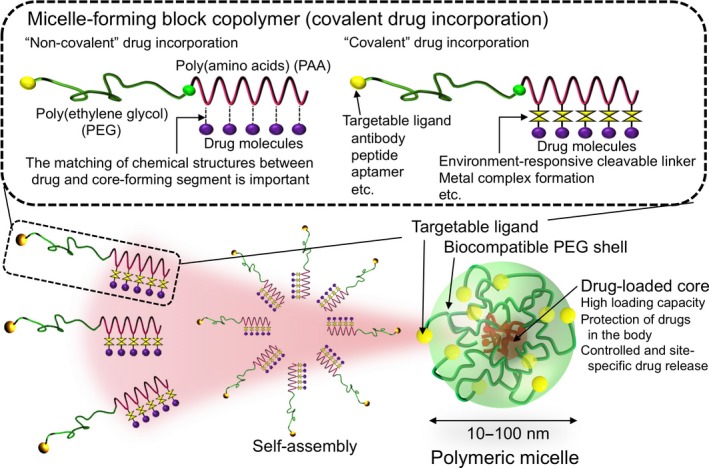
Polymeric micelles constructed through the self‐assembly of block copolymers as nano‐scaled drug carriers. Polymeric micelles have a core–shell structure, where the drug‐loaded core is surrounded by a biocompatible PEG shell, with a narrowly distributed size controllable in the range of 10–100 nm. Engineering the micelle‐forming block copolymers endowed polymeric micelles with on‐demand and smart functionalities such as environment‐sensitivity and targetability.
Design Rationale of Polymeric Micelles
Optimization of block copolymers and in vivo behavior of polymeric micelles
As aforementioned, AB‐type block copolymers assemble into polymeric micelles with characteristic core–shell structures and narrow size distributions in the range of 10–100 nm (Fig. 1).3, 4, 5, 6, 7, 8, 9, 10 The dense PEG palisade protecting the drug‐loaded core can effectively hinder interaction with plasma proteins and cells, avoiding the recognition of the micelle by the reticuloendothelial system in the bloodstream.20, 21 Therefore, polymeric micelles can display prolonged circulation with a half‐life longer than 10 h.3, 4, 5, 6, 7, 14, 15 Note that polymeric micelles finally dissociate into the constituent block copolymers, the size of which is below the threshold of glomerular excretion, thereby avoiding long‐term accumulation in the body. Long‐circulating polymeric micelles effectively accumulate in solid tumors3, 4, 5, 6, 7, 14, 15 due to the augmented leakiness of tumor neovasculature and impaired lymphatic drainage, which is known as the enhanced permeability and retention (EPR) effect.22 Subsequently, polymeric micelles release incorporated drugs in a sustained or microenvironment‐responsive manner.3, 4, 5, 6, 7, 14, 15 Thus, polymeric micelles can achieve tumor‐selective drug activity while minimizing side‐effects in normal tissues.3, 4, 5, 6, 7, 8, 10 Depending on the formulations, polymeric micelles are internalized by cancer cells, and then exert the drug effect in an organelle‐specific manner.5, 6, 7, 9 In such a way, polymeric micelles potentially circumvent drug efflux or intracellular detoxification mechanisms, overcoming drug resistance in cancer cells.23, 24
Polymeric micelles are formed from various block copolymers. Poly(ethylene glycol) is widely used as a shell‐forming polymer due to its hydration property and large excluded volume effect preventing interaction with serum proteins.25 The micellar core is composed of a variety of synthetic polymers, which critically affect the critical properties of polymeric micelles as drug vehicles, including size, association number, critical micelle concentration, drug loading and release, and stability in the bloodstream.4, 5, 6, 7, 13, 14, 15 Biodegradable polyesters such as poly(D,L‐lactide‐co‐glycolide),26 poly(D,L‐lactide),13 poly(ε‐caprolactone),27, 28 and long‐chain alkyl derivatives29 appear to be widely used as a core‐forming polymer. We have used PEG‐b‐poly(amino acids) (PEG‐b‐PAA) copolymers due to freedom of the choice of amino acids and versatile side chain modification to optimize the micelles’ properties.3, 4, 5, 6, 7, 8, 9, 10 Compared with aforementioned polyesters and long‐chain alkyl derivatives, PAA can form stabilized core structures due to various intermolecular interactions such as van der Waals interaction, hydrogen‐bonding, ion‐pair interaction, and dipole interaction.5, 6 Formation of secondary structures of PAA also improves the stability of polymeric micelles. Recently, we reported that the formation of α‐helix of poly(L‐glutamate) might greatly contribute to prolonged blood circulation and effective tumor accumulation of cisplatin‐loaded micelles,15 which are under phase III clinical evaluation (described below). Considering that the plasma clearance of drug vehicles in humans is approximately four times slower than that in mice,30 the micellar structure and drug release rate should be respectively stabler and slower than their expectations in the evaluation using animal models.
