

Abstract
Antibody–drug conjugates deliver anticancer agents selectively and efficiently to tumor tissue and have significant antitumor efficacy with a wide therapeutic window. DS‐8201a is a human epidermal growth factor receptor 2 (HER2)‐targeting antibody–drug conjugate prepared using a novel linker‐payload system with a potent topoisomerase I inhibitor, exatecan derivative (DX‐8951 derivative, DXd). It was effective against trastuzumab emtansine (T‐DM1)‐insensitive patient‐derived xenograft models with both high and low HER2 expression. In this study, the bystander killing effect of DS‐8201a was evaluated and compared with that of T‐DM1. We confirmed that the payload of DS‐8201a, DXd (1), was highly membrane‐permeable whereas that of T‐DM1, Lys‐SMCC‐DM1, had a low level of permeability. Under a coculture condition of HER2‐positive KPL‐4 cells and negative MDA‐MB‐468 cells in vitro, DS‐8201a killed both cells, whereas T‐DM1 and an antibody–drug conjugate with a low permeable payload, anti‐HER2‐DXd (2), did not. In vivo evaluation was carried out using mice inoculated with a mixture of HER2‐positive NCI‐N87 cells and HER2‐negative MDA‐MB‐468‐Luc cells by using an in vivo imaging system. In vivo, DS‐8201a reduced the luciferase signal of the mice, indicating suppression of the MDA‐MB‐468‐Luc population; however, T‐DM1 and anti‐HER2‐DXd (2) did not. Furthermore, it was confirmed that DS‐8201a was not effective against MDA‐MB‐468‐Luc tumors inoculated at the opposite side of the NCI‐N87 tumor, suggesting that the bystander killing effect of DS‐8201a is observed only in cells neighboring HER2‐positive cells, indicating low concern in terms of systemic toxicity. These results indicated that DS‐8201a has a potent bystander effect due to a highly membrane‐permeable payload and is beneficial in treating tumors with HER2 heterogeneity that are unresponsive to T‐DM1.
Keywords: Antibody‐drug conjugate, bystander killing, HER2, T‐DM, topoisomerase I inhibitor
Antibody–drug conjugates (ADCs) represent a promising class of drugs with a wider therapeutic window than conventional chemotherapeutic agents, owing to efficient and specific drug delivery to antigen‐expressing tumor cells. A first‐generation ADC, gemtuzumab ozogamicin, an anti‐CD33 antibody conjugated with a DNA alkylating agent calicheamicin, was approved by the FDA in 2000 for the treatment of CD33‐positive relapsed AML.1 However, 10 years after its approval, gemtuzumab ozogamicin was withdrawn from the market owing to a lack of clinical benefit in a post‐marketing clinical trial.2 Two ADCs have been approved and are currently in the market; brentuximab vedotin, a CD30‐targeting antibody in conjugation with a tubulin polymerization inhibitor monomethyl auristatin E, was approved in 2011 for the treatment of relapsed or refractory Hodgkin's lymphoma and systemic anaplastic large cell lymphoma.3, 4, 5 The introduction of trastuzumab emtansine (T‐DM1), a human epidermal growth factor receptor 2 (HER2)‐targeting ADC with a tubulin polymerization inhibitor DM1, was approved in 2013 for HER2‐positive breast cancer,6, 7, 8 proved that ADCs could target solid tumors in addition to hematological malignancies. More than 30 kinds of ADC programs are currently in clinical trials, and most of them are conjugated with the same type of payload, tubulin polymerization inhibitor.9 However, in the clinical trials of these ADCs, several dose‐limiting toxicities were observed, and some were considered to be mediated by the free drugs in plasma.10 Currently, the development of a new linker‐payload system focusing on different types of drugs has received attention11 as improvements in terms of different antitumor spectrum, overcoming drug resistance to the conventional systems, and providing a greater safety profile would be much desired.
DS‐8201a is a novel HER2‐targeting ADC prepared using our original linker‐payload system, which is composed of a humanized anti‐HER2 antibody and a novel topoisomerase I inhibitor, exatecan derivative (DX‐8951 derivative, DXd), by enzymatically cleavable peptide‐linker.12 It showed potent antitumor activity that was attributable the combination of pharmacological effects of the antibody component and topoisomerase I inhibition. The stability in plasma was favorable and the safety profiles in rats and monkeys were well tolerated. Moreover, DS‐8201a was effective against T‐DM1‐insensitive tumors owing to the different mechanism of action of the conjugated drug and higher drug‐to‐antibody ratio (DAR) than that of T‐DM1. Based on these promising preclinical profiles, a phase I dose escalation study was initiated in August 2015 in Japan (trial registration ID: NCT02564900).
The ADCs in both clinical trials and market target hematological malignancies; solid tumors are difficult to target as the target antigen in tumor tissue is expressed heterogeneously.9 Recently, preclinical studies of some ADCs showed that “bystander killing” may be a solution for this issue, which could benefit not only antigen‐expressing tumor cells but also adjacent antigen‐negative cells through the transfer of released payload from the antigen‐expressing cells to the neighboring antigen‐negative cells.13, 14, 15, 16 However, several reports showed that T‐DM1 did not have a bystander killing effect in vitro.15, 16 This could be another differentiation point between DS‐8201a and T‐DM1.
