

Abstract
In this study, we evaluated the clinical utility of detecting KRAS mutations in circulating cell‐free (ccf)DNA of metastatic colorectal cancer patients. We prospectively recruited 94 metastatic colorectal cancer patients. Circulating cell‐free DNA was extracted from plasma samples and analyzed for the presence of seven KRAS point mutations. Using the Invader Plus assay with peptide nucleic acid clamping method and digital PCR,KRAS mutations were detected in the ccfDNA in 35 of 39 patients previously determined to have primary tumors containing KRAS mutations using the Luminex method, and in 5 of 55 patients with tumors containing wild‐type KRAS. Curative resection was undertaken in 7 of 34 patients with primary and ccfDNA KRAS mutations, resulting in the disappearance of the mutation from the cell‐free DNA in five of seven patients. Three of these patients had tumor recurrence and KRAS mutations in their ccfDNA reappeared. Epidermal growth factor receptor blockade was administered to 24 of the KRAS tumor wild‐type patients. Of the 24 patients with wild‐type KRAS in their primary tumors, three patients had KRAS mutations in their ccfDNA and did not respond to treatment with epidermal growth factor receptor (EGFR) blockade. We also detected a new KRAS mutation in five patients during chemotherapy with EGFR blockade, before disease progression was detectable with imaging. The detection of KRAS mutations in ccfDNA is an attractive approach for predicting both treatment response and acquired resistance to EGFR blockade, and for detecting disease recurrence.
Keywords: Anti‐EGFR antibody, circulating cell‐free DNA, circulating tumor DNA, EGFR blockade, KRAS
Epidermal growth factor receptor (EGFR) blockade has improved the outcome of unresectable colorectal cancers (CRC).1 KRAS codon 12 or 13 mutations in exon 2 have been widely reported to be a major predictive biomarker for resistance to EGFR blockade in patients with metastatic CRC (mCRC).2 Mutations in other members of the RAS family may also confer resistance to EGFR blockade in patients without KRAS exon 2 mutations.3 Other oncogenic mutations, such as BRAF or PIK3CA mutations have also been presented as promising predictors for treatment resistance in these patients, although their predictive value has not yet been established.4
Thus, it is important to examine KRAS mutation status in patients with CRC. To date, KRAS mutation status has been examined using primary tumor samples, even when EGFR blockade is given for the treatment of metastases. However, colorectal tumors are heterogeneous in nature, and tumor heterogeneity and mutational selection are generated by tumor progression. Thus, there are many discordant patients (i.e., patients who show genetic differences between their primary tumors and their metastases)5, 6 and non‐responders, with discordance of KRAS mutations observed in 8% of mCRC cases.7
Epidermal growth factor receptor blockade induces the selection of pre‐existing mutant clones and leads to de novo acquisition of KRAS mutations.8 In the past, these two phenomena have not been clinically examined because it is difficult and invasive to collect samples from metastases deep within the body, such as from the lungs or liver. Circulating tumor cells (CTC) and circulating cell‐free DNA (ccfDNA) were recently identified in the plasma of patients with malignant disease and are now used for diagnosis, treatment selection, and therapy evaluation.9 However, CTC cannot always be used to detect KRAS mutations because it is difficult to extract sufficiently high CTC yields. Two studies have analyzed KRAS mutations using CTC, but both displayed very low sensitivity.10, 11
Circulating cell‐free DNA shows tumor‐specific sequence alterations, and advances in sequencing technologies have enabled the rapid identification of somatic genomic alterations.12 However, both the small number of circulating mutant gene fragments compared with the number of circulating wild‐type DNA fragments,13 and the small amount of ccfDNA able to be extracted in a clinical setting make it difficult to detect mutations, requiring high‐sensitivity detection systems. In this study, we evaluated the clinical utility of a highly sensitive PCR‐based method for detecting KRAS mutations in the ccfDNA of mCRC patients.
Materials and Methods
Patients and study design
We prospectively recruited 94 patients with histologically confirmed mCRC with distant metastases. Inclusion criteria for this study were age >20 years and patient performance status of 0 or 1. This study was carried out in accordance with the Declaration of Helsinki, and the study protocol was approved by the Ethics Review Committee of Nippon Medical School (Tokyo, Japan). Written informed consent was obtained from all participants.
Analysis of KRAS mutations in primary colorectal tumor tissue
All patients had pathological diagnosis from primary colorectal tumors. DNA was purified from formalin‐fixed, paraffin‐embedded specimens using a QIAamp DNA Mini kit (Qiagen, Limburg, the Netherlands) according to the manufacturer's recommendations. KRAS mutation analysis of the primary tumors was undertaken using the MEBGEN‐Luminex method (MBL, Nagoya, Japan) as previously reported.14
Blood collection and ccfDNA extraction
Blood samples were collected at the first visit from patients with synchronous metastases. Blood samples were also collected when metastatic sites were detected in patients with metachronous metastases, and every 2–3 months from patients undergoing chemotherapy.
Plasma was prepared by centrifugation of 8 mL EDTA‐treated blood at 800 g for 10 min at 4°C, followed by transfer to a fresh tube and re‐centrifugation at 16 000 g for 10 min at 4°C. The supernatants were then either immediately used for DNA extraction or stored at −80°C until use. Circulating cell‐free DNA was extracted from 1 mL plasma with the QIAamp Circulating Nucleic Acid kit (Qiagen) according to the manufacturer's plasma and serum fluid protocol. DNA concentrations were determined with the PicoGreen Assay (Quant‐iT PicoGreen dsDNA Assay Kit; Thermo Fisher Scientific, Tokyo, Japan).
Invader Plus assay with peptide nucleic acid clamping for KRAS mutations
We detected KRAS mutations in ccfDNA using the Invader Plus assay with peptide nucleic acid clamping (Inv‐Clamp assay; Hologic, Inc. (Marlborough, MA, USA) Figs S1–S3).15 KRAS mutation status was blinded to investigators. To evaluate the assay's sensitivity for mutation detection, a titration study of plasmid DNA was initially carried out using plasmid DNA (0.15 fg) containing seven KRAS mutations (G12V, G12A, G12D, G12S, G12C, G12R, and G13D) mixed with 150 fg wild‐type DNA. Oligonucleotide sequences used in this study are shown in Table S1. These seven somatic KRAS mutations were detected by the Inv‐Clamp assay with a positive threshold ratio for all mutations defined as a 0.1% mutant to 99.9% wild‐type mixture of plasmid DNA as per previous reports.16 These mutant and wild‐type DNAs were PCR‐amplified and cloned into plasmid pCR2.1 (Thermo Fisher Scientific); the synthesized mutant and wild‐type templates were verified with sequencing.