Drug loading and controlled drug release
Enabling on‐demand drug incorporation is one of the most prominent features characterizing polymeric micelles. The method of drug incorporation can be classified into “non‐covalent” and “covalent” manners.5 In the non‐covalent drug loading, water‐insoluble compounds are physically entrapped into the micellar core by the dialysis, ultrasound‐aided dispersion or oil in water emulsion methods. A relatively high drug loading capacity of approximately 20% can be achieved without chemical modification of drug molecules. For successful drug incorporation, the compatibility (the matching of chemical structures) between drug molecules and the core‐forming segments should be taken into consideration. Also, the properties of the core‐forming segments such as hydrophobicity, the glass transition temperature, the degree of crystallinity, and secondary structure (e.g., α‐helix formation) should be critical factors. These properties critically affect the efficiency and capacity of drug loading as well as its release behavior. In the covalent drug loading, drug molecules are chemically conjugated to the core‐forming segments. For drug conjugation, the environment‐responsive cleavable linkage is exploited to ensure the drug release at the target site.5, 7, 31, 32 Because the tumor microenvironment is known to develop an acidic condition due to the production of lactate by predominant anaerobic glycolysis in cancer cells (Warburg effect),33 the acid‐cleavable linkage such as hydrazone bond is useful for tumor‐selective drug release.5, 7, 31, 32
The PEG‐b‐PAA copolymers are useful for aforementioned on‐demand drug incorporation due to freedom of the choice of amino acids and versatile side chain modification. For the “non‐covalent” drug incorporation, we reported that chemical conjugation of doxorubicin (DOX) to the side chain of PAA by a stable amide linkage resulted in loss of cytotoxic activity of conjugated DOX but contributed to stable physical entrapment of free DOX through the π–π interaction between the anthracycline structures of conjugated and unconjugated drugs.3, 5 Thus, optimization of chemical structures of the micellar core‐forming segments depending on drug molecules is feasible. Meanwhile, as an example of “covalent” drug incorporation, DOX was conjugated to the core‐forming segments through the hydrazone bond between the carbonyl group at C13 of DOX and the hydrazide group introduced to poly(D,L‐aspartate).7, 34 The DOX‐conjugated block copolymers formed polymeric micelles, which showed acidic pH‐responsive DOX release. Currently, the micellar formulation of a less cardiotoxic epimer, epirubicin (code name NC‐6300/ K‐912) is under phase I clinical study.35, 36 In addition to hydrophilic molecules, metal complexes can be incorporated into PEG‐b‐PAA micelles. cis‐Damminedichloroplatinum(II) (cisplatin, CDDP) and (trans‐l‐1,2‐diaminocyclohexane) platinum(II) (DACHPt, an active complex of oxaliplatin) were complexed with PEG‐b‐poly(L‐glutamate) through Pt(II)–carboxylate complex formation, leading to the formation of narrowly distributed micelles with the size of 30 nm.37, 38 In these systems, the reversible ligand exchange reaction of Pt(II) enables the preferable release of active platinum complexes from the micelles, ensuring their potent cytotoxic activities. After systemic administration, CDDP and DACHPt‐loaded micelles were revealed to show prolonged circulation and effective tumor accumulation, achieving remarkable in vivo antitumor efficacies with reduced side‐effects.37, 39, 40, 41 Currently, CDDP and DACHPt‐loaded micelles (code names NC‐6004 and NC‐4016) are under phase III and I clinical studies, respectively.8, 10
Intracellular drug release using the vehicles may enhance the drug potency. For instance, N‐(2‐hydroxypropyl) methacrylamide copolymer–Dox conjugate and pH‐responsive Dox‐loaded polymeric micelles were reported to overcome the Dox‐resistance in cancer cells as a result of intracellular drug release.23, 42 As we noticed that DACHPt‐loaded micelles accelerate the drug release in the pH and [Cl−] conditions mimicking the late endosomal environment, we tested their in vitro and in vivo efficacies against oxaliplatin‐resistant cancer cells.24 Confocal microscopic observation revealed that DACHPt‐loaded micelles achieved intracellular drug release under both in vitro and in vivo conditions.24 Consequently, DACHPt‐loaded micelles showed remarkable in vitro and in vivo antitumor activities against oxaliplatin‐resistant cancer cells (Fig. 2a).24 We assume that DACHPt‐loaded micelles might circumvent detoxification of DACHPt by metallothionein and methionine synthase overexpressed in the cytoplasm of oxaliplatin‐resistant cancer cells. Thus, polymeric micelles with the function of intracellular drug delivery may behave like a nanoscale Trojan horse, thereby potentially overcoming drug resistance (Fig. 2b).
Figure 2.
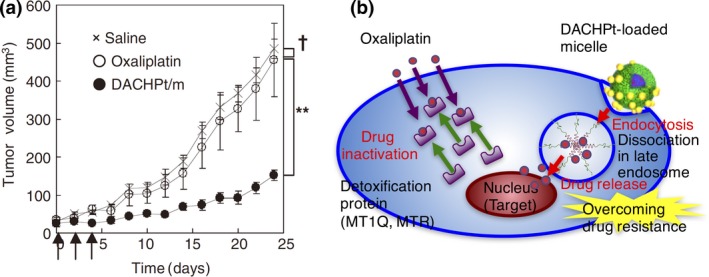
(a) In vivo antitumor activity of (trans‐l‐1,2‐diaminocyclohexane) platinum(II) (DACHPt)‐loaded micelles (DACHPt/m) against oxaliplatin‐resistant human colon adenocarcinoma HT29 (HT29/ox) tumors. (×) Saline; (○) oxaliplatin (8 mg/kg); (●) DACHPt/m (4 mg/kg); (↑) injection of oxaliplatin and DACHPt/m. †P > 0.1; **P < 0.01. Data are expressed as mean ± SEM (n = 4). Reprinted from Murakami et al.,24 with permission from [AAAS, Washington, DC, USA]. (b) Hypothetic mechanism of overcoming oxaliplatin resistance by DACHPt/m. DACHPt/m are assumed to be internalized through the endocytic pathway, and reach the late endosomes and lysosomes closed to the nuclei, leading to facilitated drug release. Therefore, DACHPt/m may bypass the cytoplasmic detoxification pathways consisting of metallothionein (MT1Q) and methionine synthase (MTR), which are overexpressed in HT29/ox cells.
Nucleic acids‐based drugs such as plasmid, antisense DNA, and siRNA can be incorporated into polymeric micelles through polyion complex formation between negatively charged nucleic acids and positively charged PEG‐b‐PAA copolymers.43, 44 Polymeric micelles greatly improve the stability of nucleic acids‐based drugs under in vivo conditions, leading to prolonged blood circulation.16 Integration of endosome escape and organelle‐selective release functionalities led to remarkably enhanced in vitro and in vivo efficacies of nucleic acids‐based drugs in various disease models.16 For details of the delivery of nucleic acids‐based drugs, refer to other expert review papers.9, 45, 46
Design of Polymeric Micelles for Targeting Intractable Cancers
Optimization of the size of polymeric micelles
The clinically approved PEGylated liposomes such as Doxil (Janssen Pharmaceutical Co.) and albumin nanoparticles termed Abraxane have the size of approximately 100 nm. This size might be adequate for effective tumor accumulation based on the EPR effect. Although the transvascular transport of nanoparticles depends on the origin of tumor cells and the microenvironment, solid tumors have a pore cut‐off size larger than 200 nm except for some intractable cancers such as glioblastoma (GBM).47 In this regard, pancreatic cancers and diffuse‐type gastric cancers (scirrhous gastric cancers) have characteristic histological features characterized by less permeable vasculature with pericyte coverage and thick fibrosis (Fig. 3a), which might be an obstacle to extravasation and penetration of nanoparticles.48 Indeed, we have shown that PEGylated liposomes show heterogeneous accumulation in tumor stroma and cannot reach tumor nests in a s.c. model of human pancreatic cancer BxPC3 cells (Fig. 3b). This result motivated us to study the accumulation and penetration of polymeric micelles with different sizes ranging from 30 to 100 nm in BxPC3 tumors.14 As a result, the accumulation of 30‐nm micelles is twofold higher than that of 50‐nm micelles and fourfold higher than that of 70‐ and 100‐nm micelles.14 Note that all the micelles of 30–100 nm displayed similar accumulation levels in a s.c. model of murine colon carcinoma C26 cells, in which the pericyte coverage and tumor stroma are minimal. Co‐administration of 30 and 70 nm micelles in mice bearing BxPC3 tumors revealed that 30‐nm micelles show a uniform intratumoral microdistribution while 70‐nm micelles show heterogeneous localization at perivascular regions (Fig. 3c).14 These results strongly suggest that the accumulation and penetration of polymeric micelles in pancreatic cancer models largely depend on their size, that is, the 30‐nm micelles can bypass the barriers in transvascular transport and penetrate tumor stroma, deeply penetrating tumor nests. Such enhanced tumor accumulation of 30‐nm micelles was observed in spontaneous murine pancreatic tumors in transgenic mice expressing SV40 T antigen and luciferase regulated by the elastase‐1 promoter.41 The 30‐nm DACHPt‐loaded micelles showed remarkably prolonged survival in mice bearing clinically relevant tumors (Fig. 3d).41 Furthermore, a similar size effect was observed in an orthotopic model of human diffuse‐type gastric cancer OCUM‐MLN cells.49
Figure 3.