In this report, we investigated the bystander killing potential of DS‐8201a and compared with that of T‐DM1 in vitro and in vivo using a unique method that supports more accurate evaluation than the methods reported previously. Moreover, we assessed the effect of membrane permeability of payloads on bystander killing based on a hypothesis that the ability of cell penetration of released payload is correlated to the intensity of bystander killing. Notably, DS‐8201a has high membrane‐permeable drug payload and showed more potent bystander killing than T‐DM1 and anti‐HER2 ADC with low cell membrane‐permeable payload of topoisomerase I inhibitor, suggesting its potential in targeting tumors with HER2 heterogeneity, such as gastric cancer.
Materials and Methods
Antibody–drug conjugates and chemotherapeutics
DS‐8201a (anti‐HER2‐DXd (1)) and anti‐HER2‐DXd (2) were synthesized according to the conjugation procedure described in another report17 by using the anti‐HER2 Ab produced with reference to the same amino acid sequence of trastuzumab and its DAR is approximately 7 to 8. The IgG‐DXd (1) and IgG‐DXd (2) were synthesized using a method similar to that for DS‐8201a and anti‐HER2‐DXd (2) with purified human IgG (Bethyl Laboratories, Montgomery, TX, USA.), resulting in comparable DAR. T‐DM1 was purchased from Genentech Roche (San Francisco, CA, USA), and DXds (1) and (2) and Lys‐SMCC‐DM1 were synthesized in our laboratory.
Cell lines
The human gastric carcinoma cell line NCI‐N87 and the human breast adenocarcinoma cell line MDA‐MB‐468 were purchased from ATCC (Manassas, VA, USA). The human breast cancer cell line KPL‐4 was provided by Dr. Kurebayashi at the Kawasaki Medical University (Kurashiki, Japan). MDA‐MB‐468‐Luc cells were established by transfecting them with the lentivirus vector (Valent BioSciences Corporation, Libertyville, IL, USA) carrying the luciferase gene. All cell lines were cultured with RPMI 1640 Medium (Thermo Fisher Scientific Inc. Waltham, MA, USA) supplemented with 10% heat‐inactivated FBS at 37°C under 5% CO2 atmosphere.
Distribution coefficient (log D, pH 7.4)
Equal amounts of n‐octanol and PBS (pH 7.4) were mixed, shaken, and left for more than 12 h. Each compound solution in DMSO (10 mM) was added to the solution (final 100 μM containing DMSO 1%) and shaken vigorously for 30 min at room temperature. After centrifugation, the concentration of compound in each phase (upper, n‐octanol phase; lower, aqueous phase) was measured by an LC‐MS/MS system (API 4000; AB Sciex, Framingham, MA, USA). Log D was calculated using the following equation: Log D = log (peak area in n‐octanol phase / peak area in aqueous phase).
Parallel artificial membrane permeability assay
Parallel artificial membrane permeability assay (PAMPA) was carried out using a Freedom EVO200 system (Tecan, Männedorf, Switzerland). The filter membrane of the acceptor plate (Stirwell PAMPA Sandwich; Pion, Billerica, MA, USA) was coated with GIT‐0 lipid solution (Pion). Each compound solution in DMSO (10 mM) was added to Prisma HT buffer (Pion) to obtain 5‐μM donor solutions (containing 0.05% DMSO, pH 5.0 and pH 7.4), and then placed on a donor plate. The acceptor plate was filled with an acceptor sink buffer (Pion). The donor plate was stacked onto the acceptor plate and incubated for 4 h at 25°C. After incubation, the concentrations of compounds in both plates were measured by an LC‐MS/MS system (API 4000). The permeability coefficient (Peff; 10−6 cm/s) was calculated using PAMPA Evolution DP software (Pion).
In vitro cell growth assay
Cells were seeded in a 96‐well plate at 1000 cells/well for KPL‐4 and 2000 cells/well for MDA‐MB‐468. After overnight incubation, a serially diluted solution of each ADC was added. Cell viability was evaluated after 5 days using a CellTiter‐Glo luminescent cell viability assay from Promega (Madison, WI, USA) according to the manufacturer's instructions. For coculture study, KPL‐4 and MDA‐MB‐468 cells were seeded in a 6‐well plate at 1 × 105 cells and 3 × 105 cells, respectively, in 2 mL/well culture medium. After overnight incubation, the supernatant was removed from the plate and each ADC diluent (10 nM) was added at 6 mL/well. Viable cells were detached from the plate after 5 days of culture, and the cell number in each well was determined using a cell counter. In order to determine the ratio of KPL‐4 and MDA‐MD‐468 cells of the total viable cells, the cells were stained with anti‐HER2/nue FITC (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and incubated on ice for 20 min. After washing, fluorescent signals of 2 × 104 stained cells were measured using a flow cytometer. Based on the number and ratio of HER2‐positive and HER2‐negative cells in each treatment well, the number of KPL‐4 or MDA‐MB‐468 cells was calculated.