Validation with digital PCR
We also detected KRAS mutations in ccfDNA using digital PCR (dPCR). Digital PCR was carried out using a QuantStudio 3D Digital PCR System platform comprising a Gene Amp 9700 PCR machine including a chip adapter kit, an automatic chip loader, and the QuantStudio 3D Instrument (Thermo Fisher Scientific). The primers were synthesized by Thermo Fisher Scientific and the assay identification numbers of the primers were: G12C, AH0JEUD; G12S, AHI14FA; G12R, AHMSYXY; G12V, AHX1IHY; G12D, AH6R5PI; G12A, AHPAVDP; and G13D, AHD2BW0. We prepared 18 reaction mixtures containing 9 μL of 2× QuantStudio 3D Digital PCR Master Mix (Thermo Fisher Scientific), 0.45 μL of 40× TaqMan Assay by Design primer‐probe mix, and 8.55 μL diluted sample genomic DNA (110–40 300 ng/mL). We loaded 14.5 μL of each reaction mixture onto a QuantStudio 3D Digital PCR 20K Chip (Thermo Fisher Scientific) using the automatic chip loader according to the manufacturer's instructions. Loaded chips underwent amplification in the Gene Amp 9700 PCR System under the following conditions: 96°C for 10 min, 39 cycles at 56°C for 2 min and at 98°C for 30 s, followed by a final extension step at 60°C for 2 min. After amplification, the chips were imaged on the QuantStudio 3D Instrument, which assesses raw data and calculates the estimated concentration of the nucleic acid sequence targeted by the FAM and VIC dye‐labeled probes according to the Poisson distribution. The resulting data were reported in copies/μL together with the results of the data quality assessment metrics. For deeper analysis of the chip data, QuantStudio 3D Analysis Suite Cloud Software (Thermo Fisher Scientific) was used for relative and quantitative data analysis. An illustration of KRAS rare allele quantification by digital PCR is shown in Figure S4.
Treatment plan
Twenty‐four patients with KRAS wild‐type primary tumors were treated with chemotherapy including anti‐EGFR antibody therapy. Ten patients were treated with mFOLFOX6 (day 1, oxaliplatin 85 mg/m², folinic acid 400 mg/m², and fluorouracil 400 mg/m² i.v. bolus, followed by 2400 mg/m2 over 46 h of continuous infusion) and cetuximab (400 mg/m2 on day 1). Eight patients were treated with mFOLFOX6 and panitumumab (6 mg/kg on day 1). Three patients were treated with FOLFIRI (day 1, irinotecan 150 mg/m2, folinic acid 400 mg/m2, and fluorouracil 400 mg/m2 i.v. bolus, followed by 2400 mg/m2 over 46 h of continuous infusion) and panitumumab (6 mg/kg on day 1). Two patients were treated with irinotecan (150 mg/m2 on day 1) and panitumumab (6 mg/kg on day 1) and one patient was treated with the De Gramont regimen (day 1, folinic acid 400 mg/m2 and fluorouracil 400 mg/m2 i.v. bolus, followed by 2400 mg/m2 over 46 h of continuous infusion) and panitumumab (6 mg/kg on day 1).
Evaluation of clinical response
Tumor response was assessed with computed tomography (CT) according to the Response Evaluation Criteria in Solid Tumors version 1.1. We also measured carcinoembryonic antigen (CEA) and CA19‐9 levels every month throughout chemotherapy. A “normal” level of CEA falls under 5.0 ng/mL, while that of CA19‐9 falls under 37 U/mL.
Results
KRAS status and ccfDNA yield
Using the MEBGEN‐Luminex method previously reported,14 39 of the 94 patients recruited were determined to have colon cancers with mutated KRAS in their primary tumors (primary KRAS mutations) and 55 patients had colon cancers with wild‐type KRAS in their primary tumors (primary KRAS wild‐type). We obtained ccfDNA from all 94 patients with a median yield of 790 ng/mL (Fig. 1a). No significant difference was observed between the yield from patients with or without KRAS mutations (Fig. 1b; P = 0.26). In addition, ccfDNA yield did not correlate with CEA (P = 0.24) or CA19‐9 (P = 0.50) levels (data not shown). No significant difference was observed in the yield between the KRAS‐detectable patients and the non‐detectable patients (Fig. 1c; P = 0.47).
Figure 1.
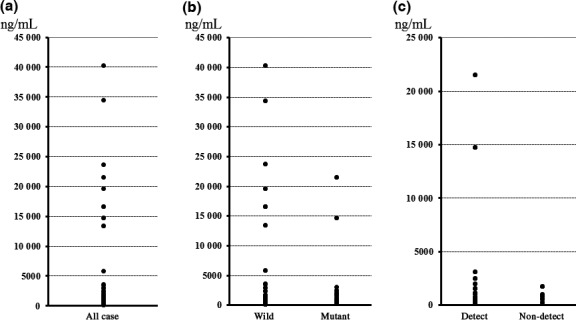
Circulating cell‐free (ccf)DNA yield for KRAS mutation analysis. (a) Median yield of ccfDNA from all 94 patients with metastatic colorectal cancer was 790 (110–40 300) ng/mL; the mean yield was 3343 ± 7398 ng/mL. (b) Median yield of ccfDNA from 39 patients with primary KRAS mutations was 720 (230–40 300) ng/mL; the median yield from the 55 patients without primary KRAS mutations was 885 (110–21 500) ng/mL (P = 0.26). (c) KRAS mutations were detected with the Inv‐Clamp assay in the ccfDNA of 34 of the 39 patients with primary tumor KRAS mutations. Median yield of ccfDNA in KRAS‐detectable cases (Detect) was 706 ng/mL, whereas that for the non‐detectable (Non‐detect) cases was 830 ng/mL (P = 0.47).