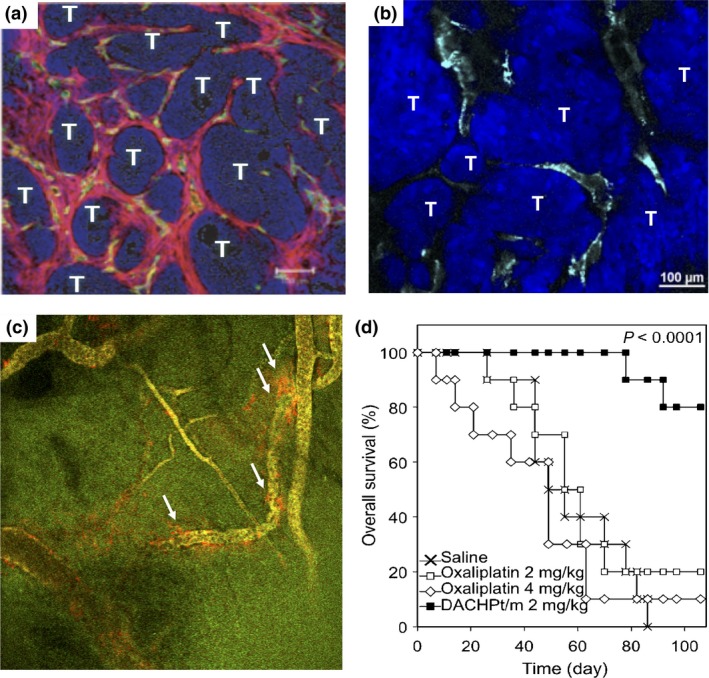
(a) Histology of s.c. inoculated human pancreatic BxPC3 tumors. Red, α‐smooth muscle actin; green, platelet/endothelial cell adhesion molecule‐1; blue, nucleus. T, tumor cells. Reprinted from Kano et al.,48 with permission from [pnas, Washington, DC, USA]. (b) Microdistribution of PEGylated liposomes (cyan) in BxPC3 tumors. (c) Microdistribution of fluorescently labeled 30‐nm (green) and 70‐nm (red) micelles 1 h after injection into BxPC3 tumors. Yellow, colocalization; white arrows, 70‐nm micelles localizing at perivascular regions. Reprinted from Cabral et al.,14 with permission from [NPG, London, UK]. (d) Overall survival of EL1‐luc/TAg mice developing spontaneous pancreatic tumors without treatment, treated with oxaliplatin at 2 mg/kg and 4 mg/kg, and treated with (trans‐l‐1,2‐diaminocyclohexane) platinum(II) (DACHPt)‐loaded micelles (DACHPt/m). Drugs were injected weekly. P‐value calculated using the log–rank test. Reprinted from Cabral et al.,41 with permission from [pnas, Washington, DC, USA].
The size of polymeric micelles is also important for targeting tumor metastasis. Lymph nodes are a common route for metastasis in some tumors. It is known that local administration of nanoparticles to primary tumors leads to accumulation in a neighboring lymph node though the lymphatic vessels, which is clinically applied in the sentinel lymph node biopsy.50 However, such local administration cannot be applicable to systemic delivery of antitumor drugs due to the lack of selectivity, especially when targeting cancer cells are disseminated in the body. Recently, we found that polymeric micelles can accumulate selectively in lymph node metastases through the blood vascular route, which is believed to be specific to active recruitment of lymphocytes to lymph nodes.51 We evaluated the accumulation of systemically injected 30‐ and 70‐nm micelles and 100‐nm PEGylated liposomes in branchial metastatic lymph nodes formed by inoculation of murine melanoma B16‐F10‐luc cells to the left forepaw of mice. As a result, only 30‐nm micelles accumulated and penetrated in the metastatic focus, whereas 70‐nm micelles and PEGylated liposomes did not (Fig. 4a). The 30‐nm micelles did not accumulate in contralateral healthy lymph nodes, suggesting selective accumulation of 30‐nm micelles in metastatic lymph nodes. Importantly, 30‐nm micelles showed comparable accumulation in metastatic lymph nodes and following tumor growth inhibition by antitumor drugs in mice with their primary tumors resected. This result suggests the targeting of lymph node metastasis though the blood vascular route (Fig. 4b). Thus, 30‐nm micelles may be used for systemic delivery of antitumor drugs for the treatment of lymph node metastasis.
Figure 4.
