

Abstract
The development of inhibitors for phospholipases A2 (PLA2s) is important in elucidating their implication in various biological pathways. PLA2 enzymes are an important pharmacological target implicated in various inflammatory diseases. Computational chemistry, organic synthesis and in vitro assays were employed to develop potent and selective inhibitors for Group VIA calcium-independent PLA2. A set of fluoroketone inhibitors, were studied for their binding mode with two human cytosolic PLA2 enzymes: Group IVA cPLA2 and Group VIA iPLA2. New compounds were synthesized and tested against three major PLA2 enzyme. This study led to the development of four potent and selective thioether fluoroketone inhibitors as well as a thioether keto-1,2,4-oxadiazole inhibitor for GVIA iPLA2, which will serve as lead compounds for future development and studies. The keto-1,2,4-oxadiazole functionality with a thioether is a novel structure and it will be used as a lead to develop inhibitors with higher potency and selectivity towards GVIA iPLA2.
Graphical abstract
Introduction
Phospholipase A2 (PLA2) constitutes a superfamily of enzymes that currently consists of 16 groups and several subgroups, and can be classified into the following six types: cytosolic (cPLA2), secreted (sPLA2), calcium-independent (iPLA2), platelet-activating factor acetylhydrolase (PAF-AH) also known as lipoprotein-associated PLA2 (Lp-PLA2), lysosomal PLA2 (LPLA2), and adipose-PLA (AdPLA).1 PLA2s catalyze the release of free fatty acids including arachidonic acid (AA) which is the initial and rate limiting step for eicosanoid biosynthesis.2 Through their catalytic activity, Group IVA cytosolic (GVIA cPLA2), Group VIA calcium-independent (GVIA iPLA2), and Group V secreted (GV sPLA2) enzymes are implicated in many inflammatory diseases.1 Thus, the development of potent and selective inhibitors for each of these three enzymes should lead to the development of novel pharmaceutical agents for different inflammatory conditions.3
GIVA cPLA2 was cloned and sequenced in 19914–6 and its crystal structure was reported in 1999.7 This enzyme utilizes a catalytic dyad of Ser/Asp, and it exhibits high specificity for membrane phospholipids containing AA at the sn-2 position. Thus, it is the main AA provider for the cyclooxygenase (COX) and lipoxygenase (LOX) pathways.8 Therefore, this enzyme can be considered a key enzyme for mediating production of eicosanoids which are implicated in numerous inflammatory diseases.1
GVIA iPLA2 was purified from macrophages in 1994,9, 10 and its crystal structure has not been solved yet. The human gene of this enzyme expresses multiple splice variants.11, 12 Among them, the transcript variant 1 is one of the two active isoforms in humans. GVIA iPLA2 also utilizes a catalytic dyad of Ser/Asp, and it is not known to exhibit specificity for the sn-2 fatty acid of the membrane phospholipids. It was found to be involved in membrane homeostasis and remodeling involved in the basic metabolic functions within the cell.1, 13 GVIA iPLA2 is involved in signaling and other pathological conditions including diabetes14 and Barth syndrome.15
GV sPLA2 was expressed and characterized in 1998,16 and its crystal structure has not been reported either. This enzyme is also implicated in several inflammatory diseases, and its activity was found to be interrelated with that of GIVA cPLA2.17 GV sPLA2 utilizes a catalytic dyad of His/Asp, and it was detected in rheumatoid arthritis synovial fluids, but its expression was significantly lower than GIIA sPLA2, which is the major enzyme secreted in rheumatoid arthritis.18
A variety of diverse small molecule inhibitors have been reported and their structures are summarized in review articles.19, 20 Our groups have developed some novel classes of inhibitors including 2-oxoamides for GIVA cPLA2,21, 22 amides for GV sPLA2,23 and fluoroketones for GVIA iPLA2.24–26 We have now explored potent and selective inhibitors for GVIA iPLA2 using structure-based design and in vitro mixed micelle assays.27 Even though there is no available crystal structure for this enzyme, we have developed a robust homology model based on hydrogen/deuterium exchange mass spectrometry (DXMS) experimental data and molecular dynamics (MD) simulations.28–30 The 3D structure of GVIA iPLA2 was used for molecular docking calculations and MD simulations with previously synthesized inhibitors in an effort to establish a structure-activity relationship (SAR). Based on our SAR model, new fluoroketone compounds were designed, synthesized and tested in vitro, and we were able to identify four new fluoroketones that potently inhibit GVIA iPLA2 and they are quite selective relative to GIVA cPLA2 and GV sPLA2. In addition, a novel potent and selective GVIA iPLA2 inhibitor based on a keto-1,2,4-oxadiazole functionality with a thioether was identify.
Results and Discussion
Structure-based design protocol
Fluoroketones represent a unique class of PLA2 inhibitors that are highly potent and selective toward GVIA iPLA2.24–26 Understanding the binding mode (conformation of the inhibitor as well as its interactions with the binding pocket of the enzyme) of these compounds is crucial in designing new compounds with improved inhibitory properties. To do so, a data set of 27 PLA2 inhibitors was used in order to develop a SAR for designing, synthesizing and testing new compounds (Table 1).24, 25 Figure 1 schematically represents the structure-based design approach adopted in this study with the goal to design, synthesize and test new fluoroketone compounds for their inhibitory activity against representatives of three major PLA2 groups.
Table 1.