Mutation detection in ccfDNA of patients with primary KRAS mutations
Using the Inv‐Clamp assay and dPCR, we were able to detect mutant KRAS in ccfDNA from 35 of the 39 patients with primary KRAS mutations (87%; Table 1). Patient characteristics of all individuals with primary KRAS mutations are shown in Table 2. The genotype of the primary tumor was coincident with the genotype of the ccfDNA in all cases except for five non‐detectable patients (cases 4, 8, 16, 33, and 34). All discordant patients had small metastases (Fig. 2). Their CEA (<5 ng/mL) and CA19‐9 (<37 U/mL) levels were normal (Table 2). Four of the five patients were treated with FOLFOX/bevacizumab and all patients obtained an objective clinical response. Three of these four patients (cases 4, 16, and 33) had a complete response (CR), whereas the other patient (case 8) showed an extended partial response (PR) over 18 months. For 6 of the 11 out of 39 patients with primary KRAS mutations but normal CEA and CA19‐9 levels, KRAS mutations were also detected in the ccfDNA (Table 2).
Table 1.
Detection rate of KRAS mutations in circulating cell‐free (ccf)DNA of patients with stage IV colorectal cancer using the Inv‐Clamp Assay and digital PCR (dPCR)
| ccfDNA KRAS wild‐type (%) | ccfDNA KRAS mutants (%) | |
|---|---|---|
| Primary KRAS wild‐type (n = 55) | 91 | 9 |
| Primary KRAS mutants (n = 39) | 13 | 87 |
Using the Inv‐Clamp assay and dPCR, we were able to detect mutant KRAS in ccfDNA from 34 of the 39 patients (87%) with primary KRAS mutations. Using the Inv‐Clamp assay, a detection rate of 85% (33/39) was obtained, whereas dPCR yielded a detection rate of 80% (31/39). KRAS mutations in the ccfDNA were detected in 5 of the 55 (9%) patients determined to have wild‐type KRAS in their primary tumors by both methods (Inv‐Clamp assay and dPCR).
Table 2.
Characteristics of patients with stage IV colorectal cancer with primary KRAS mutations
| Case | Sex | Age, years | Primary site | Metastatic site | KRAS, primary | KRAS, ccfDNA | Mutation rate, % | CEA, <5.0 ng/mL | CA19‐9, <37 U/mL |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 67 | A‐colon | Liver | G12V | G12V | 16.30 | 7.8 | 8.6 |
| 2 | M | 70 | A‐colon | Liver | G12D | G12D | 0.33 | 26.5 | 3726.0 |
| 3 | M | 67 | Rectum | Liver, lung | G12D | G12D | 0.00 | 231.0 | 1991.2 |
| 4 | M | 69 | Rectum | Lung | G12V | Wild | 0.00 | 3.5 | 9.2 |
| 5 | F | 68 | T‐colon | Peritoneum bone | G13D | G13D | 0.39 | 3.5 | 3.9 |
| 6 | F | 66 | Rectum | PA‐LN | G12A | G12A | 0.29 | 2.0 | 10.3 |
| 7 | M | 78 | S‐colon | Liver, lung | G12D | G12D | 4.63 | 61.5 | 44.8 |
| 8 | M | 59 | Rectum | Lung | G12V | Wild | 0.00 | 2.4 | 16.5 |
| 9 | F | 69 | Rectum | Peritoneum | G12V | G12V | 0.00 | 55.2 | 38.7 |
| 10 | M | 82 | Rectum | Lung | G12V | G12V | 0.67 | 467.0 | 636.6 |
| 11 | F | 88 | Cecum | Liver | G12V | G12V | 48.10 | 433.7 | 8.1 |
| 12 | F | 67 | Rectum | Liver, lung | G12A | G12A | 0.00 | 2.3 | 15.4 |
| 13 | F | 65 | A‐colon | Lung | G13D | G13D | 0.36 | 7.3 | 7.9 |
| 14 | F | 80 | Cecum | Liver | G12V | G12V | 24.70 | 34.7 | 153.6 |
| 15 | F | 81 | A‐colon | Peritoneum | G12D | G12D | 4.21 | 34.3 | 44.6 |
| 16 | M | 74 | A‐colon | Lung | G12D | Wild | 0.00 | 3.3 | 9.1 |
| 17 | M | 76 | A‐colon | Liver, lung | G12V | G12V | 0.49 | 90.0 | 645.3 |
| 18 | M | 62 | Rectum | Lung | G12V | G12V | 3.49 | 14.2 | 25.9 |
| 19 | M | 70 | A‐colon | Peritoneum | G12D | G12D | 0.78 | 3.5 | 35.5 |
| 20 | F | 62 | A‐colon | Liver, lung | G13D | G13D | 18.22 | 965.6 | 4277.2 |
| 21 | M | 57 | S‐colon | Liver | G12D | G12D | 2.25 | 3.9 | 203.4 |
| 22 | M | 69 | S‐colon | PA‐LN | G12D | G12D | 0.05 | 87.5 | 85.1 |
| 23 | M | 68 | Rectum | Peritoneum | G13D | G13D | 0.03 | 39.9 | 70.9 |
| 24 | M | 78 | A‐colon | Liver, peritoneum | G12V | G12V | 0.46 | 142.5 | 474.0 |
| 25 | F | 74 | S‐colon | Liver, lung, bone | G12A | G12A | 28.57 | 39.9 | 288.6 |
| 26 | M | 64 | S‐colon | Liver, lung | G12V | G12V | 0.05 | 58.8 | 230.5 |
| 27 | F | 53 | S‐colon | Liver | G12D | G12D | 0.68 | 54.2 | 2.0 |
| 28 | F | 82 | Cecum | Liver | G12D | G12D | 3.06 | 43.3 | 245.8 |
| 29 | M | 72 | A‐colon | Liver | G12D | G12D | 2.18 | 8.0 | 23.4 |
| 30 | M | 56 | Rectum | Liver | G12D | G12D | 40.03 | 298.5 | 2.0 |
| 31 | M | 69 | S‐colon | Lung, peritoneum | G12D | G12D | 0.05 | 87.5 | 85.1 |
| 32 | M | 67 | Rectum | Lung, bone | G13D | G13D | 3.95 | 98.0 | 1368.4 |
| 33 | F | 72 | Cecum | Lung | G12V | Wild | 0.00 | 4.9 | 2.0 |
| 34 | M | 84 | S‐colon | Liver | G13D | Wild | 0.00 | 2.6 | 4.0 |
| 35 | M | 84 | Rectum | Liver | G12S | G12S | 3.98 | 2.6 | 2.0 |
| 36 | M | 82 | Rectum | Liver | G12V | G12V | 0.26 | 11.0 | 44.3 |
| 37 | M | 83 | S–colon | Liver | G12V | G12V | 35.52 | 6.5 | 7.1 |
| 38 | M | 70 | S‐colon | Liver, bone | G12V | G12V | 41.72 | 2500.2 | 4219.0 |
| 39 | M | 73 | S‐colon | Liver | G12D | G12D | 0.97 | 4.9 | 5.3 |
Using the Inv‐Clamp assay and digital PCR, we were able to detect mutant KRAS in circulating cell‐free (ccf)DNA from 35 of the 39 patients with primary KRAS mutations (87%). The genotype of the primary tumor was coincident with the genotype of the ccfDNA in all cases except five non‐detectable patients. A‐colon, ascending colon; CEA, carcinoembryonic antigen; F, female; M, male; PA‐LN, para‐aortic lymph node; S‐colon, sigmoid colon; T‐colon, transverse colon.