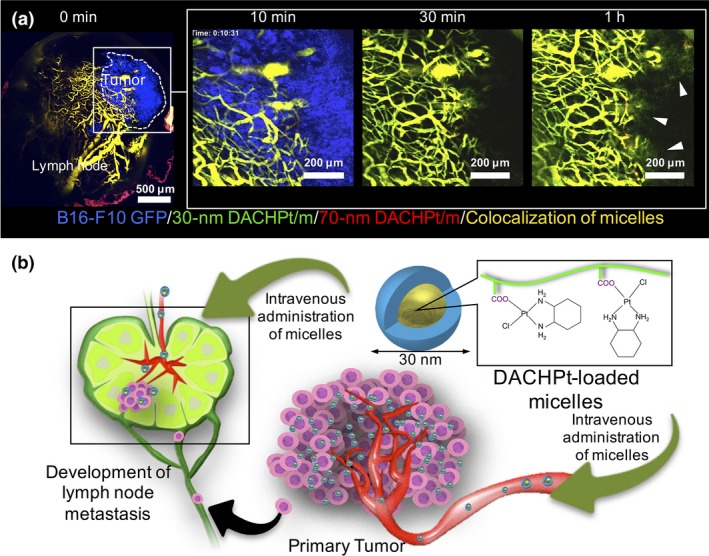
(a) Effect of the size of (trans‐l‐1,2‐diaminocyclohexane) platinum(II)‐loaded micelles (DACHPt/m) on their targeting against lymph node metastasis of murine melanoma B16F10‐GFP cells. Time‐lapse intravital microscopies of metastatic lymph nodes after co‐injection of fluorescent‐labeled 30‐nm (green) and 70‐nm (red) DACHPt/m. Blue, B16F10‐GFP metastasis; yellow, micelles colocalization. To facilitate observation, the tumor region was magnified (white square) and the tumor signal was removed in 30‐min and 1‐h snapshots. White arrowheads indicate extravasation for 30‐nm DACHPt/m. The results suggest that 30‐nm micelles penetrated the metastatic focus. (b) Hypothetic mechanism of the accumulation of DACHPt/m in lymph node metastasis. DACHPt/m are assumed to accumulate in lymph node metastasis through not only the lymphatic route from primary tumors but also the blood vascular route. Reprinted from Cabral et al.,51 with permission from [American Chemical Society, Washington, DC, USA].
Furthermore, we studied the targeting of liver metastases of colon carcinoma C26 cells.52 In this study, we investigated the accumulation of the micelles in different stages of tumor metastasis (from day 2 to day 10 after inoculation). Interestingly, polymeric micelles accumulated not only in overt liver metastases, where the EPR effect is highly expected, but also in early‐stage pre‐angiogenic metastases on day 3. Polymeric micelles showed prolonged retention in the whole metastatic niche (cluster of C26‐GFP cells), where α‐SMA positive stellate cells and CD68‐positive Kupffer cells were recruited. Administration of COX‐2 inhibitor (celecoxib) resulted in significant decrease in accumulation of polymeric micelles in pre‐angiogenic metastases; therefore, the inflammatory microenvironment seems to be a mechanism for the retention of micelles in the metastatic niche. Note that the targeting pre‐angiogenic metastases of C26 cells was not affected by the size of polymeric micelles. Thus, polymeric micelles can target early‐stage pre‐angiogenic metastases.
Actively targetable polymeric micelles
As mentioned, GBM is characterized by limited vascular permeability.47 The pore cut‐off size in GBM is reported to range from 7 to 100 nm, whereas that in other tumors is beyond 200 nm.47 Such limited vascular permeability in GBM is known as the blood–brain tumor barrier (BBTB),53 attenuating the efficacy of some antitumor drugs and nano‐scaled drug vehicles. The integration of targetable ligands on drug vehicles is a promising approach for overcoming BBTB. Recently, we developed DACHPt‐loaded micelles modified with cyclic Arg‐Gly‐Asp (cRGD) peptide.17 The cRGD peptide has a high affinity to αvβ3 integrin overexpressed on the tumor vasculature,54 and has been approved in Europe as an orphan drug named Cilengitide (Merck KGaA, Darmstadt, Germany) for the anti‐angiogenic treatment of GBM.55 The cRGD‐conjugated micelles showed more than twofold higher accumulation in a s.c. model of human GBM U87MG cells compared with non‐targeted micelles having cyclic Arg‐Ala‐Asp ligand, thereby achieving remarkably enhanced efficacy of platinous antitumor drug in orthotopic U87MG tumors.17 The observation by intravital confocal laser scanning microscopy revealed that cRGD‐conjugated micelles show very fast transvascular movement to tumor tissues (Fig. 5), which may not be explained by the passive targeting based on the EPR effect.17 We hypothesize that cRGD‐conjugated micelles might bypass BBTB through the active transvascular transport system, such as transcytosis.
Figure 5.
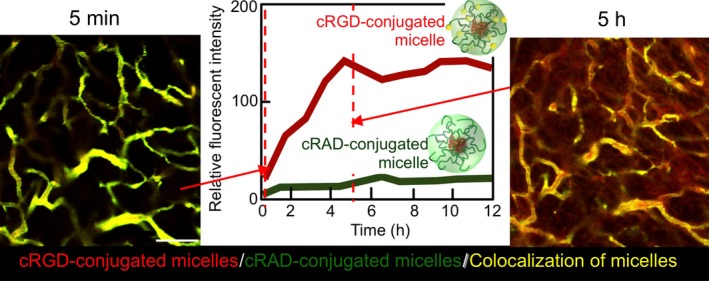
Time‐lapse intravital microscopies of human glioblastoma U87MG tumors after co‐injection of non‐targeted cyclic Arg‐Ala‐Asp‐conjugated (green) and targeted Arg‐Gly‐Asp (cRGD)‐conjugated (red) (trans‐l‐1,2‐diaminocyclohexane) platinum(II) (DACHPt)‐loaded micelles. Yellow, micelles colocalization. Microdistribution of cRGD‐conjugated micelles (red) at 5 h postadministration and quantitative analysis of the number of micelles in tumor sites (center) revealed that cRGD‐conjugated micelles show very fast translocation from the vasculature to tumor tissues. Reprinted from Miura et al.,17 with permission from [American Chemical Society, Washington, DC, USA].
Antibody and its fragments are also useful as targetable ligands to design actively targetable micelles. Several antibody‐conjugated micelles (immunomicelles) have been reported for delivery of antitumor drugs.29, 56, 57, 58 These immunomicelles were conjugated with antibodies targeting osteopontin,29 epidermal growth factor receptor,57 human epidermal growth factor receptor‐2,58 and others. Recently, we reported immunomicelles conjugated with anti‐tissue factor (TF) antibody fragments.18, 19 Tissue factor is known as a primary initiator of blood coagulation, and plays an important role in tumor proliferation, invasion, and metastasis.59 It is reported that the expression level of TF on cancers is associated with patient prognosis.59 We have reported that anti‐TF antibody fragment‐conjugated micelles incorporating epirubicin and DACHPt were efficiently internalized by TF‐overexpressing cancer cells and showed superior in vitro and in vivo antitumor activity to non‐targeted micelles.18, 19 Recently, antibody–drug conjugates (ADCs) have been receiving great attention and many ADCs are under clinical evaluation.60 However, ADCs can load only two to four cytotoxic drugs per antibody, leading to limitation of versatile use, potential side‐effects due to overdose, and rising drug prices. In contrast, immunomicelles can deliver hundreds of drug molecules per antibody, expanding the choice of anticancer drugs and functional design. Thus, immunomicelles are expected to be more versatile platforms for drug delivery than current ADC formulations.