Data set of fluoroketones used to generate SAR for GVIA iPLA2
| # | Structure | XI(50) | log XI(50) | XP GScore (GVIA iPLA2) | XP GScore (GIVA cPLA2) |
|---|---|---|---|---|---|
| 1a |
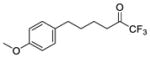
|
0.0001 | −4.00 | −8.9 | −7.1 |
| 2a |
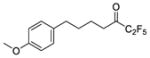
|
0.0001 | −4.00 | −8.9 | −6.7 |
| 3b |
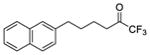
|
0.0002 | −3.70 | −8.9 | −7.8 |
| 4a |
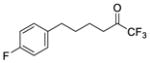
|
0.0002 | −3.70 | −8.1 | −6.9 |
| 5a |
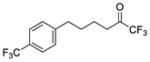
|
0.0002 | −3.70 | −8.6 | −7.9 |
| 6a |
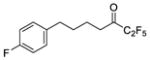
|
0.0003 | −3.52 | −6.5 | … |
| 7a |
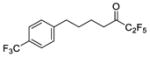
|
0.0003 | −3.52 | −8.4 | … |
| 8 b |
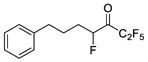
|
0.0005 | −3.30 | −6.7 | … |
| 9b |
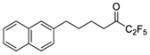
|
0.0006 | −3.22 | −8.7 | … |
| 10b |
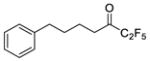
|
0.0014 | −2.85 | −7.7 | −6.7 |
| 11a |
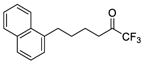
|
0.0018 | −2.74 | −8.6 | … |
| 12a |
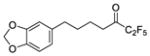
|
0.0019 | −2.72 | −6.7 | … |
| 13b |
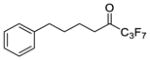
|
0.0022 | −2.66 | ND* | … |
| 14a |
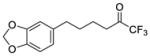
|
0.0025 | −2.60 | −8.00 | … |
| 15b |
|
0.0030 | −2.52 | −7.4 | … |
| 16a |
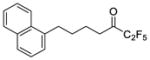
|
0.0034 | −2.47 | −7.8 | … |
| 17b |
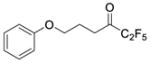
|
0.0036 | −2.44 | −7.8 | … |
| 18b |
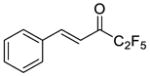
|
0.0066 | −2.18 | −5.3 | … |
| 19b |
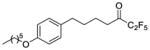
|
0.0084 | −2.08 | −7.0 | … |
| 20a |
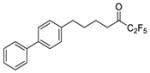
|
0.0134 | −1.87 | −6.1 | −9.8 |
| 21a |
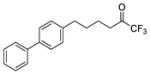
|
0.0189 | −1.72 | −7.3 | −9.2 |
| 22b |
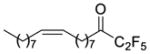
|
0.0192 | −1.72 | −8.4 | … |
| 23b |
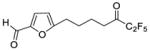
|
0.0262 | −1.58 | −6.4 | … |
| 24a |
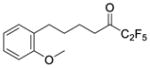
|
0.0263 | −1.58 | −8.2 | … |
| 25b |
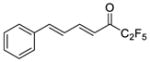
|
0.0313 | −1.50 | −5.4 | … |
| 26b |
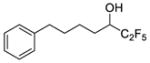
|
0.0390 | −1.41 | ND* | … |
| 27a |
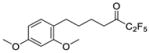
|
0.0553 | −1.26 | −6.9 | … |
Figure 1. Structure-based design protocol.

Fluoroketone inhibitors were docked in GVIA iPLA2 and GIVA cPLA2 binding site. Each enzyme-inhibitor complex was placed on a POPC membrane patch, solvated, minimized, equilibrated and subjected in 300 ns MD simulation. Suitable docking conformations of each complex were identifying using clustering analysis.
Three compounds were initially selected and docked in the binding pocket of both GVIA iPLA2 and GIVA cPLA2. GV sPLA2 was not included in the structure-based design protocol because none of the fluoroketone compounds exhibited significant activity towards this enzyme.24, 25 Compounds 2 (GK187)25 and 3 (FKGK18)24 are potent inhibitors of GVIA iPLA2 that showed similar inhibitory activity (Table 1). Compound 2 is a pentafluoro ketone containing a methoxy phenyl group, while compound 3 is a trifluoromethyl ketone with a naphthalene group at the hydrophobic chain. These two compounds were used to initiate our studies because they are structurally different in terms of the fluoro carbonyl as well as the hydrophobic chain even though they have comparable inhibitory potency.24, 25 Compound 21 (GK174)25 is a trifluoromethyl ketone with two phenyl groups at the hydrophobic chain and is the only compound in this class that showed high inhibitory activity against GIVA cPLA2.25 Each compound was docked in the binding pocket of the relevant enzyme to create an optimized enzyme-inhibitor complex in terms of conformation and binding energy. The resulting enzyme-inhibitor complexes were then placed on a POPC membrane patch based on previously published models,28 and the systems (enzyme-inhibitor-membrane-water) were minimized, equilibrated and subjected to a 300 ns simulation each. Clustering analysis was performed to identify the most abundant conformation for each complex. These complexes were used in docking calculations using a set of published fluoroketone inhibitors24, 25 with the goal to create reliable models to assist the structure-based design of new compounds.
Binding mode of fluoroketone inhibitors based on Induced Fit Docking (IFD)
The IFD calculations were performed using the homology model of GVIA iPLA228 and the crystal structure for GIVA cPLA2.7 Residues Gly486, Gly487, Lys489, Ser519, Val548, Phe549, Leu560, Tyr643, Phe644, Asp652, Lys729, and Leu770 were selected to define the binding pocket of GVIA iPLA2 in the IFD calculations. These residues were found to show decreased “on exchange” rates in DXMS studies upon binding of a GVIA iPLA2 inhibitor, which belongs to the same fluoroketone class of compounds.30 For GIVA cPLA2 residues Gly197, Gly198, Phe199, Arg200, Ser228, Trp232, Pro263, Phe295, Leu298, Ile299, Thr302, Leu303, Phe397, Met417, Asp549, Asn555, Thr680 and Phe683 were chosen to define the binding pocket of the enzyme in the IFD calculations. These residues showed decreased “on exchange” rates in DXMS studies upon binding with pyrrophenone, which is a potent GIVA cPLA2 inhibitor.31
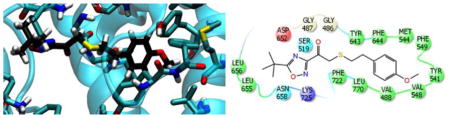
Figure 2A and 2C show the binding mode of compound 2 and 3 in the binding pocket of GVIA iPLA2 generated by the IFD calculations. The fluoroketone group is located in the hydrophilic region of the binding pocket. The oxygen atom of the carbonyl group is near to residues Gly486/Gly487 which constitute the “oxyanion hole”. These two glycine residues were found to interact with the sn-2 carbonyl group of the phospholipid according to our previously published structural studies.28 The same oxygen atom is in spatial proximity to the catalytic Ser519. The fluorine atoms are located near to the residues Asn658 and Lys725. These two residues were found to interact with the phosphate group according to our previous studies.28 The methoxy phenyl group in compound 2 and the naphthalene group in 3 is located near to the entrance of the binding pocket, and it interacts with aromatic and aliphatic residues including Val548, Phe549, Leu560, Tyr643, Phe644 and Leu770 (Fig. 2A and 2C).
Figure 2. Binding mode of compound 2 and 3 in GVIA iPLA2 binding pocket.