In vivo xenograft studies
All in vivo studies were carried out in accordance with the local guidelines of the Institutional Animal Care and Use Committee. Specific pathogen‐free female CAnN.Cg‐Foxn1nu/CrlCrlj mice (BALB/c nude mice) aged 5 weeks were purchased from Charles River Laboratories Japan Inc. (Yokohama, Japan) All models were established by s.c. inoculation in the flanks of the mice. NCI‐N87 and MDA‐MB‐468‐Luc models were established by injecting 5 × 106 and 1 × 107 cells suspended in a Matrigel matrix (Corning Inc. Corning, NY, USA), respectively. After 6 days for NCI‐N87, and 9 days for MDA‐MB‐468‐Luc models, the tumor‐bearing mice were randomized into treatment and control groups based on the tumor volume, and dosing initiated (day 0). Each ADC was given i.v. to the mice at a dose of 3 or 10 mg/kg, and a volume of 10 mL/kg. As a vehicle, ABS buffer (10 mM acetate buffer, 5% sorbitol, pH 5.5) was given at the same volume as the ADCs. The tumor volume was defined as 1/2 × length × width2.
In vivo luciferase imaging
Seven days after inoculating the mixture of 5 × 106 NCI‐N87 cells and 1 × 107 MDA‐MB‐468‐Luc cells suspended in the Matrigel matrix into the right flank of the mice at a total volume of 100 μL, the tumor‐bearing mice were randomized into treatment and control groups based on the tumor volume, and dosing initiated (day 0). Each ADC or the vehicle was given i.v. to the mice. Luciferase activity of each mouse was measured using an in vivo imaging system (IVIS 200 Imaging System; PerkinElmer Waltham, MA, USA) twice a week in parallel with the measurement of tumor length 10 min after administering 150 mg/kg luciferin (Promega) i.v. The amount of luminescence was analyzed using analysis software (Living Image Software version 4.3.1; PerkinElmer) as average radiance (p/s/cm2/sr). For another study, MDA‐MB‐468‐Luc cells at a density of 1.5 × 107 cells were inoculated into the left flank of the mice in addition to the inoculation of the mixture into the right flank. Seven days after inoculation, dosing and evaluation were undertaken in a similar manner as described above.
Immunohistochemistry
Fourteen days after drug treatment, tumors were collected and fixed in formalin. Immunohistochemistry for HER2 was carried out by using formalin‐fixed, paraffin‐embedded tumor samples and a HercepTest II kit (Dako A/S) by SRL Medisearch Inc. (Tokyo, Japan).
Statistical analysis
All statistical analyses carried out using SAS System Release 9.2 (SAS Institute, Cary, NC, USA). The IC50 values were determined by sigmoid Emax model.
Results
Structure of anti‐HER2 DXd and T‐DM1 and cell membrane permeability of payloads
The structures of HER2‐targeting ADCs are shown in Figure 1. DS‐8201a (anti‐HER2‐DXd (1)) and anti‐HER2‐DXd (2) are structurally composed of an anti‐HER2 antibody, a tetrapeptide linker, and topoisomerase I inhibitors, DXds. The linker‐payload was conjugated to the antibody through cysteine residues following the reduction of the disulfide bonds. DS‐8201a and anti‐HER2‐DXd (2) were designed to be cleaved by lysosomal enzymes such as cathepsins B and L, which are highly expressed in tumor cells.18, 19, 20 DS‐8201a has a self‐immolative moiety between the tetrapeptide linker and payload part, which is rapidly hydrolyzed, resulting in the release of DXd (1) having a primary hydroxyl group, whereas anti‐HER2‐DXd (2) simply releases DXd (2) having an amino group. T‐DM1 is an FDA‐approved HER2‐targeting ADC and is composed of trastuzumab, a non‐cleavable linker, and a tubulin polymerization inhibitor, DM1. In tumor cells, T‐DM1 is cleaved and releases Lys‐SMCC‐DM1, a linker‐payload component that has the potential to inhibit tubulin polymerization.21, 22, 23 The cell membrane permeability of these released payloads was assessed based on Log D and Peff values. The Log D and Peff values of each compound are shown in Table 1. The Log D of DXd (1) was 2.3, which was higher than those of DXd (2) and Lys‐SMCC‐DM1 (both 0.2). In addition, the Peff of DXd (1) was the highest at both pH 5.0 and pH 7.4, and cytotoxicity of DXd (1) was the most potent owing to its high membrane permeability (Table 1). These results suggest that the membrane permeability of DXd (1) is higher than that of DXd (2) and Lys‐SMCC‐DM1; therefore, after DS‐8201a is internalized into HER2‐positive cells and releases DXd (1) into the cytoplasm, DXd (1) could penetrate to adjacent cells, hypothesizing a bystander effect, whereas it would be difficult for DXd (2) and Lys‐SMCC‐DM1 to penetrate to adjacent cells.
Figure 1.