Figure 2.
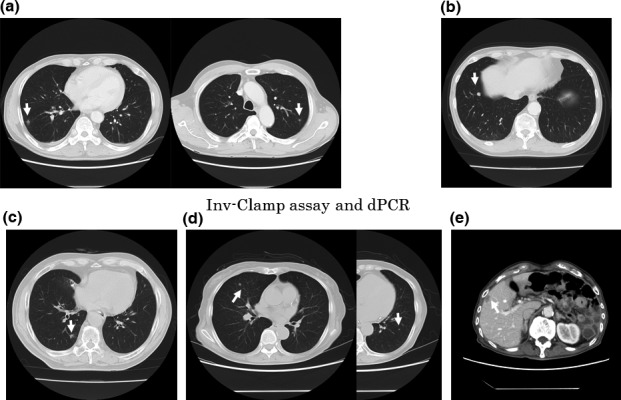
Computed tomography of five patients with stage IV colorectal cancer in whom KRAS was not detectable. White arrows indicate the metastatic tumors. In all five cases, the size and the number of metastatic tumors were very small (<1.5 cm) (arrows). (a) Case 4. (b) Case 8. (c) Case 16. (d) Case 33. (e) Case 34.
Using the Inv‐Clamp assay, a detection rate of 85% (34/39) was obtained, and dPCR yielded a detection rate of 80% (31/39). Case 26 was the only case that was detected by dPCR but not the Inv‐Clamp assay. Cases 3, 9, and 12 were all detected by the Inv‐Clamp assay but not by dPCR. Digital PCR analysis was also used to determine the number of mutations present in the ccfDNA (compared with wild‐type DNA); this varied from 0.00% to 48.10%, with a median of 0.67%. For 15 patients, <1.00% of ccfDNA was determined to contain KRAS mutations (Table 2).
Analysis of ccfDNA KRAS mutations following curative resection
Curative resection of primary and metastatic lesions was carried out for 7 of the 34 patients (cases 2, 6, 7, 27, 35, 36, and 39) with primary KRAS mutations and ccfDNA KRAS mutations. Resection in five of the seven patients (cases 2, 6, 7, 27, and 36) resulted in the disappearance of the KRAS mutations from the ccfDNA within the first month following resection.
Three of these patients had tumor recurrence (cases 2, 6 and 7) and KRAS mutations in their ccfDNA reappeared before detection with any other methods (i.e., CT imaging or CEA and CA19‐9 biomarker levels). Two of these patients (cases 27 and 36) had no tumor recurrence and no detectable KRAS mutations in their ccfDNA after resection.
For the remaining two patients (cases 35 and 39), the ccfDNA KRAS mutations had not disappeared 1 month subsequent to resection, but a reduction in the mutation rate was observed (case 35, 3.98% to 0.85%; case 59, 0.97% to 0.64%); tumor recurrence was still not detected 3 months after resection.
Case 6 had rectal cancer with large lymphatic metastases including the para‐aortic lymph nodes. The clinical course and CT imaging for this patient is shown in Figure 3(a). Two months after the curative surgery, no KRAS mutations were detected in the ccfDNA. However, 8 months after the operation, a KRAS mutation was detected in the ccfDNA. Two months after detecting the KRAS mutation in the ccfDNA, a CT scan revealed a liver metastasis (Fig. 3b), and ccfDNA yield and the percentage of ccfDNA with KRAS mutations continued to increase. Lung metastases subsequently developed (Fig. 3c). Carcinoembryonic antigen was within normal limits until identification of the lung metastases, whereas CA19‐9 (data not shown) remained within normal limits after identification of the lung metastases.
Figure 3.