(A) GVIA iPLA2-2 complex generated by IFD. (B) GVIA iPLA2-2 complex after MD simulation (Movie 1). (C) GVIA iPLA2-3 complex generated by IFD. (D) GVIA iPLA2-3 complex after MD simulation (Movie 2).
Figure 3A represents the binding mode of compound 21 in the binding pocket of GIVA cPLA2 from the IFD calculations. The fluoroketone group is located near to the “oxyanion hole” (Gly197/Gly198) and to the catalytic Ser228, with fluorine atoms near Asn555. It seems that the binding of the fluoroketone group in both enzymes involves identical residues. These residues were also found to interact either with the sn-2 carbonyl group (Gly197/Gly198) or the phosphate group (Asn555) of the phospholipid molecule in our previous studies.28 The two phenyl groups are placed in the hydrophobic region of the active site near residues Trp232, Pro263, Phe295, Leu298, Ile299, Thr302, Leu303, Phe397 and Phe683. This is the region where the fatty acyl chains of the phospholipid molecule were bound according to our previous studies.28
Figure 3. Binding mode of compound 21 in GIVA cPLA2 binding pocket.

(A) GIVA cPLA2-21 complex generated by IFD. (B) GIVA cPLA2-21 complex after MD simulation (Movie 3).
Binding mode of fluoroketone inhibitors after MD
Each enzyme-inhibitor complex was placed on the surface of a POPC membrane patch (Fig. 1) minimized, equilibrated and subjected to a 300 ns simulation using NAMD 2.9.32 The RMSD for the enzyme backbone atoms was stabilized at ~2.5 Å relative to the starting structure indicating that each simulation was reasonably converged (Fig. S1).
The binding mode of compound 2 and 3 after the MD simulation is shown in Figure 2B and 2D. The fluoroketone group remains in the hydrophilic region of the enzyme binding pocket with the oxygen atom of each carbonyl group interacting periodically with Gly486/Gly487 the so called “oxyanion hole” (Movie 1 and 2 or Fig. 2B and 2D). The same oxygen atom does not show any interactions with the catalytic Ser519. The fluorine atoms participate in H-bonding interactions with residue Asn658 but no interactions were observed with residue Lys725. In both simulations, the fluoroketone group shows an identical interaction pattern while the number of fluorine atoms does not seem to play a significant role in the binding. The simulations showed an interesting movement of the methoxy phenyl group in 2 and the naphthalene group in 3 from the entrance of the active site into the hydrophobic region of the binding pocket (Movie 1 and 2). The final pose of the MD simulations shows the two compounds in a horizontal orientation (Fig. 2B and 2D) rather than the vertical one suggested by the IFD complexes (Fig. 2A and 2C). This hydrophobic region was found to accommodate the sn-2 fatty acyl chain during our previously published studies on a PAPC substrate.28 On their final binding mode, the two groups were found to interact with residues like Val488, Ile523, Tyr541, Met544, Val548, Phe549, Leu560, Tyr643, Phe644 and Leu770. It is worth mentioning that the oxygen atom of the methoxy phenyl group participates in H-bonding with Tyr541 during the simulation (Movie 1).
The MD simulation of the GIVA cPLA2–21 complex (Fig. 3B) shows that the oxygen atom of the fluoroketone carbonyl group interacts with Gly197/Gly198, the so called “oxyanion hole” (Movie 3 or Fig. 3B). The fluorine atoms are in spatial proximity with Asn555 but no H-bonding interactions were observed during the simulation. The distance between the nitrogen atom of Asn555 and the fluorine atoms ranges between 3.0 to 3.5 Å, but never drops below 2.8 which is the threshold for the formation of an H-bond. The two phenyl groups remain in the same hydrophobic region of the deep binding channel indicated by the IFD complex and they do not appear to change their binding mode significantly during the simulation (Movie 3 or Fig. 3B).
Structure-activity relationships (SAR)
A representative enzyme-inhibitor complex was selected from each of the five clusters (Fig. 1) for docking calculations. Each complex was optimized using the Protein Preparation Wizard (PPW),33 and the already bound inhibitor was redocked in the active site. Enzyme-inhibitor complexes that belong to the most abundant clusters received the highest (absolute value) theoretical binding score, named herein as the XP GScore (Extra-Precision Glide Score). Compound 2 and 3 received similar XP GScore towards each MD complex with 3 showing slightly more optimized H-Bonding interactions. Thus, 3D complex of GVIA iPLA2 with compound 3 was used for conducting docking calculations for the SAR studies. All the fluoroketone compounds of Table 124, 25 were docked in GVIA iPLA2 binding site in order to examine if the structural models can give a good correlation between inhibitory activity and XP GScore. Table 1 summarizes the structures, the XI(50), the log XI(50), and the XP GScore for the selected compounds. XI(50) is the mole fraction of the compound that is required for 50% inhibition of the enzyme. Mole fraction is a dimensionless number derived by dividing the number of moles of inhibitor by the total number of moles of surface (moles of substrate plus inhibitor plus detergent).34, 35 XI(50) values can be converted to molar concentration by considering that 0.091 mole faction of inhibitor corresponds to 50 μM concentration. Thus, an XI(50) of 0.0001 corresponds to 55 nM.
An initial linear regression analysis did not reveal a correlation between the XI(50) and XP GScore values (Fig. 4A, Table 1). However, further analysis of the results showed that compounds 6, 8 and 12 give false negative predictions, while compounds 22 and 24 give false positive predictions according to the XP Glide scoring function. Outliers are typical of docking calculations since empirical scoring functions are not completely accurate in calculating theoretical binding scores.36 By excluding these five compounds from the linear regression, the linearity was significantly improved (Fig. 4B). It is worth noting that the compounds with high XI(50) values tend to have higher XP GScore against GVIA iPLA2, with our lead compounds 2 and 3 ranked among the highest ones. Another interesting compound is 21 which is the only compound exhibiting significant inhibitory activity against GIVA cPLA2 (XI(50) = 0.0074). This compound ranked with the highest XP GScore against GIVA cPLA2 in comparison with the lower scored 2 and 3 which showed no significant activity against GIVA cPLA2 (Table 1).
Figure 4. Correlation between the inhibitory activity (log XI(50)) and the XP GScore.

(A) All of the fluoroketone compounds were included. (B) Five outlier compounds were excluded (compounds 6, 8, 12, 22 and 24 in Table 1).