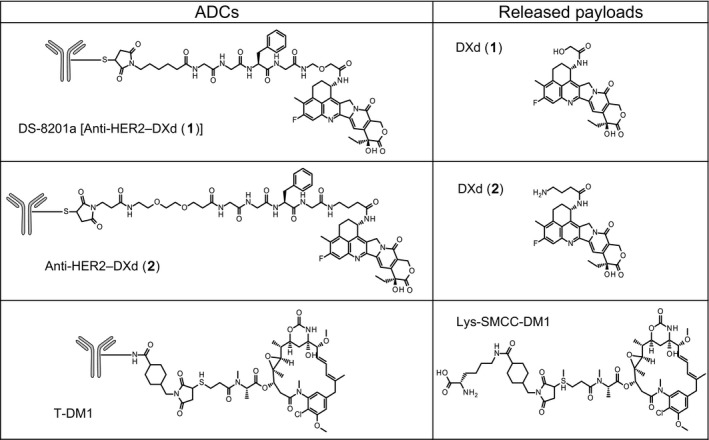
Structures of antibody–drug conjugates (ADCs) and released payloads. DXd, DX‐8951 derivative; HER2, human epidermal growth factor receptor 2; T‐DM1, trastuzumab emtansine.
Table 1.
Physicochemical parameters of antibody–drug conjugate payloads
| DXd (1) | DXd (2) | Lys‐SMCC‐DM1 | ||
|---|---|---|---|---|
| Molecule weight | 493.5 | 520.6 | 1103.7 | |
| Log D | pH 7.4 | 2.3 | 0.2 | 0.2 |
| PAMPA, Peff | pH 5.0 | 13 | <0.1 | <0.1 |
| pH 7.4 | 12.2 | <0.1 | <0.1 | |
| Cytotoxicity, IC50, nM | KPL‐4 | 4.0 | 33.5 | 24.8 |
| MDA‐MB‐468 | 0.5 | 9.7 | 40.5 | |
DXd, DX‐8951 derivative; PAMPA, parallel artificial membrane permeability assay; Peff, permeability coefficient; T‐DM1, trastuzumab emtansine.
In vitro cell growth inhibitory activity
The cell growth inhibitory activities of DS‐8201a, anti‐HER2‐DXd (2), and T‐DM1 were evaluated. As negative control, IgG‐DXd (1) and IgG‐DXd (2) were also evaluated. All HER2‐targeting ADCs were effective against the HER2‐positive KPL‐4 cell line; however, control IgG ADCs were not (Fig. 2a). The IC50 values of DS‐8201a, anti‐HER2‐DXd (2), and T‐DM1 were 109.7, 22.1, and 18.5 pM, respectively. None of the ADCs inhibited the growth of the HER2‐negative MDA‐MB‐468 cell line (IC50 values > 10 000 pM, Fig. 2a). As a reference, all released payloads showed growth inhibition in both KPL‐4 and MDA‐MB‐468 cells (Table 1). Thus, HER2‐specific cell growth inhibitory activity of all HER2‐targeting ADCs was confirmed.
Figure 2.
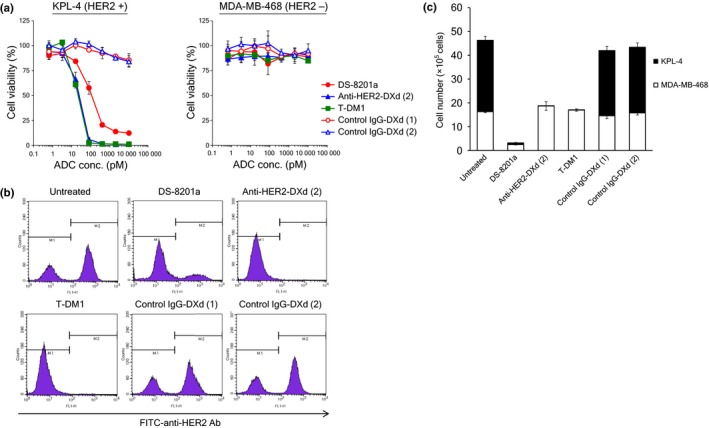
Bystander killing effect of antibody–drug conjugates (ADCs) in coculture conditions in vitro. (a) Growth inhibitory activity of ADCs against KPL‐4 and MDA‐MB‐468 cells. Tumor cells were treated with ADCs for 5 days and cell viability (%) was calculated. Each point represents the mean and SD (n = 3). KPL‐4 and MDA‐MB‐468 cells were cocultured and treated with 10 nM ADCs for 5 days. After collecting adherent cells, cell number and ratio of human epidermal growth factor receptor 2 (HER2)‐positive and HER2‐negative cells were determined by a cell counter and a flow cytometer, respectively. (b) Data of flow cytometric analysis. (c) Numbers of KPL‐4 and MDA‐MB‐468 viable cells. Each bar represents the mean and SD (n = 3). DXd, DX‐8951 derivative; T‐DM1, trastuzumab emtansine.