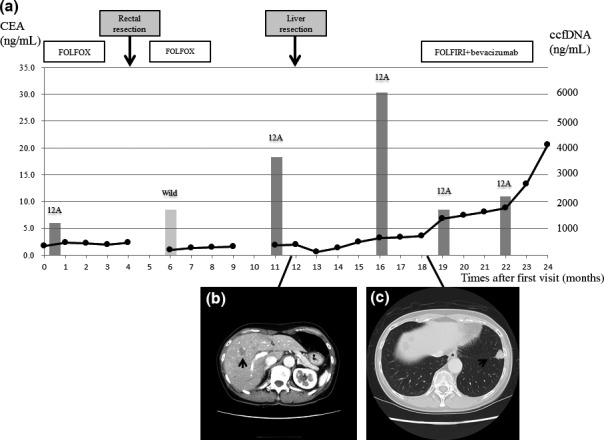
Clinical course of a patient with metastatic colorectal cancer with a KRAS mutation that disappeared from the circulating cell‐free (ccf)DNA following curative resection but subsequently re‐appeared with tumor recurrence. (a) Carcinoembryonic antigen (CEA) (black circles), KRAS status, and ccfDNA yield (gray columns: light gray, wild‐type; dark gray, mutated). Curative resection was carried out after six courses of mFOLFOX6 (oxaliplatin, folinic acid, and fluorouracil). (b) A computed tomography scan shows liver metastasis (arrow) 8 months later, leading to partial liver resection. (c) Lung metastasis (arrow) was detected 6 months after the resection, and mFOLFOX6 with bevacizumab was then administered. A KRAS mutation in the ccfDNA was detectable at the primary visit; this disappeared after curative resection but reappeared after recurrence of the liver metastasis. The ccfDNA yield (1050–6110 ng/mL) and mutation rate (0.29% to 2.64%) increased after the second resection. CEA was negative until detection of the lung metastasis.
Case 7 had rectal cancer with liver and lung metastases. Low anterior resection was carried out. Treatment with FOLFOX and bevacizumab was given and the lung metastases disappeared (data not shown). Liver resection was subsequently carried out, and the KRAS mutation in the ccfDNA disappeared within 1 month after surgery. A KRAS mutation was detected in the ccfDNA 3 months after resection, but CEA and CA19‐9 levels were within normal limits at that time. Recurrence of liver metastases was later detected with imaging 2 months after detecting the KRAS mutation in the ccfDNA.
Detection of KRAS mutations in ccfDNA of patients with wild‐type primary tumors
Using Inv‐Clamp assay and dPCR, KRAS mutations in the ccfDNA were detected in 5 of the 55 (9%) patients determined to have wild‐type KRAS in their primary tumors (Table 1).
Chemotherapy with EGFR blockade was given to 24 of the 55 patients with wild‐type primary tumors; these patient characteristics are shown in Table 3. Cases 47, 56, and 63, who had a KRAS mutation detected in their ccfDNA, did not show treatment response. Case 59 was the only patient who had no KRAS mutation in the ccfDNA and did not show treatment response. All other patients presented an objective response to therapy (1 CR, 19 PR).
Table 3.
Characteristics of patients with KRAS‐wild‐type primary colorectal tumors treated with epidermal growth factor receptor blockade
| Case | Sex | Age | Primary site | Metastatic site | ccfDNA | Regimen | Best response | CEA | CA19‐9 |
|---|---|---|---|---|---|---|---|---|---|
| 40 | M | 54 | S‐colon | Liver | Wild | FOLFIRI/Pmab | CR | 2.3 | 11.9 |
| 41 | F | 70 | S‐colon | Liver | Wild | FOLFOX/Cmab | PR | 19.7 | 85.2 |
| 42 | F | 79 | Rectum | Liver | Wild | FOLFOX/Pmab | PR | 2.3 | 4.4 |
| 43 | M | 66 | A‐colon | Liver, lung | Wild | FOLFIRI/Pmab | PR | 22.3 | 169.8 |
| 44 | M | 68 | Rectum | Liver | Wild | FOLFOX/Pmab | PR | 24.4 | 24.2 |
| 45 | F | 66 | T‐colon | Liver | Wild | FOLFOX/Cmab | PR | 15 000.0 | 693.2 |
| 46 | M | 60 | Rectum | Liver | Wild | FOLFOX/Cmab | PR | 9.6 | 45.1 |
| 47 | M | 71 | Rectum | Liver | G13D | FOLFOX/Cmab | SD | 9.7 | 25.9 |
| 48 | M | 73 | Rectum | Lung | Wild | Irinotecan/Pmab | PR | 39.4 | 53.1 |
| 49 | M | 79 | Rectum | Liver | Wild | DeGramont/Pmab | PR | 18.6 | 13.0 |
| 50 | F | 60 | A‐colon | Lung | Wild | FOLFOX/Cmab | PR | 2.4 | 16.7 |
| 51 | M | 72 | S‐colon | Liver | Wild | FOLFOX/Cmab | PR | 32.4 | 11.6 |
| 52 | M | 56 | S‐colon | Liver | Wild | FOLFOX/Pmab | PR | 1223.0 | 1878.5 |
| 53 | M | 73 | Rectum | Liver | Wild | FOLFOX/Cmab | PR | 19.1 | 22.0 |
| 54 | M | 68 | Rectum | Liver | Wild | FOLFOX/Pmab | PR | 2722.0 | 11 405.8 |
| 55 | M | 69 | Rectum | Liver | Wild | FOLFOX/Pmab | PR | 403.6 | 110.8 |
| 56 | M | 51 | A‐colon | Peritoneum | G12A | FOLFOX/Cmab | SD | 6.0 | 10.3 |
| 57 | F | 66 | S‐colon | Liver | Wild | FOLFOX/Pmab | PR | 44.6 | 174.1 |
| 58 | F | 79 | S‐colon | Liver | Wild | FOLFOX/Cmab | PR | 15 000.0 | 609.4 |
| 59 | M | 74 | S‐colon | Liver | Wild | FOLFOX/Pmab | PD | 467.7 | 155.4 |
| 60 | F | 60 | S‐colon | Lung | Wild | DeGramont/Cmab | PR | 13.1 | 22.7 |
| 61 | M | 60 | Rectum | Lung | Wild | Irinotecan/Pmab | PR | 17.4 | 35.8 |
| 62 | F | 37 | Rectum | Liver | Wild | FOLFIRI/Pmab | PR | 127.4 | 401.8 |
| 63 | F | 64 | Rectum | Liver, lung | G12R | FOLFOX/Cmab | SD | 38.5 | 21.5 |
Chemotherapy with epidermal growth factor receptor blockade was given to 24 of 55 patients with wild‐type primary tumors. Cases 47, 56, and 63, who had a KRAS mutation detected in their circulating cell‐free (ccf)DNA, did not show any treatment response. Case 59 was the only patient who had no KRAS mutation in the ccfDNA and did not show treatment response. All other patients presented an objective response to therapy (1 complete response [CR], 19 partial response [PR]). A‐colon, ascending colon; CEA, carcinoembryonic antigen; Cmab, cetuximab; F, female; FOLFIRI, irinotecan, folinic acid, and fluorouracil; FOLFOX, oxaliplatin, folinic acid, and fluorouracil; M, male; PD, progressive disease; Pmab, panitumumab; S‐colon, sigmoid colon; SD, stable disease; T‐colon, transverse colon.