Design and synthesis of thioether fluoroketone and keto-1,2,4-oxadiazole inhibitors using the SAR model
In order to test the predictive ability of our SAR models, we have designed four new fluoroketone compounds based on the structures of compound 1 (GK177),25 2, 3 and 10 (FKGK11, Table 1).24, 25 These new analogues (28, 29, 30, and 31 in Table 2) contain a sulfur atom at the beta position to the carbonyl group taking into consideration the following literature data. During the ‘80s, Hammock and coworkers demonstrated that in some cases the presence of a thioether beta to the carbonyl group of trifluoromethyl ketones increased the inhibitory potency on juvenile hormone esterase37 and mammalian carboxylesterase.38 In addition, polyunsaturated trifluoromethyl ketones containing sulfur or oxygen atoms at the beta position were synthesized starting from EPA and DHA as potential PLA2 inhibitors.39 Recently, it was demonstrated that such sulfur derivatives directly inhibit GIVA cPLA2 and suppress PGE2 formation in mesangial cells.40 Given the substantial structural similarity of the new compounds with their original analogues we also synthesized two novel compounds (32 and 33 in Table 2) containing a 1,2,4-oxadiazole ring in order to test the ability of the SAR model to predict structurally different compounds. Oxadiazoles compounds were developed in the past as inhibitors of various enzymes including HIV integrase and they have been proven to exhibit a wide range of biological activities.41, 42
Table 2.
XP GScore and XI(50) values for fluoroketone and keto-1,2,4-oxadiazole inhibitors
| # | Structure | XP GScore (GVIA iPLA2) | XP GScore (GIVA cPLA2) | GVIA iPLA2 | GIVA cPLA2 | GV sPLA2 | |
|---|---|---|---|---|---|---|---|
| % Inh. | XI(50) | % Inh. | % Inh. | ||||
| 28 |
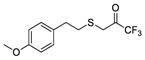
|
−8.9 | −6.8 | 96 ± 1 | 0.00009 ± 0.00001 | 60 ± 4 | ND* |
| 29 |
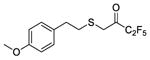
|
−8.9 | −6.8 | 100 ± 1 | 0.00012 ± 0.00001 | 61 ± 4 | 28 ± 5 |
| 30 |
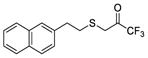
|
−8.8 | −7.4 | 99 ± 1 | 0.0002 ± 0.00006 | 88 ± 1 | 32 ± 2 |
| 31 |
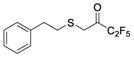
|
−8.4 | −6.1 | 99 ± 1 | 0.00015 ± 0.00004 | ND* | ND* |
| 32 |
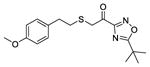
|
−7.4 | −6.9 | 98 ± 0.4 | 0.0057 ± 0.0012 | 70 ± 2 | 38 ± 4 |
| 33 |
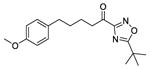
|
−4.3 | −6.0 | 64 ± 4 | … | 52 ± 6 | 35 ± 6 |
Compound exhibited less than 25% or no detectable inhibition.
Two different routes were employed to synthesize fluoroketones containing a sulfur atom at the beta position to activated carbonyl. Thiols 34a–c were reacted with commercially available 3-bromo-1,1,1-trifluoroacetone or 1-bromo-3,3,4,4,4-pentafluorobutan-2-one to give directly the target compounds 28, 30, 31 (Scheme 1). Pentafluoro derivative 29 was prepared by conversion of carboxylic acid 35 to the corresponding Weinreb amide, followed by treatment with (pentafluoroethyl)lithium (Scheme 1). Both routes led to products in high yield and in conclusion the synthesis of these new thioether fluoroketones requires fewer steps in comparison to the synthesis of compound 1, 2 and 3, where the synthesis starts from the Horner-Emmons reaction of an appropriate aromatic aldehyde and triethyl phosphonocrotonate.
Scheme 1a.

aReagents and conditions: (a) BrCH2COCF3 or BrCH2COCF2CF3, CCl4; (b) N,O-dimethylhydroxylamine, DMAP, NMM, WSCI, CH2Cl2;(c) MeLi.LiBr, CF3CF2I, −78 °C.
The synthesis of keto-1,2,4-oxadiazoles is depicted in Scheme 2. Alcohols 36a,b were oxidized to aldehydes and treated with tert-butyldimethylsilyl cyanide (TBDMSCN) in the presence of KCN and 18-crown-6 to produce nitriles 37a,b. Then, amidoximes 38a,b were prepared and coupled with pivalic acid using N,N’-dicyclocarbodiimide (DCC) as the coupling agent to give compounds 39a,b. The formation of the 1,2,4-oxadiazole ring was accomplished by treatment with tetrabutylammonium fluoride (TBAF) under microwave irradiation. Under such conditions the silyl protecting group was removed and compounds 40a,b were finally oxidized to target compounds 32, 33 using the Dess-Martin reagent.
Scheme 2a.

aReagents and conditions: (a) i. Dess-Martin periodinane, CH2Cl2; ii. TBDMSCN, 18-crown-6, KCN, CH2Cl2; (b) 50% aq. NH2OH, 50W; (c) pivalic acid, DCC, CH2Cl2; (d) TBAF, toluene, 90W; (e) Dess-Martin periodinane, CH2Cl2.
As shown in Table 2, our SAR model ranked compounds 28 (GK407), 29 (GK388) and 30 (GK408) with an XP GScore as high as the ones for 1, 2 and 3 against GVIA iPLA2, and as low as the ones against GIVA cPLA2. These compounds were tested in vitro against GVIA iPLA2 and GIVA cPLA2 and they indeed showed a comparable inhibitory activity and selectivity against GVIA iPLA2 as their precursor compounds. They also showed negligible inhibitory activity against the GV sPLA2, as expected for this class of fluoroketones. It is worth mentioning that compound 3 and 30 gave a higher XP GScore against GIVA cPLA2 than 1, 2, 28 and 29 and they also showed a higher percentage of inhibition against GIVA cPLA2 (inhibition 80.8% against GIVA cPLA2 was reported for compound 3).24 The importance of the sulfur atom for the potency of these inhibitors indicated by dramatic improvement of the inhibitory activity of compound 31 (Table 2) which is 10-fold more potent than its non-thio analogue 10 (Table 1). This compound shows high potency and selectivity for GVIA iPLA2 and it was ranked among the highly active compounds by receiving an XP GScore of 8.4. Finally, the potency of compounds 32 (GK392) and 33 (GK367) towards GVIA iPLA2 was satisfactorily predicted by the SAR model even though the compounds are structurally different from fluoroketones. Compound 32 received an XP score of 7.4 and an XI(50) of 0.0057 in contrast with it non-thio analogue 33 which received and XP of 4.8 and insignificant inhibition towards GVIA iPLA2. The selectivity of 32 towards GVIA iPLA2 was also predicted by receiving low XP GScore towards GIVA cPLA2.