Bystander killing effect of DS‐8201a in vitro
In order to confirm whether DS‐8201a induced bystander killing, a coculture cell killing assay was carried out. The HER2‐positive KPL‐4 cells and the HER2‐negative MDA‐MB‐468 cells were mixed and cultured at an appropriate ratio overnight. The cells were treated with ADC for 5 days, and then the number of living cells was determined. Concurrently, in order to determine the ratio of KPL‐4 and MDA‐MB‐468 cells, HER2‐expressing living cells were detected by flow cytometric analysis. As shown in Figure 2(b), the living cells clearly showed two peaks indicative of KPL4 and MDA‐MB‐468 cells. Based on the number of living cells and ratio of KPL‐4 and MDA‐MB‐468 cells in each well, the number of KPL‐4 or MDA‐MB‐468 cells was calculated. As a result, although anti‐HER2‐DXd (2) and T‐DM1 killed only HER2‐positive KPL‐4 cells completely, DS‐8201a killed both KPL‐4 and MDA‐MB‐468 cells (Fig. 2c). Negative control ADCs did not induce cell killing compared to untreated wells. As DS‐8201a has HER2‐specific cytotoxicity, MDA‐MB‐468 cell killing could be caused by the released payload, DXd (1), suggesting the bystander killing effect of DS‐8201a. However, anti‐HER2‐DXd (2) and T‐DM1 did not show a bystander killing effect against HER2‐negative cells, and it was supposed that the membrane permeability of the payload is an important factor for bystander killing.
Bystander killing effect of DS‐8201a in vivo
A novel in vivo evaluation system was established for in vivo assessment of the bystander killing observed in the in vitro study. MDA‐MB‐468‐Luc cells were generated to detect the existence of HER2‐negative cells in a mixed tumor composed of HER2‐positive cells and HER2‐negative cells using an in vivo imaging system. First, the antitumor activity of each HER2‐targeting ADC was evaluated against a HER2‐positive model or a HER2‐negative model. As the HER2‐positive model, NCI‐N87 cells were used. All HER2‐targeting ADCs at single doses induced tumor regression in the NCI‐N87 model, but not in the MDA‐MB‐468‐Luc model, indicating HER2‐dependent efficacy of the ADCs (Fig. 3a). In both models, control IgG‐DXds were not effective. Second, we confirmed the heterogeneity of the mixed tumor generated by inoculating a mixture of NCI‐N87 and MDA‐MB‐468‐luc cells through immunohistochemistry of HER2. As shown in Figure 3(b), both HER2‐positive and HER2‐negative cancer cells were observed in the mixed tumor; only HER2‐positive cancer cells and host‐derived stromal cells were observed in an NCI‐N87 tumor. Then, each HER2‐targeting ADC or control ADC was administered to the co‐inoculated xenograft mice, and tumor volume and luciferase activity of each mouse were measured. In the DS‐8201a group, an apparent reduction in luciferase signal was observed, indicating that MDA‐MB‐468‐Luc cells were completely eradicated by DS‐8201a (Fig. 4a,b). Similar results were not observed with other HER2‐targeting ADCs or control ADCs. In terms of tumor volume changes, anti‐HER2‐DXd (2) and T‐DM1 inhibited tumor growth but were less potent than DS‐8201a (Fig. 4c), which could be attributed to the limited elimination of tumor, in particular only the HER2‐positive population by anti‐HER2‐DXd (2) and T‐DM1. In the anti‐HER2‐DXd (2)‐treated and T‐DM1‐treated groups, the immunohistochemical analysis of treated tumors showed eradication of HER2‐positive cells, and HER2‐negative cancer cells occupied a large part of tumor tissue (Fig. S1). In the DS‐8201a‐treated group, almost all of the HER2‐positive and HER‐negative cells disappeared and there were little or no cancer cells remaining in the tumors. From these results, it was confirmed that DS‐8201a showed antitumor activity against not only HER2‐positive tumors but also HER2‐negative tumors under the co‐inoculated condition, whereas T‐DM1 did not affect HER2‐negative tumors.
Figure 3.
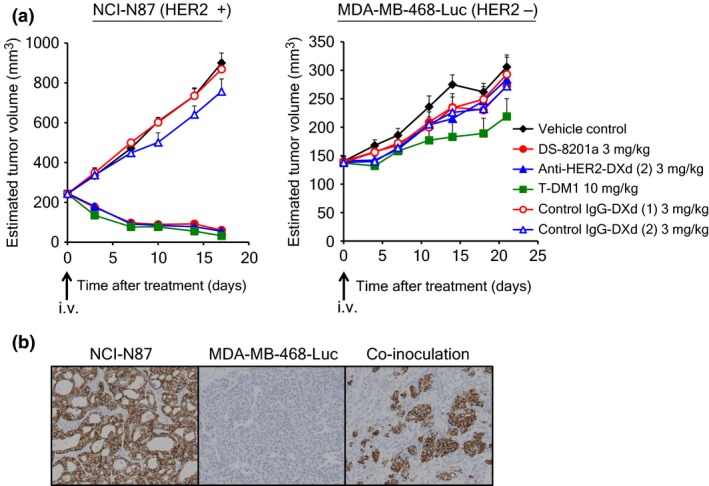
Antitumor activity of antibody–drug conjugates (ADCs) and model establishment for bystander evaluation. (a) Antitumor activity of ADCs in NCI‐N87 and MDA‐MB‐468‐Luc xenograft models. The mean estimated tumor volume and SE (n = 6) are represented on the graph. (b) Human epidermal growth factor receptor 2 (HER2) expression in co‐inoculated tumor. Eight days after s.c. inoculation, each tumor was collected and fixed with 10% neutral‐buffered formalin. Immunohistochemistry of HER2 protein was carried out using the HercepTest II kit according to manufacturer's instructions. DXd, DX‐8951 derivative; T‐DM1, trastuzumab emtansine.