Case 40 represents an instance wherein our assay was able to predict continued response to EGFR blockade. For this patient, a complete response lasted for over 18 months (data not shown). After 18 months of chemotherapy, treatment was stopped; 6 months later, a metastatic liver tumor was detected. Wild‐type KRAS in the ccfDNA continued to be present; therefore, treatment with FOLFIRI and panitumumab was restarted. Three months after re‐initiation of chemotherapy, the metastatic liver tumor had markedly decreased in size. The presence of wild‐type KRAS in the ccfDNA continued throughout treatment (Fig. S5).
Eight of the 24 patients acquired resistance to EGFR blockade, and KRAS mutations in the ccfDNA were detected in five of these eight patients 2–3 months before detection of disease progression was possible with CT. The clinical course and CT imaging of case 41 is shown in Figure 4. In this case, PR to chemotherapy was observed for 12 months (Fig. 4a–c). The G12C KRAS mutation was then detected in the ccfDNA, but no tumor size increase was noted and both CEA and CA19‐9 levels remained within normal limits. Two months after detecting the mutation, the liver tumor increased in size (Fig. 4d). Treatment with FOLFIRI and bevacizumab was given, but no response occurred. The patients died 9 months after the initial detection of the KRAS mutation.
Figure 4.
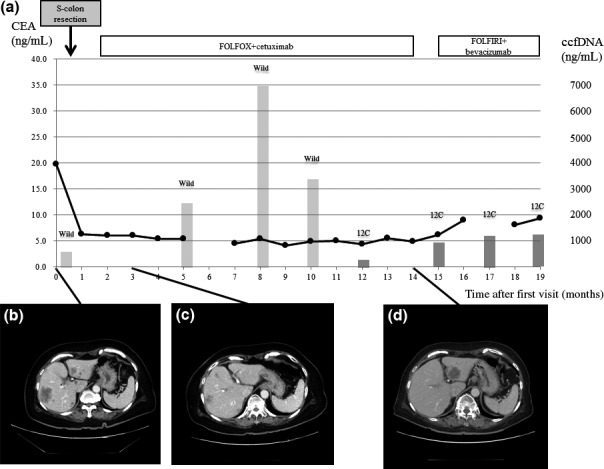
Clinical course of a patient with stage IV colorectal cancer with a detected KRAS mutation in circulating cell‐free (ccf)DNA before acquired resistance to epidermal growth factor receptor blockade. (a) Carcinoembryonic antigen (CEA) (black circles), KRAS status, and ccfDNA yield (gray columns: light gray, wild‐type; dark gray, mutated). (b) Sigmoidectomy and treatment with mFOLFOX6 (oxaliplatin, folinic acid, and fluorouracil) and cetuximab were used for treatment of the liver metastasis. (c) The metastatic tumor decreased in size 3 months after starting chemotherapy. (d) Progression of the liver tumor was detected 14 months after initiation of chemotherapy, leading to treatment with FOLFIRI (irinotecan, folinic acid, and fluorouracil) and bevacizumab. No KRAS mutations in the ccfDNA were detected at the first visit or at 8 months after starting chemotherapy. We detected the G12C KRAS mutation in the ccfDNA 12 months after starting chemotherapy, followed by progression of the metastatic tumor 2 months after detecting the KRAS mutation in the ccfDNA.
Discussion
In this study, we showed the utility of KRAS mutation detection in the ccfDNA of patients with tumors containing KRAS mutations or wild‐type KRAS. Many studies have reported the utility of detecting KRAS mutations in the ccfDNA of patients with KRAS‐mutated tumors. However, few studies have reported its utility in patients with KRAS‐wild‐type tumors, and no studies have reported its utility as a biomarker for monitoring disease progression in patients with KRAS‐mutated tumors. Based on the above results, it is apparent that KRAS mutations in ccfDNA could be used as a biomarker for the early detection of recurrence in mCRC patients with primary KRAS mutations. Moreover, this approach can provide real‐time assessment of patients' mutation status and enable both prediction of the efficacy of EGFR blockade and analysis of acquired resistance to this blockade before clinical resistance is observed.
Using the Inv‐Clamp assay and dPCR, we detected KRAS mutations in the ccfDNA of mCRC patients with primary KRAS mutations at a high rate (87%). Previous studies have reported detection rates for mutant KRAS in ccfDNA of 36–92%.17, 18, 19, 20, 21, 22 Our study was blinded but many of these previous studies were non‐blinded (paired formalin‐fixed, paraffin‐embedded tumors and plasma). Detection rates can be affected by the detection method, tumor extent, and patient selection. Our patients included only stage IV patients, and our detection rate is second only to that reported by Thierry et al. (92%).21 In that study, researchers recruited 106 patients but excluded 11 patients (10.4%) with insufficient sample quality. In our study, all recruited patients were included in the analysis, which may account for the slight difference in detection rates.
The reason for lack of detection in certain patients is unclear; however, it does not appear to be related to the yield of ccfDNA. In general, 1000 ng ccfDNA includes approximately 150 000 copies of the genome. In early studies, the ccfDNA concentrations obtained varied from 0 to >1000 ng/mL blood, with an average of 180 ng/mL.23, 24 However, advances in technology have allowed increased ccfDNA extraction yields.25 In our study, the median concentration of ccfDNA extracted was 790 ng/mL. In the five cases we observed with non‐detectable primary KRAS mutations (i.e., when using the Inv‐Clamp assay and dPCR), extraction yielded 275–1760 ng/mL ccfDNA. Of note, however, is that patients who had lower amounts of extracted ccfDNA (230, 234, 266, and 270 ng/mL) still had detectable KRAS mutations. Indeed, Taly et al.22 reported that KRAS mutations can be detected in ccfDNA with a concentration as low as 100 ng/mL.