Understanding the binding of the fluoroketone group
Our structural model gives insight into the binding mode of fluoroketone inhibitors as potent and selective inhibitors of GVIA iPLA2. Starting with the fluoroketone group it is clear that the number of fluorine atoms does not play a central role in the inhibitory activity. Both trifluoromethyl and pentafluoroethyl ketone gave the same activity (1 and 2 in Table 1).25 Simulations of both compounds 2 and 3 showed one residue Asn658 in GVIA iPLA2 interacting with the fluorine atoms of both pentafluoroethyl and trifluoromethyl groups (Movie 1 and 2). According to our proposed catalytic cycle, the “oxyanion hole” stabilizes the tetrahedral intermediate formed after the Ser519 attacks the carbon atom of the sn-2 carbonyl group.28 It is likely that the fluorine atoms add to the polarity of the carbonyl group and the catalytic Ser519 attacks the carbon atom forming a reversible hemiacetal, while residues Gly486/Gly487 stabilize the negatively charged oxygen atom. Based on our mechanistic assumptions, it appears that our model works well. However, the possibility of the formation of a covalent bond cannot be excluded, but that would not change the conclusion significantly. It is worth mentioning that the interactions of the fluoroketone group in GIVA cPLA2 (Movie 3) are identical with the ones in GVIA iPLA2.
Understanding the binding of the hydrophobic chain
The hydrophobic chain appears to play a critical role in the inhibitory activity of these compounds. The aromatic rings participate in pi-pi stacking while the bulky naphthalene ring in compound 3 reduces the inhibitory activity two-fold in contrast to the methoxy phenyl group in 2. The simulations (Movie 1 and 2) show that the methoxy phenyl group is much more flexible during its accommodation in the hydrophobic region of the pocket and, thus, optimizes its interactions with the surrounding residues. Moreover, the oxygen atom forms an H-bond with residue Tyr541 making the binding of 2 more favorable (Movie 1). The hydrophobic chain also plays an important role in the selectivity of these compounds. Short compounds like 2 show high selectivity toward GVIA iPLA2, while they tend to lose selectivity when the chain becomes bulkier (compound 3 exhibits less selectivity) and longer (compound 21 shows no selectivity). Our studies on the binding of a PAPC molecule,28 indicated that GIVA cPLA2 has a much deeper channel-like binding pocket than GVIA iPLA2. As a result, fluoroketones with a long hydrophobic chain will complement better the deep pocket and they will have higher inhibitory potency.
In summary, our studies show that the same fluoroketone functional group (activated carbonyl group) can be used to develop inhibitors with selectivity for either GVIA iPLA2 or GIVA cPLA2, which share the same catalytic mechanism both utilizing a catalytic dyad of Ser/Asp. However, the size of the hydrophobic chain is very critical for both activity and selectivity. Short-chain compounds tend to prefer GVIA iPLA2 while long-chain compounds also inhibit GIVA cPLA2.
Thioether enhances inhibitor binding and potency
MD simulations on compounds 32 and 33 revealed the binding mode of these two compounds and how the sulfur atom improves the inhibitory potency of the thio-analogues. The binding mode of 32 that occur during the simulation (Fig. 5A) showed that this compound exhibits tighter binding than 33 (Fig. 5B). It is obvious that the phenyl ring of 32 is closer to the residues of the hydrophobic pocket of GVIA iPLA2. The MD simulations showed that 32 (Movie 4) is much more stable in the binding site of the enzyme in comparison with 33 (Movie 5). The carbonyl group is constantly interacting with the “oxyanion hole” (Gly486/Gly487) while theoxadiazole ring is forming H-Bonds with Asn658. The same interactions do not occur in the case of 33 making its binding weaker and as a results is very unstable in the binding site of the enzyme during the simulation (Movie 5). The sulfur atom is also a very versatile atom in terms of its interactions with the residues of an enzyme. Its electron cloud can be involved in pi-pi stacking interactions and other interaction with aromatic or non-aromatic residues.43 In the case of 32 the sulfur atom is in spatial proximity to Tyr643, Phe722, Leu770 as well as the “oxyanion hole” (Gly486/Gly487) constantly interacting with this residues (Movie 4). According to the XI(50) value of the thio versus non-thio inhibitors, it is obvious that the presence of the beta-thioether enhances the binding and potency of these compounds and the MD simulations clearly explain the beneficial effect of the sulfur atom upon binding of 32 (Fig. 5A and Movie 4).
Figure 5.

Binding mode occur during the MD simulation for (A) compounds 32 (Movie 4) and for (B) compounds 33 (Movie 5) in GVIA iPLA2 binding site.
Conclusion
Fluoroketones exhibit unique inhibitory properties against PLA2 enzymes that belong to serine hydrolases and, thus, are attractive synthetic compounds for elucidating the binding mode with each particular enzyme and performing structure-activity relationship studies. Our computational studies allow us to understand clearly how fluoroketones interact with either GVIA iPLA2 or GIVA cPLA2, and what structural features an inhibitor should possess in order to present selectivity for GVIA iPLA2 or GIVA cPLA2. These studies constitute a valuable guide indicating that inhibitors of GVIA iPLA2 should be small-size molecules. Increase of the size and the lipophilicity of the inhibitor lead to decrease of selectivity for GVIA iPLA2. An SAR model was established and new compounds were synthesized and tested that show high inhibitory activity against GVIA iPLA2 while showing the validity of our structural and SAR models. In conclusion, this study introduces a new class of thioether fluoroketones that have the potential for further development in order to identify potent and selective inhibitors not only for GVIA iPLA2 but for GIVA cPLA2 as well. In addition, a novel structure of GVIA iPLA2 inhibitor combining the keto-1,2,4-oxadiazole functionality with a thioether was identify.