Polymeric Micelles Under Clinical Evaluation
Paclitaxel (PTX)‐loaded polymeric micelles formed from PEG‐b‐poly(D,L‐lactide) (Genexol‐PM) (Samyang Biopharm Co., Seoul, Korea) have been approved for the treatment of breast cancer, non‐small‐cell lung cancer, and ovarian cancer.61, 62 Expansion of clinical applications of Genexol‐PM to other cancers is under clinical evaluation.63, 64
In our system, five micellar formulations incorporating PTX (NK105), cisplatin (NC‐6004), SN‐38 (NK102), dachplatin (active complex of oxaliplatin) (NC‐4016), and epirubicin (NC‐6300/K‐912) are currently under clinical evaluation.8, 10 Among them, NC‐6004 progressed to phase III study and approval application of NK105 will be undertaken in 2016.
NK105 was reported to show enhanced antitumor activity and reduced PTX‐induced peripheral neuropathy in a preclinical study.65 Also, NK105 can solubilize PTX without Cremophor EL (CEL) (BASF Co., Ludwigshafen, Germany), preventing CEL‐induced hypersensitive reactions. In a phase I study, NK105 was given by i.v. infusion for 1 h without anti‐allergic premedication.66 Of 19 patients, only one experienced allergic reactions. A partial response in one pancreatic cancer patient and stable disease lasting 10 to seven courses depending on the patients with colon or gastric cancers were observed. In a pharmacokinetic study, NK105 (at 150 mg/m2) showed 30‐fold higher plasma area under the receiver–operating characteristic curve than conventional PTX‐CEL formulation (at 210 mg/m2). Dose‐limiting toxicity (DLT) of NK105 was grade 4 neutropenia. In a phase II study against advanced stomach cancer, as a second‐line therapy, patients received NK105 by i.v. infusion for 0.5 h once every 3 weeks without anti‐allergic premedication.67 As a result, the overall response rate of NK105 accounted for 25% of patients, with two achieving complete response and 12 partial responses. The most observed grade 3/4 hematological toxicity was neutropenia; there was no grade 3/4 non‐hematological toxicity and no hypersensitive reactions without anti‐allergic premedication. Importantly, grade 3 neurotoxicity was observed for only one in 57 patients (1.8%), which is in contrast with other PTX delivery systems such as Xyotax (CTI BioPharm Co., Seattle, WA, USA) and Abraxane, showing grade 3 neurotoxicity in 10–15% of patients. Since 2012, the phase III study of NK105 for comparison with conventional PTX‐CEL formulation has started in multiple countries including Japan, Taiwan, and Korea, and patient registration was completed in 2014. Pending the successful outcome of the phase III study, approval application of NK105 will be undertaken in 2016.
NC‐6004 has undergone clinical studies in different countries. In a preclinical study, NC‐6004 was shown to prevent renal toxicity, DLT of CDDP, because 30‐nm micelles do not undergo glomerular filtration, and decreasing C max of CDDP in the proximal tubule.39 NC‐6004 also prevented CDDP‐induced ototoxicity.40 As a result, the phase I study of NC‐6004 in the UK was carried out using 1‐h infusion once every three weeks and hydration with 1000 mL saline on the day of drug treatment.68 In all the patients, NC‐6004 was given without hospitalization. Although nausea and vomiting caused by NC‐6004 were milder than conventional CDDP treatment, hypersensitive reactions were more frequently observed in NC‐6004 treatment. The maximum tolerated dose and recommended dose for the phase II study were determined to be 120 and 90 mg/m2, respectively. In a phase I/II study, NC‐6004 in combination with gemcitabine was tested in advanced pancreatic cancer patients.69 In these studies, the dose of NC‐6004 reached 90 mg/m2 without hydration, and hypersensitivity observed in phase I study was prevented by dexamethasone premedication. Currently, NC‐6004 has progressed to phase III study for comparison between the combination of NC‐6004 and gemcitabine and gemcitabine alone in advanced or metastatic pancreatic cancer in Asian countries. A phase I/II study of NC‐6004 against non‐small‐cell lung cancer patients is ongoing in the USA.
NC‐6300/K‐912 is a pH‐responsive polymeric micelle that can selectively release epirubicin under acidic conditions of intratumoral microenvironment or the endo‐/lysosomal compartment in cancer cells.34 In preclinical study, NC‐6300/K‐912 was reported to show improved accumulation in solid tumors and reduced accumulation in normal tissues.35 Importantly, NC‐6300/K‐912 prevented cardiotoxicity and DLT of anthracycline anticancer drugs, due to decreased accumulation in cardiac muscle tissues.36 Currently, a phase I study of NC‐6300/K‐912 is ongoing at the National Cancer Center in Japan.
Future Formulations and Applications of Polymeric Micelles
The preferential tumor accumulation of nano‐scaled drug vehicles has been reported not only for animal models but also for patients with various cancers including non‐small‐cell lung cancer, squamous cell lung cancer, breast cancer, and ovarian cancer.30, 70 However, it is likely that the accumulation level and intratumoral distributions of drug vehicles might depend on different types of cancers in individual patients.71 Such interpatient and intratumoral variations may change during treatment, and finally should affect the therapeutic outcome. Therefore, it might be important to evaluate such interpatient and intratumoral variations in individual patients before and during treatment with anticancer drugs formulated in nano‐scaled vehicles. This information may be helpful for estimating the therapeutic effect and designing and optimizing the therapeutic protocols for individual patients (tailor‐made nanomedicine). In this regard, we have devoted great efforts to develop polymeric micelles incorporating MRI contrast agents such as gadolinium‐diethylenetriaminepentaacetic acid (Gd‐DTPA) (for T1‐weighted images)72 and superparamagnetic iron‐oxide nanoparticles (for T2‐weighted images).73 These diagnostic micelles were shown to visualize s.c. and orthotopic models of various cancers, including pancreatic cancers. In addition, DACHPt‐loaded micelles have been integrated with imaging functionality by incorporating Gd‐DTPA through the reversible complex formation between DACHPt and Gd‐DTPA.74 Interestingly, incorporation of Gd‐DTPA into polymeric micelles drastically increased longitudinal relaxivity, r1 (from 3.5 to 80.5 mmol/L/s) due to the flexibility reduction per Gd molecule and the increase of the rotational correlation time. In the animal experiment, the accumulation of Gd‐DTPA/DACHPt‐loaded micelles in orthotopically inoculated pancreatic cancer was successfully visualized in a real‐time manner by MRI (Fig. 6). The non‐invasive monitoring of tumor volume during treatment was also feasible by Gd‐DTPA/DACHPt‐loaded micelles. Such dual diagnostic and therapeutic functions are called “theranostic”, and have been attracting increasing attention.75, 76 In the future, diagnostic and theranostic nanoparticles will evolve functions of molecular imaging to detect cellular responses to therapeutic agents and histological information. Thus, imaging functionality is expected to improve reliability and safety of targeted therapy.