Figure 4.
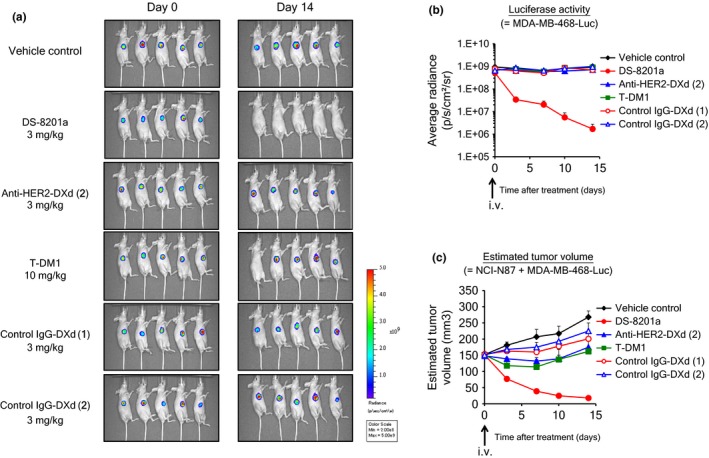
Bystander killing in co‐inoculated conditions in vivo. Seven days after inoculation of a mixture of both NCI‐N87 and MDA‐MB‐468‐Luc cells, mice were i.v. given antibody–drug conjugates (day 0). Luciferase activity was detected by in vivo imager after i.v. administration of substrate. (a) Imaging data of luciferase activity. (b) Luciferase activity. (c) Estimated tumor volume. SE (n = 5) represented on the graph. DXd, DX‐8951 derivative; HER2, human epidermal growth factor receptor 2; T‐DM1, trastuzumab emtansine.
Bystander effect on distant cells
It is still unclear whether the bystander killing of DS‐8201a is observed only in HER2‐negative cells adjacent to HER2‐positive cells. Therefore, an effect on cells distant from HER2‐positive cells was evaluated. A mixture of NCI‐N87 and MDA‐MB‐468‐Luc cells was inoculated into the right flank of mouse and only MDA‐MB‐468‐Luc cells were inoculated into the left flank of the same mouse. Fourteen days after treatment with DS‐8201a, luciferase signals in tumors of both sides were measured and compared. The signal of co‐inoculated tumor decreased, but that of mono‐inoculated tumor did not change (Fig. 5a). On day 14, the signal of mono‐inoculated tumors of DS‐8201a treated mice was comparable to that of untreated mice (Fig. 5). These results indicated that the effect of DS‐8201a on HER2‐negative cells was observed only in the case of cells adjacent to HER2‐positive cells, not distant cells.
Figure 5.
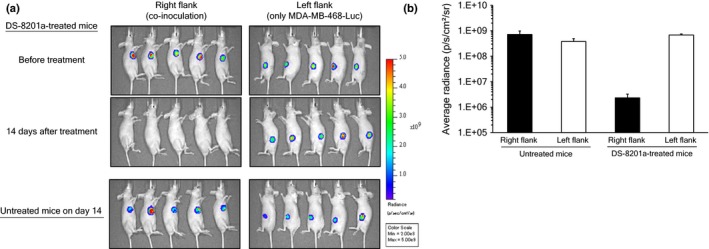
Bystander killing effect on distant tumor. Nude mice were s.c. inoculated with a mixture of both NCI‐N87 and MDA‐MB‐468‐Luc cells into the right flank, and only MDA‐MB‐468‐Luc into the left flank. Seven days after inoculation, mice were treated with i.v. DS‐8201a at 3 mg/kg. Luciferase activity was detected by an in vivo imager after i.v. administration of substrate. (a) Imaging data of luciferase activity. (b) Mean luciferase activity and SE (n = 5) are represented on the graph.
Discussions
Generally, it is reported that ADCs including approved ones, T‐DM1 and brentuximab vedotin, show antigen‐dependent cytotoxicity in vitro and antitumor efficacy in vivo.24, 25 Similarly, HER2‐dependent efficacy of DS‐8201a was confirmed in a previous report.12 In this study, we reported that DS‐8201a kills both HER2‐positive and HER2‐negative cells under coculture conditions, an effect named bystander killing. In contrast, the low permeable payload‐loaded HER2 ADC, anti‐HER2‐DXd (2), did not show any bystander killing effect in the same conditions even though the payload, DXd (2), has comparable topoisomerase I inhibitory activity (data not shown). Although the bystander effect of several ADCs has already been reported,13, 14, 15, 16 this is the first study to investigate the relationship between bystander killing and membrane permeability of payload using two different kinds of topoisomerase I inhibitors. T‐DM1 did not show evidence of bystander killing and it is supposed that the low permeability of Lys‐SMCC‐DM1, which is the active metabolite of T‐DM1, is one of the reasons for the bystander killing effect, as shown in Table 1.