Experimental Procedures
Induced Fit Docking (IFD) protocol
Each enzyme-inhibitor initial complexes were predicted using the IFD protocol implemented in Schrödinger Suite 2014.44, 45 The enzyme 3D structure (receptor) was optimized using the PPW module.33 The inhibitor 3D structures (ligand) were sketched using Maestro 9.9 sketcher,46 and they were optimized using the LigPrep 3.2 application.47 IFD protocol employs Glide 6.548 and the refinement module in Prime 3.849 to accurately predict enzyme-inhibitor complexes by incorporating side-chain flexibility for the receptor. The box center for the docking calculations was defined using the centroid of selected residues that were found to constitute the binding pocket of GVIA iPLA2 and GIVA cPLA2 using DXMS and extensive MD simulations in our previously published papers.28–30 The box size was determined automatically according to the centroid of the specified binding pocket residues. For the initial docking of the inhibitors the side-chain of the binding pocket residues were trimmed automatically based on the B-factor. The receptor van der Waals scaling was set to 0.70 and the ligand van der Waals scaling to 0.50. Twenty poses (binding modes) of each inhibitor were allowed to pass to the Prime refinement step. During the Prime refinement step, the side-chains of the residues within 6 Å the inhibitor pose were optimized in terms of conformation and potential energy. Finally, twenty enzyme-inhibitor structures were allowed to pass to the redocking step with a threshold for eliminating high-energy structures from the Prime refinement of 30 kcal/mol, and the Glide Extra-Precision (XP) scoring function.50 The final complexes were selected based on the binding mode and the score.
Molecular Dynamics (MD) simulations
MD simulations were carried out on enzyme-inhibitor complexes (compounds 2, 3, 32 and 33 for GVIA iPLA2, and compound 21 for GIVA cPLA2), which were derived from the docking calculations. Each complex was placed on a POPC membrane patch of ~100 × 100 Å2 area according to our previously published model.28–30 POPC molecules within 0.6 Å of the enzyme were removed, the system was solvated with TIP3P water molecules, and was neutralized using sodium chloride at a concentration of 100 mM. The total number of atoms of the entire system was approximately 105,000. NAMD 2.9 was employed for the MD simulations.32 The CHARMM General Force Field (CGenFF) and parameters were used for the inhibitors,51 and the CHARMM36 all-atom additive force field and parameters were used for the enzyme and the POPC membrane patch.52 The system was minimized for 20,000 steps and equilibrated for 12 ns to relieve all the unfavorable contacts. Finally, a 300 ns MD simulation was performed at a temperature of 310 K using the NPT ensemble. The trajectory files of the simulations were analyzed using the Visual Molecular Dynamics (VMD) software.53
Docking Calculations
Clustering analysis was performed on the trajectories derived from the MD simulations on the enzyme-2, -3 and -21 complexes in order to identify suitable conformations for the docking calculations. The 3D structures were optimized using the PPW.33 The 3D structures of the inhibitors were sketched using Maestro 9.9 sketcher46 and they were optimized using LigPrep 3.2.47 Glide 5.8 was used for the rigid-docking of the compounds into the enzyme binding pocket.48 The grid required for the docking procedure was generated using a scaling factor of 1.0 and partial charge cutoff of 0.25, while X, Y, Z dimensions of the inner box were set to 12 Å. For the inhibitor docking a scaling factor of 0.8 and partial charge cutoff of 0.15 were used that allow complete flexibility of the structures. The poses were selected according to the binding mode and the XP GScore. The Glide Extra-Precision (XP) scoring function50 was used for the calculations.
In vitro PLA2 activity Assay
The activities of human GVIA iPLA2, GIV cPLA2 and GV sPLA2 were determine using a group-specific mixed micelle modified Dole assay.21, 27 The substrate was prepared using slightly different conditions for each enzyme to achieve optimum activity: (i) GVIA iPLA2 mixed micelle substrate consisted of 400 μM Triton X-100, 98.3 μM PAPC, and 1.7μM arachidonyl-1–14C PAPC in a buffer containing 100 mM HEPES pH 7.5, 2 mM ATP, and 4 mM DTT; (ii) GIVA cPLA2 mixed micelle substrate consisted of 400 μM Triton X-100, 95.3 μM PAPC, 1.7 μM arachidonyl-1–14C PAPC, and 3 μM PI(4,5)P2 in a buffer containing 100 mM HEPES pH 7.5, 90 μM CaCl2, 2 mM DTT, and 0.1 mg/ml BSA; and (iii) GV sPLA2 mixed micelles substrate consisted of 400 μM Triton X-100, 98.3 μM PAPC, and 1.7 μM arachidonyl-1–14C PAPC in a buffer containing 50 mM Tris-HCl pH 8.0, and 5 mM CaCl2. The compounds were initially screened at 0.091 mole fraction (5 μL of 5 mM inhibitor in DMSO) in substrate (495 uL). XI(50) was determined for compounds exhibiting greater than 95% inhibition. Inhibition curves were generated using GraphPad Prism 5.0 and the non-linear regression by plotting percentage of inhibition vs log (mole fraction) to calculate the reported XI(50) and its associated error.
Synthesis of the inhibitors. General Methods
Chromatographic purification of products was accomplished using Merck Silica Gel 60 (70–230 or 230–400 mesh). Thin-layer chromatography (TLC) was performed on Silica Gel 60 F254 aluminum plates. Spots were visualized with UV light and/or phosphomolybdic acid in EtOH. Melting points were determined using a Bu chi 530 apparatus and were uncorrected. 1H, 13C and 19F NMR spectra were recorded on a Varian Mercury. Chemical shifts are given in ppm, and coupling constants (J), in Hz. Peak multiplicities are described as follows: s, singlet; d, doublet; t, triplet; and m, multiplet. HRMS spectra were recorded on a Bruker Maxis Impact QTOF Spectrometer. Dichloromethane was dried by standard procedures and stored over molecular sieves. All other solvents and chemicals were reagent grade and used without further purification. The purity of all compounds subjected to biological tests was determined by analytical HPLC and was found to be 95%.
Synthesis of Fluoroketones 28, 30, 31
Thiol 34a-c (1 mmol) was transferred to an oven dried flask and CCl4 (2 mL) was added under nitrogen atmosphere. After 5 minutes, 3-bromo-1,1,1-trifluoropropan-2-one (1.3 mmol, 248 mg) or 1-bromo-3,3,4,4,4-pentafluorobutan-2-one (1.3 mmol, 313 mg) was added dropwise over 15 minutes. The mixture was left stirring under nitrogen bubling at room temperature for 4 hours. Column chromatography of the crude mixture afforded the desired product [EtOAc/petroleum ether (bp 40–60 °C)].