Figure 6.
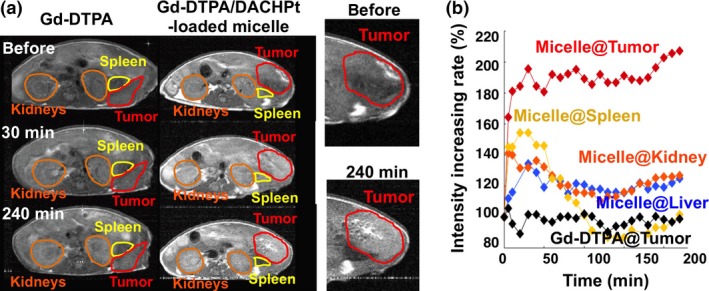
(a) Magnetic resonance images (T1‐weighted) of mice bearing an orthotopic pancreatic cancer (BxPC3) before and after injection of gadolinium‐diethylenetriaminepentaacetic acid (Gd‐DTPA) or Gd‐DTPA/(trans‐l‐1,2‐diaminocyclohexane) platinum(II) (DACHPt)‐loaded micelles. (b) Relative MRI intensity in tumor and each organ after injection of Gd‐DTPA or Gd‐DTPA/DACHPt‐loaded micelles. Micelles showed tumor‐specific enhancement of MRI signal. Reprinted from Kaida et al.,74 with permission from [AACR, Philadelphia, PA, USA].
Conclusion
Increasing numbers of important drugs have been confronting the problem of forfeiture of patent; therefore, product life cycle management has been recognized as being increasingly important. It is getting more difficult to find new drug compounds by traditional drug discovery and development technologies. Even if new lead compounds are identified, most of them show serious side‐effects in preclinical and clinical studies, as potent compounds often target the molecules in the upstream signal transduction pathways. In this regard, drug delivery systems (DDS) can improve the efficacy and safety of existing drug molecules, offering usefulness for product life cycle management. Drug delivery systems can solve the toxicity problems of potent new compounds and promote their practical applications. They can provide new functions to drug molecules by integrating functional molecules to the DDS platform. Thus, we believe that DDS will be more important in drug discovery and development. Among various DDS platforms, polymeric micelles are the most promising due to their versatile and tailor‐made designs based on polymer chemistry and nanobiotechnology. During the last two decades, polymeric micelles as drug vehicles have made rapid progress and several formulations are already being evaluated in late‐stage clinical trials. We expect the approval of these micellar anticancer drugs and generation of innovative micellar nanomedicines with smart functionalities in the near future.
Disclosure Statement
Nobuhiro Nishiyama and Kazunori Kataoka are inventors of some of polymeric micelles in clinical studies. But we transfered all the rights to the University of Tokyo. The University of Tokyo licenced to Nanocarrier Corporation.
Acknowledgments
This research was partially supported by the Center of Innovation Program from Japan Science and Technology Agency.
Cancer Sci 107 (2016) 867–874
Funding Information
Japan Science and Technology Agency
References
- 1. Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer 2006; 6: 688–701. [DOI] [PubMed] [Google Scholar]
- 2. Hubbell JA, Langer R. Translating materials design to the clinic. Nat Mater 2013; 12: 963–6. [DOI] [PubMed] [Google Scholar]
- 3. Kataoka K, Kwon GS, Yokoyama M, Okano T, Sakurai Y. Block copolymer micelles as vehicles for drug delivery. J Control Release 1993; 24: 119–32. [Google Scholar]
- 4. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev 2001; 47: 113–31. [DOI] [PubMed] [Google Scholar]
- 5. Nishiyama N, Kataoka K. Nanostructured devices based on block copolymer assemblies for drug delivery: designing structures for enhanced drug function. Adv Polym Sci 2006; 193: 67–101. [Google Scholar]
- 6. Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther 2006; 112: 630–48. [DOI] [PubMed] [Google Scholar]
- 7. Bae Y, Kataoka K. Intelligent polymeric micelles from functional poly(ethylene glycol)‐poly(amino acid) block copolymers. Adv Drug Deliv Rev 2009; 61: 768–84. [DOI] [PubMed] [Google Scholar]
- 8. Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent‐incorporating polymer micelles. Cancer Sci 2009; 100: 572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miyata K, Nishiyama N, Kataoka K. Rational design of smart supramolecular assemblies for gene delivery: chemical challenges in the creation of artificial viruses. Chem Soc Rev 2012; 41: 2562–74. [DOI] [PubMed] [Google Scholar]
- 10. Cabral H, Kataoka K. Progress of drug‐loaded polymeric micelles into clinical studies. J Control Release 2014; 190: 465–76. [DOI] [PubMed] [Google Scholar]
- 11. Kato KS, Nishiyama N, Kozaki M et al General considerations regarding the in vitro and in vivo properties of block copolymer micelle products and their evaluation. J Control Release 2015; 210: 76–83. [DOI] [PubMed] [Google Scholar]
- 12. Yokoyama M, Sugiyama T, Okano T et al Analysis of micelle formation of an adriamycin‐conjugated poly(ethylene glycol)‐poly(aspartic acid) block copolymer by gel permeation chromatography. Pharm Res 1993; 10: 895–9. [DOI] [PubMed] [Google Scholar]
- 13. Yamamoto Y, Yasugi K, Harada A, Nagasaki Y, Kataoka K. Temperature‐related change in the properties relevant to drug delivery of poly(ethylene glycol)‐poly(D, L‐lactide) block copolymer micelles in aqueous milieu. J Control Release 2002; 82: 359–71. [DOI] [PubMed] [Google Scholar]
- 14. Cabral H, Matsumoto Y, Mizuno K et al Accumulation of sub‐100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol 2011; 6: 815–23. [DOI] [PubMed] [Google Scholar]
- 15. Mochida Y, Cabral H, Miura Y et al Bundled assembly of helical nanostructures in polymeric micelles loaded with platinum drugs enhancing therapeutic efficiency against pancreatic tumor. ACS Nano 2014; 8: 6724–38. [DOI] [PubMed] [Google Scholar]
- 16. Christie RJ, Matsumoto Y, Miyata K et al Targeted polymeric micelles for siRNA treatment of experimental cancer by intravenous injection. ACS Nano 2012; 6: 5174–89. [DOI] [PubMed] [Google Scholar]
- 17. Miura Y, Takenaka T, Toh K et al Cyclic RGD‐linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood‐brain tumor barrier. ACS Nano 2013; 7: 8583–92. [DOI] [PubMed] [Google Scholar]