Another possibility for the lack of detection is that these patients had mutation rates lower than the detection limit. Using dPCR, we were able to detect KRAS mutations in the ccfDNA of 15 of 32 patients (47%) who had mutation rates <1.00%. Taly et al.22 also reported that mutation rates observable with dPCR‐based detection were <1.00% in 6 of 17 patients (35%). For case 8, we detected the KRAS mutation in the primary tumor but not in the ccfDNA. The KRAS mutation was detected in the lung metastasis obtained by CT‐guided biopsy after disease progression, but the KRAS mutation was again not detected in the ccfDNA. Particularly with case 4 in mind, another possibility may be that these patients have discordant primary and metastatic tumors; we did not obtain samples specifically from the metastatic site to confirm or exclude this hypothesis. Discordance of KRAS mutations has previously been observed in 8% of mCRC patients.7
We can use KRAS mutations in ccfDNA as a tumor marker for the prediction of poor prognosis or for early detection of recurrence in patients with KRAS mutations. Importantly, we were able to detect KRAS mutations in the ccfDNA of patients who were both CEA‐ and CA19‐9‐negative. This result is similar to that of another study that also reported detection of KRAS mutations in the serum of patients with normal CEA levels.18 Although CEA is a useful prognostic marker, its sensitivity is only 65% in stage IV CRC.26 Using CEA to identify recurrent or metastatic disease in asymptomatic patients is valuable to identify candidates for survival‐prolonging therapy;27 however, the sensitivity for recurrence is 77% with a specificity of 98%,28 indicating that the development of more sensitive biomarkers or supportive biomarkers is expected. KRAS mutations in ccfDNA may be just such a novel marker for patients with KRAS mutations because it can detect CEA‐negative recurrence.
Additionally, CRCs that show KRAS mutations are associated with poor prognosis, and KRAS status is an independent prognostic factor indicative of overall and progression‐free survival.29 KRAS mutations in ccfDNA are also reported to be predictors of poor prognosis for CRC with distant metastases.17 Thus, we believe that KRAS mutations in ccfDNA could be used as not only novel, sensitive biomarkers but also as prognostic indicators.
We can now determine the risk of recurrence in patients with primary KRAS mutations using ccfDNA. KRAS mutations in ccfDNA may disappear after removal of tumors with KRAS mutations within the short term, because ccfDNA is cleared from the blood and has a variable half‐life in the circulation ranging from 15 min to several hours.30 Thus, KRAS mutations detected in ccfDNA after surgery can indicate residual cancer in patients with KRAS mutation‐containing tumors. In this study, KRAS mutations in ccfDNA disappeared following curative resection of the primary and metastatic tumors in five of the seven patients who underwent curative resection. However, three of these five patients had tumor recurrence and KRAS mutations in the ccfDNA reappeared before detection of recurrence was possible with CT or by analyzing the CEA or CA19‐9 levels. Although this study included only three patients who had recurrence, the clinical course of these patients indicated that in addition to CEA and CA19‐9 levels, the presence of KRAS mutations in the ccfDNA may be useful for early detection of recurrence. Two patients with KRAS mutations in their ccfDNA after curative resection did not have tumor recurrence within 3 months after operation; these patients should be monitored closely.
We can also now predict EGFR blockade efficacy in patients with wild‐type KRAS primary tumors. We detected KRAS mutations in the ccfDNA of 5 of the 55 patients (9%) with wild‐type primary tumors, one of whom was later found to have a metastatic liver tumor with a G13D KRAS mutation. Another study has also reported the detection of a ccfDNA KRAS mutation in one of 37 (4%) primary KRAS wild‐type patients.18 In our study cohort, 24 patients with primary wild‐type tumors underwent EGFR blockade. Objective responses were observed in 20 of the 21 patients with wild‐type KRAS in the primary tumor and in their ccfDNA. Conversely, the remaining three patients with a wild‐type primary tumor but the presence of a KRAS mutation in their ccfDNA showed no treatment response. Although this was observed for only three cases it is highly suggestive, leading us to propose that the detection of a discordant patient by ccfDNA analysis should influence the choice of treatment.
We can predict acquired resistance to EGFR blockade by using ccfDNA before clinical resistance occurs. Diaz et al.31 reported in a retrospective study that new KRAS mutations in ccfDNA were detected in 9 of 24 (38%) patients with mCRC refractory to EGFR blockade, before radiographic evidence of disease progression was observed. Morelli et al.32 also showed in a retrospective study that new KRAS mutations in ccfDNA were detected in 27 of 62 (44%) patients with mCRC refractory to EGFR blockade. In this study, we prospectively showed that new KRAS mutations in ccfDNA were detected in five of eight patients before disease progression was detected with CT imaging. Although other reasons for acquired resistance were considered, no detection of any KRAS mutations during chemotherapy, which included EGFR blockade, occurred during 1 year of treatment. Furthermore, we did not detect KRAS mutations in the ccfDNA of another patient with wild‐type KRAS who underwent similar treatment and maintained an optimal response. These data highlight the importance of monitoring KRAS mutation status during treatment, as new mutations can lead to acquired resistance.
Our study has several limitations. First, although it is a prospective study, it is a small study. Second, it included only metastatic patients. Future studies with larger numbers of patients will be required to validate the utility of KRAS mutation detection in ccfDNA, as well as to evaluate its utility in non‐metastatic patients. Studies to determine the detection limit when using ccfDNA for metastatic cancer KRAS mutation detection for patients with small (<1 cm) metastases are also required.
In conclusion, we showed the utility of molecular diagnosis in the detection of KRAS mutations in ccfDNA, which is an attractive method for both monitoring and predicting response to EGFR blockade, as well as for the detection of recurrence or the prediction of prognosis.
Disclosure Statement
The authors have no potential conflicts of interest to declare.
Supporting information
Figure S1–S3. Invader Plus Method and the results for KRAS point mutation detection in serum and plasma.
Figure S4. The results for KRAS point mutation detection in serum and plasma by using digital PCR.
Figure S5. A patient with primary and ccfDNA KRAS wild who respond well to EGFR blockade.
Table S1. Primers to detect KRAS mutations.