1,1,1-Trifluoro-3-((4-methoxyphenethyl)thio)propan-2-one (in equilibrium with its corresponding gem-diol) (28)
Yield 88%; colourless oil; 1H NMR (200 MHz, CDCl3) : δ 7.16–7.09 (2H, m, ArH), 6.90–6.82 (2H, m, ArH), 4.09 [1H, br s, SCH2C(OH)2], 3.80 (3H, s, OMe), 3.47 (1H, br s, SCH2C=O), 3.01–2.70 (4.5H, m, 4 x CHH and 0.5 x OH), 1.81 (0.5H, br, OH); 13C NMR (50 MHz, CDCl3) : δ 185.1 (q, J = 34 Hz, C=O), 158.2, 131.7, 129.5, 122.8 (q, J = 285 Hz, CF3), 114.3, 92.4 [q, J = 32 Hz, C(OH)2], 55.2, 36.3, 35.0, 34.8, 34.7, 34.3, 33.5; 19F NMR (186 MHz, CDCl3) : δ -20.8, -30.3; HRMS (ESI) calcd for C12H12F3O2S [M-H] −, 277.0516; found, 277.0515.
1,1,1-Trifluoro-3-((2-(naphthalen-2-yl)ethyl)thio)propan-2-one (in equilibrium with its corresponding gem-diol) (30)
Yield 83%; colourless oil; 1H NMR (200 MHz, CDCl3) : δ 7.89–7.77 (3H, m, ArH), 7.67 (1H, s, ArH), 7.54–7.44 (2H, m, ArH), 7.39–7.30 (1H, m, ArH), 4.14 [1.8H, br s, SCH2C(OH)2], 3.49 (0.2H, SCH2C=O), 3.12–2.90 (4H, m, 4 x CHH), 2.98 (1.8H, br, OH); 13C NMR (50 MHz, CDCl3) : δ 137.1, 133.5, 133.4, 128.2, 127.7, 127.5, 126.9, 126.4, 126.2, 125.6, 123.0 (q, J = 282.5 Hz, CF3), 92.5 [q, J = 29 Hz, C(OH)2], 36.4, 35.8, 35.1, 34.6, 34.2, 33.4; 19F NMR (186 MHz, CDCl3) : δ −20.7, −30.2; HRMS (ESI) calcd for C15H12F3OS [M-H]−, 297.0566; found, 297.0563.
3,3,4,4,4-Pentafluoro-1-(phenethylthio)butan-2-one (in equilibrium with its corresponding gem-diol) (31)
Yield 78%; colourless oil; 1H NMR (200 MHz, CDCl3) : δ 7.40–7.12 (5H, m, ArH), 3.73 (0.2H, br s, OH), 3.50 (3H, s, OMe, SCH2), 3.28 (0.2H, br s, OH), 3.10–2.60 (4H, m, 4 x CHH); 13C NMR (50 MHz, CDCl3) : δ 187.5 (t, J = 26.1 Hz, C=O), 139.6, 139.4, 128.6, 128.5, 126.7, 117.7 (qt, J = 287.1 and 34.0 Hz, CF3), 107.0 (tq, 268.4 and 38.3 Hz, CF2), 93.9 [m, C(OH)2], 35.7, 35.4, 35.2, 34.9, 33.1, 31.9; 19F NMR (186 MHz, CDCl3) : δ −24.1, −26.7, −66.5, −71.6; HRMS (ESI) calcd for C12H12F3O2S [M-H] −, 297.0378; found, 297.0381.
3,3,4,4,4-Pentafluoro-1-((4-methoxyphenethyl)thio)butan-2-one (29)
To a solution of 2-((4-methoxyphenethyl)thio)acetic acid (3) (0.9 mmol, 203 mg) in dry dichloromethane (7 mL) DMAP (1.1 mmol, 140 mg), N,O-dimethylhydroxylamine hydrochloride (0.9 mmol, 90 mg), N-methylmorpholine (0.9 mmol, 270 mg, 0.25 mL) and WSCI (0.9 mmol, 143 mg) were added consecutively. The mixture was left stirring for 24 hours at room temperature. Then, water was added (7 mL) and the aqueous phase was extracted with dichloromethane (2 x 10 mL). The combined organic phases were washed with H2O, HCl 1N, H2O, 4% NaOH, H2O, brine (1 x 10 mL each) and dried over Na2SO4. Evaporation of the solvent afforded the corresponding Weinreb amide without further purification (78% yield, 0.70 mmol, 189 mg). In a round bottom flask containing the Weinreb amide under nitrogen atmosphere, dry Et2O (15 mL) was added. Then, the mixture was cooled down to −78 °C and CF3CF2I (3.5 mmol, 858 mg) and MeLi.LiBr (2.2M in Et2O, 3.5 mmol, 1.6 mL) were added consecutively. The mixture was left stirring at −78 °C for 2 hours and then for 1 hour at room temperature. Then, water (4 mL) and 10% KHSO4 until pH 4–5 were added. The aqueous phase was extracted with Et2O (3 x 10 mL) and the solvent was evaporated under reduced pressure. The residue was purified by flash column chromatography [EtOAc/petroleum ether (bp 40–60 °C), 1:9]. Yield 70% over two steps; colourless oil; 1H NMR (200 MHz, CDCl3) : δ 7.14 (2H, d, J = 8.7 Hz, ArH), 6.87 (2H, d, J = 8.7 Hz, ArH), 3.81 (3H, s, OMe), 3.51 (2H, s, CH2C=O), 2.90–2.68 (4H, m, 4 x CHH); 13C NMR (50 MHz, CDCl3) : δ 187.5 (t, J = 26.0 Hz, C=O), 158.3, 131.4, 129.5, 117.7 (qt, J = 287.0 Hz and 34.0 Hz, CF3), 113.9, 107.0 (tq, J = 268.4 Hz and 38.6 Hz, CF2), 55.2, 35.4, 34.3, 33.4; 19F NMR (186 MHz, CDCl3) : δ −26.8, −65.6; HRMS (ESI) calcd for C13H12F5O2S [M-H] −, 327.0484; found, 327.0482.
Synthesis of nitriles 37a,b
To a stirred solution of alcohol 36a,b (1 mmol) in dry CH2Cl2 (10 mL), Dess-Martin periodinane was added (1.2 mmol, 509 mg) and the mixture was stirred for 1 h at room temperature. The organic solvent was evaporated under reduced pressure and Et2O (30 mL) was added. The organic phase was washed with saturated aqueous NaHCO3 (20 mL) containing Na2S2O3 (1.5 g, 9.5 mmol), H2O (20 mL), dried over Na2SO4, and the organic solvent was evaporated under reduced pressure.