- 18. Ahn JY, Miura Y, Yamada N et al Antibody fragment‐conjugated polymeric micelles incorporating platinum drugs for targeted therapy of pancreatic cancer. Biomaterials 2015; 39: 23–30. [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto Y, Hyodo I, Koga Y et al Enhanced antitumor effect of anti‐tissue factor antibody‐conjugated epirubicin‐incorporating micelles in xenograft models. Cancer Sci 2015; 106: 627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stolnik S, Illum L, Davis SS. Long circulating microparticulate drug carriers. Adv Drug Deliv Rev 1995; 16: 195–214. [Google Scholar]
- 21. Mosqueria VCF, Legrand P, Gulik A et al Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG‐modified nanoparticles. Biomaterials 2001; 2001: 2967–79. [DOI] [PubMed] [Google Scholar]
- 22. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 23. Lee ES, Na K, Bae YH. Doxorubicin loaded pH‐sensitive polymeric micelles for reversal of resistant MCF‐7 tumor. J Control Release. 2005; 103: 405–18. [DOI] [PubMed] [Google Scholar]
- 24. Murakami M, Cabral H, Matsumoto Y et al Improving drug potency and efficacy by nanocarrier‐mediated subcellular targeting. Sci Transl Med 2011; 3: 64ra2. [DOI] [PubMed] [Google Scholar]
- 25. Harris JM. Introduction to biotechnical and biomedical applications of poly(ethylene glycol) (Ch 1) In: Harris JM, ed. Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications. New York: Plenum, 1992; 1–13. [Google Scholar]
- 26. Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long‐circulating polymeric nanospheres. Science 1994; 263: 1600–3. [DOI] [PubMed] [Google Scholar]
- 27. Soleymani Abyaneh H, Vakili MR, Zhang F, Choi P, Lavasanifar A. Rationale design of block copolymer micelles to control burst drug release at a nanoscale dimension. Acta Biomater 2015; 24: 127–39. [DOI] [PubMed] [Google Scholar]
- 28. Fonge H, Huang H, Scollard D, Reilly RM, Allen C. Influence of formulation variables on the biodistribution of multifunctional block copolymer micelles. J Control Release 2012; 157: 366–74. [DOI] [PubMed] [Google Scholar]
- 29. Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos‐Sternberg B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci USA 2003; 100: 6039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harrington KJ, Mohammadtaghi S, Uster PS et al Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin Cancer Res 2001; 7: 243–54. [PubMed] [Google Scholar]
- 31. Ulbrich K, Subr V. Polymeric anticancer drugs with pH‐controlled activation. Adv Drug Deliv Rev 2004; 56: 1023–50. [DOI] [PubMed] [Google Scholar]
- 32. Gillies ER, Fréchet JM. A new approach towards acid sensitive copolymer micelles for drug delivery. Chem Commun 2003; 14: 1640–1. [DOI] [PubMed] [Google Scholar]
- 33. Vander H, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bae Y, Nishiyama N, Fukushima S, Koyama H, Matsumura Y, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH‐triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem 2005; 16: 122–30. [DOI] [PubMed] [Google Scholar]
- 35. Harada M, Bobe I, Saito H et al Improved anti‐tumor activity of stabilized anthracycline polymeric micelle formulation, NC‐6300. Cancer Sci 2011; 102: 192–9. [DOI] [PubMed] [Google Scholar]
- 36. Takahashi A, Yamamoto Y, Yasunaga M et al NC‐6300, an epirubicin‐incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin. Cancer Sci 2013; 104: 920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishiyama N, Okazaki S, Cabral H et al Novel cisplatin‐incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res 2003; 63: 8977–83. [PubMed] [Google Scholar]
- 38. Cabral H, Nishiyama N, Okazaki S et al Preparation and biological properties of dichloro(1,2‐diaminocyclohexane) platinum (II) loaded polymeric micelles. J Control Release 2005; 101: 223–32. [DOI] [PubMed] [Google Scholar]
- 39. Uchino H, Matsumura Y, Negishi T et al Cisplatin‐incorporating polymeric micelles (NC‐6004) can reduce nephrotoxicity and neurotoxicity of cispatin in rats. Br J Cancer 2005; 93: 678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baba M, Matsumoto Y, Kashio A et al Micellization of cisplatin (NC‐6004) reduces its ototoxicity in guinea pigs. J Control Release 2012; 157: 112–17. [DOI] [PubMed] [Google Scholar]
- 41. Cabral H, Murakami M, Hojo H et al Targeted therapy of spontaneous murine pancreatic tumors by polymeric micelles prolongs survival and prevents peritoneal metastasis. Proc Natl Acad Sci USA 2013; 110: 11397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Minko T, Kopeckova P, Kopecek J. Efficacy of the chemotherapeutic action of HPMA copolymer‐bound doxorubicin in a solid tumor model of ovarian carcinoma. Int J Cancer 2000; 86: 108–17. [DOI] [PubMed] [Google Scholar]
- 43. Katayose S, Kataoka K. Water‐soluble polyion complex associates of DNA and poly(ethylene glycol)‐poly(L‐lysine) block copolymer. Bioconjug Chem 1997; 8: 702–7. [DOI] [PubMed] [Google Scholar]
- 44. Kataoka K, Togawa H, Harada A, Yasugi K, Matsumoto T, Katayose S. Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline. Macromolecules 1996; 29: 8556–7. [Google Scholar]
- 45. Lee Y, Kataoka K. Delivery of nucleic acid drugs. Adv Polym Sci 2012; 249: 95–134. [Google Scholar]
- 46. Itaka K, Kataoka K. Progress and prospects of polyplex nanomicelles for plasmid DNA delivery. Curr Gene Ther 2011; 11: 457–65. [DOI] [PubMed] [Google Scholar]
- 47. Hobbs SK, Monsky WL, Yuan F et al Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 1998; 95: 4607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kano MR, Bae Y, Iwata C et al Improvement of cancer‐targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF‐signaling. Proc Natl Acad Sci USA 2007; 104: 3460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rafi M, Cabral H, Kano MR et al Polymeric micelles incorporating (1,2‐diaminocyclohexane)platinum (II) suppress the growth of orthotopic scirrhous gastric tumors and their lymph node metastasis. J Control Release 2012; 159: 189–96. [DOI] [PubMed] [Google Scholar]