Recently, Roche announced that the phase II/III Gatsby trial, a clinical trial designed to support a new indication for T‐DM1 in second‐line treatment of HER2‐positive advanced gastric cancer, did not meet its primary endpoint. The heterogeneity of HER2 expression in gastric cancer has been reported,26 and it could be a potential reason for the failure. Bystander killing could address this limitation for this issue and DS‐8201a can overcome the heterogeneity to show a significant clinical response in gastric cancer patients. Even in breast cancer, which is an approved indication of T‐DM1, HER2 heterogeneity was reported27 and a higher objective response rate of DS‐8021a in a breast cancer patient group than T‐DM1 is expected. Altogether, we conclude that the bystander killing potential of DS‐8201a is a significant point of differentiation from T‐DM1.
Most previous reports regarding the bystander effect of ADCs indicated only the results of in vitro experiments, which are closed systems that provide inadequate evaluation of cells adjacent to antigen‐positive cells because the released payload could accumulate in the culture supernatant. Additionally, the reports of in vivo evaluation are limited owing to the difficulty of detecting antigen‐negative cells. In this report, we undertook a novel in vivo imaging study and for the first time achieved a time‐course observation of antigen‐negative cells under co‐inoculated conditions. The combination of in vivo imaging and the assessment of HER2 expression made it possible to detect both HER2‐positive and HER‐negative cells in one experiment. It helped us to understand that anti‐HER2‐DXd (2) and T‐DM1 killed only HER2‐positive cells in vivo (Fig. 4). However, it was difficult to control HER2 heterogeneity in vitro and in vivo; therefore we could use only KPL‐4 cells for in vitro assays and NCI‐N87 cells for in vivo assays. An improvement of experimental method is still required for assessment of bystander killing by various ADCs.
Moreover, the evaluation of the effect of released payload on distant cells, and not only neighboring cells, was also a breakthrough of this report. Safety concerns of the bystander effect have to be considered in addition to its advantages of efficacy; however, this was not evaluated in previous reports. Here, we clearly proved that the bystander killing of DS‐8201a was observed only on cells neighboring HER2‐positive cells, not distant cells, indicating that systemic toxicity or toxicity in distant normal tissues from tumor sites would be low. However, effects on tumor‐adjacent normal tissues are still unknown. There is one hypothesis that the safety concerns against these tissues, except for bone marrow cells and hematocytes, would be low because topoisomerase I inhibitors are effective against proliferative cells like cancer cells.
We showed that ADC conjugated with low‐permeable payload did not provoke bystander killing. On the contrary, this type of ADC might be valuable for targeting hematological cancers because bystander killing could not be observed in circulating cancer cells and the released payloads with low permeability could accumulate in the cells, resulting in potent cytotoxicity. In fact, the cytotoxicity of anti‐HER2‐DXd (2) was more potent than that of DS‐8201a (Fig. 2a). We may need to select an appropriate type of payload based on the target indication of ADC for potent antitumor efficacy.
The results of this study showed the potent bystander killing effect of DS‐8201a owing to its high membrane‐permeable payload and the potential of DS‐8201a to target tumors with HER2 heterogeneity, such as gastric cancer, whereas T‐DM1 does not show clinical benefit.
Disclosure Statement
All the authors are employees of Daiichi Sankyo Co., Ltd.
Supporting information
Fig. S1. Human epidermal growth factor receptor 2 (HER2) expression status after antibody–drug conjugate (ADC) treatment.
Acknowledgments
The authors thank Takashi Nakada, Ichiro Hayakawa, and Koji Morita for the synthesis of the ADCs and payloads, and Akira Okubo for technical support.