Acknowledgment
This work was supported by the Japan Society for the Promotion of Science (KAKENHI Grant No. 26462030).
Cancer Sci 107 (2016) 936–943
Funding Information
Japan Society for the Promotion of Science (26462030).
References
- 1. Cunningham D, Humblet Y, Siena S et al Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan‐refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337–45. [DOI] [PubMed] [Google Scholar]
- 2. Lievre A, Bachet JB, Le Corre D et al KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006; 66: 3992–5. [DOI] [PubMed] [Google Scholar]
- 3. Douillard JY, Oliner KS, Siena S et al Panitumumab‐FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369: 1023–34. [DOI] [PubMed] [Google Scholar]
- 4. Moroni M, Veronese S, Benvenuti S et al Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 2005; 6: 279–86. [DOI] [PubMed] [Google Scholar]
- 5. Bouchahda M, Karaboue A, Saffroy R et al Acquired KRAS mutations during progression of colorectal cancer metastases: possible implications for therapy and prognosis. Cancer Chemother Pharmacol 2010; 66: 605–9. [DOI] [PubMed] [Google Scholar]
- 6. Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 2010; 16: 790–9. [DOI] [PubMed] [Google Scholar]
- 7. Mao C, Wu XY, Yang ZY et al Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep 2015; 5: 8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Misale S, Yaeger R, Hobor S et al Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 2012; 486: 532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan KC, Leung SF, Yeung SW, Chan AT, Lo YM. Persistent aberrations in circulating DNA integrity after radiotherapy are associated with poor prognosis in nasopharyngeal carcinoma patients. Clin Cancer Res 2008; 14: 4141–5. [DOI] [PubMed] [Google Scholar]
- 10. Mostert B, Jiang Y, Sieuwerts AM et al KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J Cancer 2013; 133: 130–41. [DOI] [PubMed] [Google Scholar]
- 11. Fabbri F, Carloni S, Zoli W et al Detection and recovery of circulating colon cancer cells using a dielectrophoresis‐based device: KRAS mutation status in pure CTCs. Cancer Lett 2013; 335: 225–31. [DOI] [PubMed] [Google Scholar]
- 12. Murtaza M, Dawson SJ, Tsui DW et al Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497: 108–12. [DOI] [PubMed] [Google Scholar]
- 13. Diehl F, Li M, Dressman D et al Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA 2005; 102: 16368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bando H, Yoshino T, Tsuchihara K et al KRAS mutations detected by the amplification refractory mutation system‐Scorpion assays strongly correlate with therapeutic effect of cetuximab. Br J Cancer 2011; 105: 403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lyamichev V, Mast AL, Hall JG et al Polymorphism identification and quantitative detection of genomic DNA by invasive cleavage of oligonucleotide probes. Nat Biotechnol 1999; 17: 292–6. [DOI] [PubMed] [Google Scholar]
- 16. Kitano S, Myers J, Nakamura J et al A novel fully automated molecular diagnostic system (AMDS) for colorectal cancer mutation detection. PLoS ONE 2013; 8: e62989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trevisiol C, Di Fabio F, Nascimbeni R et al Prognostic value of circulating KRAS2 gene mutations in colorectal cancer with distant metastases. Int J Biol Markers 2006; 21: 223–8. [DOI] [PubMed] [Google Scholar]
- 18. Ryan BM, Lefort F, McManus R et al A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: strong prognostic indicator in postoperative follow up. Gut 2003; 52: 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yen LC, Yeh YS, Chen CW et al Detection of KRAS oncogene in peripheral blood as a predictor of the response to cetuximab plus chemotherapy in patients with metastatic colorectal cancer. Clin Cancer Res 2009; 15: 4508–13. [DOI] [PubMed] [Google Scholar]
- 20. Spindler KL, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell‐free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res 2012; 18: 1177–85. [DOI] [PubMed] [Google Scholar]
- 21. Thierry AR, Mouliere F, El Messaoudi S et al Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med 2014; 20: 430–5. [DOI] [PubMed] [Google Scholar]
- 22. Taly V, Pekin D, Benhaim L et al Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem 2013; 59: 1722–31. [DOI] [PubMed] [Google Scholar]
- 23. Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 1983; 51: 2116–20. [DOI] [PubMed] [Google Scholar]
- 24. Lecomte T, Berger A, Zinzindohoue F et al Detection of free‐circulating tumor‐associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer 2002; 100: 542–8. [DOI] [PubMed] [Google Scholar]
- 25. Umetani N, Kim J, Hiramatsu S et al Increased integrity of free circulating DNA in sera of patients with colorectal or periampullary cancer: direct quantitative PCR for ALU repeats. Clin Chem 2006; 52: 1062–9. [DOI] [PubMed] [Google Scholar]
- 26. Louhimo J, Carpelan‐Holmstrom M, Alfthan H, Stenman UH, Jarvinen HJ, Haglund C. Serum HCG beta, CA 72‐4 and CEA are independent prognostic factors in colorectal cancer. Int J Cancer 2002; 101: 545–8. [DOI] [PubMed] [Google Scholar]
- 27. Locker GY. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J Clin Oncol 2006; 24: 5313–27. [DOI] [PubMed] [Google Scholar]
- 28. Glover C, Douse P, Kane P et al Accuracy of investigations for asymptomatic colorectal liver metastases. Dis Colon Rectum 2002; 45: 476–84. [DOI] [PubMed] [Google Scholar]
- 29. Lievre A, Bachet JB, Boige V et al KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26: 374–9. [DOI] [PubMed] [Google Scholar]
- 30. Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer–a survey. Biochim Biophys Acta 2007; 1775: 181–232. [DOI] [PubMed] [Google Scholar]
- 31. Diaz LA Jr, Williams RT, Wu J et al The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486: 537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morelli MP, Overman MJ, Dasari A et al Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti‐EGFR treatment. Ann Oncol 2015; 26: 731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1–S3. Invader Plus Method and the results for KRAS point mutation detection in serum and plasma.
Figure S4. The results for KRAS point mutation detection in serum and plasma by using digital PCR.
Figure S5. A patient with primary and ccfDNA KRAS wild who respond well to EGFR blockade.
Table S1. Primers to detect KRAS mutations.