- 50. Harisinghani MG, Barentsz J, Hahn PF et al Noninvasive detection of clinically occult lymph‐node metastases in prostate cancer. New Eng J Med 2003; 348: 2491–9. [DOI] [PubMed] [Google Scholar]
- 51. Cabral H, Makino J, Matsumoto Y et al Systemic targeting of lymph node metastasis through the blood vascular system by using size‐controlled nanocarriers. ACS Nano 2015; 9: 4957–67. [DOI] [PubMed] [Google Scholar]
- 52. Wu H, Cabral H, Toh K et al Polymeric micelles loaded with platinum anticancer drugs target preangiogenic micrometastatic niches associated with inflammation. J Control Release 2014; 189: 1–10. [DOI] [PubMed] [Google Scholar]
- 53. Groothuis DR. The blood‐brain and blood tumor barriers: a review of strategies for increasing drug delivery. Neuro Oncol 2000; 2: 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Danhier F, Breton AL, Préat V. RGD‐based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm 2012; 9: 2961–73. [DOI] [PubMed] [Google Scholar]
- 55. Scaringi C, Minniti G, Caporello P, Enrici RM. Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer Res 2012; 32: 4213–23. [PubMed] [Google Scholar]
- 56. Sawant RR, Jhaveri AM, Torchilin VP. Immunomicelles for advancing personalized therapy. Adv Drug Deliv Rev 2012; 64: 1436–46. [DOI] [PubMed] [Google Scholar]
- 57. Noh T, Kook YH, Park C et al Block copolymer micelles conjugated with anti‐EGFR antibody for targeted delivery of anticancer drug. J Polym Sci Pol Chem 2008; 46: 7321–31. [Google Scholar]
- 58. Li W, Zhao H, Qian W et al Chemotherapy for gastric cancer by finely tailoring anti‐Her2 anchored dual targeting immunomicelles. Biomaterials 2012; 33: 5349–62. [DOI] [PubMed] [Google Scholar]
- 59. van den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood 2012; 119: 924–32. [DOI] [PubMed] [Google Scholar]
- 60. Mullard A. Maturing antibody–drug conjugate pipeline hits 30. Nat Rev Drug Discov 2013; 12: 329–32. [DOI] [PubMed] [Google Scholar]
- 61. Lee KS, Chung HC, Im SA et al Multicenter phase II trial of Genexol‐PM, a Cremophor‐free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat 2008; 108: 241–50. [DOI] [PubMed] [Google Scholar]
- 62. Kim DW, Kim SY, Kim HK et al Multicenter phase II trial of Genexol‐PM, a novel Cremophor‐free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non‐small‐cell lung cancer. Ann Oncol 2007; 18: 2009–14. [DOI] [PubMed] [Google Scholar]
- 63. Lee JL, Ahn JH, Park SH et al Phase II study of a cremophor‐free, polymeric micelle formulation of paclitaxel for patients with advanced urothelial cancer previously treated with gemcitabine and platinum. Invest New Drugs 2012; 30: 1984–90. [DOI] [PubMed] [Google Scholar]
- 64. Kim HS, Lee JY, Lim SH et al A prospective phase II study of cisplatin and Cremophor EL‐free paclitaxel (Genexol‐PM) in patients with unresectable thymic epithelial tumors. J Thorac Oncol 2015; 10: 1800–6. [DOI] [PubMed] [Google Scholar]
- 65. Hamaguchi T, Matsumura Y, Suzuki M et al NK105, a paclitaxel‐incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer 2005; 92: 1240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hamaguchi T, Kato K, Yasui H et al A phase I and pharmacokinetic study of NK105, a paclitaxel‐incorporating micellar nanoparticle formulation. Br J Cancer 2007; 97: 170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kato K, Chin K, Yoshikawa K et al Phase II study of NK105, a paclitaxel‐incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer. Invest New Drugs 2012; 30: 1621–7. [DOI] [PubMed] [Google Scholar]
- 68. Plummer R, Wilson RH, Calvert H et al A phase I clinical study of cisplatin‐incorporated polymeric micelles (NC‐6004) in patients with solid tumors. Br J Cancer 2011; 104: 593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Su WC, Chen LT, Li CP et al A novel micellar formulation of cisplatin, in combination with gemcitabine in patients with pancreatic cancer in Asia ‐ Results of Phase I. ESMO 2012; Abstract 863. [Google Scholar]
- 70. Seymour LW, Ferry DR, Kerr DJ et al Phase II studies of polymer‐doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int J Cancer 2009; 34: 1629–36. [DOI] [PubMed] [Google Scholar]
- 71. Stapleton S, Milosevic M, Allen C et al A mathematical model of the enhanced permeability and retention effect for liposome transport in solid tumors. PLoS ONE 2013; 8: e81157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mi P, Kokuryo D, Cabral H et al Hydrothermally synthesized PEGylated calcium phosphate nanoparticles incorporating Gd‐DTPA for contrast enhanced MRI diagnosis of solid tumors. J Control Release 2014; 174: 63–71. [DOI] [PubMed] [Google Scholar]
- 73. Kumagai M, Sarma TK, Cabral H et al Enhanced in vivo magnetic resonance imaging of tumors by PEGylated iron‐oxide‐gold core‐shell nanoparticles with prolonged blood circulation properties. Macromol Rapid Commun 2010; 31: 1521–8. [DOI] [PubMed] [Google Scholar]
- 74. Kaida S, Cabral H, Kumagai M et al Visible drug delivery by supramolecular nanocarriers directing to single‐platformed diagnosis and therapy of pancreatic tumor model. Cancer Res 2010; 70: 7031–41. [DOI] [PubMed] [Google Scholar]
- 75. Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res 2011; 44: 1029–38. [DOI] [PubMed] [Google Scholar]
- 76. Cabral H, Nishiyama N, Kataoka K. Supramolecular nanodevices: from design validation to theranostic nanomedicine. Acc Chem Res 2011; 44: 999–1008. [DOI] [PubMed] [Google Scholar]