Cancer Sci 107 (2016) 1039–1046
Funding Information
Daiichi Sankyo Co., Ltd., Japan
References
- 1. Bross PF, Beitz J, Chen G et al Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 2001; 7: 1490–6. [PubMed] [Google Scholar]
- 2. Petersdorf SH, Kopecky KJ, Slovak M et al A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013; 121: 4854–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gopal AK, Ramchandren R, O'Connor OA et al Safety and efficacy of brentuximab vedotin for Hodgkin lymphoma recurring after allogeneic stem cell transplantation. Blood 2012; 120: 560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Younes A, Gopal AK, Smith SE et al Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol 2012; 30: 2183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogura M, Tobinai K, Hatake K et al Phase I/II study of brentuximab vedotin in Japanese patients with relapsed or refractory CD30‐positive Hodgkin's lymphoma or systemic anaplastic large‐cell lymphoma. Cancer Sci 2014; 105: 840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burris HA 3rd, Rugo HS, Vukelja SJ et al Phase II study of the antibody drug conjugate trastuzumab‐DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)‐positive breast cancer after prior HER2‐directed therapy. J Clin Oncol 2011; 29: 398–405. [DOI] [PubMed] [Google Scholar]
- 7. Verma S, Miles D, Gianni L et al Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med 2012; 367: 1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krop IE, Kim SB, Gonzalez‐Martin A et al Trastuzumab emtansine versus treatment of physician's choice for pretreated HER2‐positive advanced breast cancer (TH3RESA): a randomised, open‐label, phase 3 trial. Lancet Oncol 2014; 15: 689–99. [DOI] [PubMed] [Google Scholar]
- 9. Hamilton GS. Antibody‐drug conjugates for cancer therapy: the technological and regulatory challenges of developing drug‐biologic hybrids. Biologicals 2015; 43: 318–32. [DOI] [PubMed] [Google Scholar]
- 10. Hinrichs MJ, Dixit R. Antibody drug conjugates: nonclinical safety considerations. AAPS J 2015; 17: 1055–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sapra P, Shor B. Monoclonal antibody‐based therapies in cancer: advances and challenges. Pharmacol Ther 2013; 138: 452–69. [DOI] [PubMed] [Google Scholar]
- 12. Ogitani Y, Aida T, Hagihara K et al DS‐8201a, a novel HER2‐targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising anti‐tumor efficacy with differentiation from T‐DM1. Clin Cancer Res 2016; doi: 10.1158/1078‐0432.CCR‐15‐2822. [DOI] [PubMed] [Google Scholar]
- 13. Kovtun YV, Audette CA, Ye Y et al Antibody‐drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res 2006; 66: 3214–21. [DOI] [PubMed] [Google Scholar]
- 14. Golfier S, Kopitz C, Kahnert A et al Anetumab ravtansine: a novel mesothelin‐targeting antibody‐drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther 2014; 13: 1537–48. [DOI] [PubMed] [Google Scholar]
- 15. van der Lee MM, Groothuis PG, Ubink R et al The preclinical profile of the duocarmycin‐based HER2‐targeting ADC SYD985 predicts for clinical benefit in low HER2‐expressing breast cancers. Mol Cancer Ther 2015; 14: 692–703. [DOI] [PubMed] [Google Scholar]
- 16. Li JY, Perry SR, Muniz‐Medina V et al A biparatopic HER2‐targeting antibody‐drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2‐targeted therapy. Cancer Cell 2016; 29: 117–29. [DOI] [PubMed] [Google Scholar]
- 17. Nakada T, Masuda T, Naito H et al Novel antibody drug conjugates containing exatecan derivative‐based cytotoxic payloads. Bioorg Med Chem Lett 2016; 26: 1542–5. [DOI] [PubMed] [Google Scholar]
- 18. Shiose Y, Ochi Y, Kuga H, Yamashita F, Hashida M. Relationship between drug release of DE‐310, macromolecular prodrug of DX‐8951f, and cathepsins activity in several tumors. Biol Pharm Bull 2007; 30: 2365–70. [DOI] [PubMed] [Google Scholar]
- 19. Niedergethmann M, Wostbrock B, Sturm JW, Willeke F, Post S, Hildenbrand R. Prognostic impact of cysteine proteases cathepsin B and cathepsin L in pancreatic adenocarcinoma. Pancreas 2004; 29: 204–11. [DOI] [PubMed] [Google Scholar]
- 20. Aggarwal N, Sloane BF. Cathepsin B: multiple roles in cancer. Proteomics Clin Appl 2014; 8: 427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun X, Widdison W, Mayo M et al Design of antibody‐maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug Chem 2011; 22: 728–35. [DOI] [PubMed] [Google Scholar]
- 22. Erickson HK, Lewis Phillips GD, Leipold DD et al The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther 2012; 11: 1133–42. [DOI] [PubMed] [Google Scholar]
- 23. Kovtun YV, Audette CA, Mayo MF et al Antibody‐maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res 2010; 70: 2528–37. [DOI] [PubMed] [Google Scholar]
- 24. Lewis Phillips GD, Li G, Dugger DL et al Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody‐cytotoxic drug conjugate. Cancer Res 2008; 68: 9280–90. [DOI] [PubMed] [Google Scholar]
- 25. Francisco JA, Cerveny CG, Meyer DL et al cAC10‐vcMMAE, an anti‐CD30‐monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003; 102: 1458–65. [DOI] [PubMed] [Google Scholar]
- 26. Lee HE, Park KU, Yoo SB et al Clinical significance of intratumoral HER2 heterogeneity in gastric cancer. Eur J Cancer 2013; 49: 1448–57. [DOI] [PubMed] [Google Scholar]
- 27. Bethune GC, Mullen JB, Chang MC. Detecting intratumoral heterogeneity in routine breast‐HER2 testing: low yield of testing multiple blocks. Ann Diagn Pathol 2015; 19: 385–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Human epidermal growth factor receptor 2 (HER2) expression status after antibody–drug conjugate (ADC) treatment.