To a stirred mixture of tert-butyldimethylsilyl cyanide (TBDMSCN) (1.0 mmol, 141 mg), potassium cyanide (0.2 mmol, 13 mg), and 18-crown-6 (0.4 mmol, 106 mg) in CH2Cl2 (15 mL) at 0 °C, a solution of the aldehyde (1.0 mmol) derived from alcohol 36a,b, was added dropwise over 30 min. After addition was completed, the mixture was stirred overnight at room temperature. The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C), 0.5:9.5].
2-(tert-Butyldimethylsilyloxy)-6-(4-methoxyphenyl)hexanonitrile (37a)
Yield 76%; colourless oil; 1H NMR (200 MHz, CDCl3): δ 7.12 (2H, d, J = 10.0 Hz), 6.87 (2H, d, J = 8.0 Hz), 4.43 (1H, t, J = 6.0 Hz), 3.80 (3H, s), 2.62 (2H, t, J = 8.0 Hz), 1.89- 1.75 (2H, m), 1.71–1.48 (4H, m), 0.98 (9H, s), 0.25 (3H, s), 0.16 (3H, s); 13C NMR (50 MHz, CDCl3): δ 157.4, 133.6, 128.9, 119.7) 113.4) 61.4, 54.7, 35.8, 34.4, 30.7, 25.2, 23.7, 17.7, −5.5, −5.7; MS (ESI) m/z (%): 351.2 [(M+NH4)+, 100].
2-(tert-Butyldimethylsilyloxy)-3-(4-methoxyphenethylthio)propanenitrile (37b)
Yield 74%; colourless oil; 1H NMR (200 MHz, CDCl3): δ 7.10 (2H, d, J = 8.6 Hz), 6.82 (2H, d, J = 8.6 Hz), 4.91–4.60 (2H, m), 4.53–4.32 (1H, m), 3.77 (3H, s), 2.80–2.62 (4H, m), 0.94 (9H, s), 0.17 (3H, s), 0.11 (3H, s); 13C NMR (50 MHz, CDCl3): δ 157.9, 132.8, 129.5, 119.8, 113.8, 61.7, 55.1, 38.9, 35.2, 34.6, 25.6, 18.0, −5.2, −5.1; MS (ESI) m/z (%): 369.2 [(M+NH4)+, 100].
Synthesis of amidoximes 38a,b
Compound 37a,b (1 mmol) was placed in a microwave vessel and a 50% aqueous solution of NH2OH (4.0 mmol, 0.5 mL) was added. The reaction mixture was left stirring under microwave irradiation (initial setting at 50W) for 30 minutes at 120 °C. Then, water was added (10.0 mL) and the mixture was extracted with ether (2 x 10 mL). The combined extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The product was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C), 2:8].
(Z)-2-(tert-Butyldimethylsilyloxy)-N'-hydroxy-6-(4-methoxyphenyl)hexanimidamide (38a)
Yield 76%; colourless oil; 1H NMR (200 MHz, CDCl3): δ 7.13 (2H, d, J = 8.0 Hz), 6.86 (2H, d, J = 8.0 Hz), 4.85 (2H, s), 4.20 (1H, t, J = 6.0 Hz), 3.81 (3H, s), 2.60 (2H, t, J = 6.0 Hz), 1.85–1.61 (4H, m), 1.54–1.34 (2H, m), 0.94 (9H, s), 0.14 (6H, s); 13C NMR (50 MHz, CDCl3): δ 157.3, 155.5, 134.4, 129.0, 113.4, 70.7, 54.8, 36.5, 34.7, 31.2, 25.5, 24.6, 17.8, −5.2, −5.3; MS (ESI) m/z (%): 367.6 [(M+H)+, 100].
(Z)-2-(tert-Butyldimethylsilyloxy)-N'-hydroxy-3-(4-methoxyphenethylthio)propanimidamide (38b)
Yield 64%; colourless oil; 1H NMR (200 MHz, CDCl3): δ 7.11 (2H, d, J = 8.6 Hz), 6.83 (2H, d, J = 8.6 Hz), 4.89–4.62 (2H, m), 4.33–4.18 (1H, m), 3.78 (3H, s), 2.89–2.65 (6H, m), 0.90 (9H, s), 0.13 (3H, s), 0.09 (3H, s); 13C NMR (50 MHz, CDCl3): δ 158.0, 154.8, 132.5, 129.4, 113.8, 70.8, 55.2, 39.1, 35.3, 34.6, 25.7, 18.1, −5.0, −5.1; MS (ESI) m/z (%): 385.2 [(M+H)+, 100].
Synthesis of 39a,b
To a stirred solution of amidoxime 38a,b (1.0 mmol) in dry CH2Cl2 (20 mL), pivalic acid (1 mmol, 102 mg) and DCC (1.1 mmol, 227 mg) were added. The reaction mixture was stirred overnight at room temperature. The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C), 2:8].
(Z)-2-((tert-Butyldimethylsilyloxy)-N'-(pivaloyloxy)-6-(4-methoxyphenyl)hexanimidamide (39a)
Yield 76%; colourless oil; 1H NMR (200 MHz, CDCl3): δ 7.07 (2H, d, J = 8.0 Hz), 6.80 (2H, d, J = 8.0 Hz), 4.87 (2H, s, NH2), 4.32 (1H, t, J = 6.0 Hz), 3.77 (3H, s), 2.54 (2H, t, J = 8.0 Hz), 1.83–1.60 (6H, m), 1.28 (9H, s), 0.88 (9H, s), 0.07 (6H, s); 13C NMR (50 MHz, CDCl3): δ 174.8, 160.2, 157.5, 134.4, 129.1, 113.5, 70.0, 55.1, 38.7, 37.4, 34.7, 31.1, 27.4, 25.6, 24.6, 17.9, −5.1; MS (ESI) m/z (%): 451.8 [(M+H)+, 100].
(Z)-2-(tert-Butyldimethylsilyloxy)-3-(4-methoxyphenethylthio)-N'-(pivaloyloxy)propanimidamide (39b)
Yield 73%; colorless oil; 1H NMR (200 MHz, CDCl3): δ 7.06 (2H, d, J = 8.6 Hz), 6.78 (2H, d, J = 8.6 Hz), 5.15–4.84 (2H, m), 4.55–4.43 (1H, m), 3.74 (3H, s), 2.89–2.70 (6H, m), 1.24 (9H, s), 0.88 (9H, s), 0.11 (3H, s), 0.07 (3H, s); 13C NMR (50 MHz, CDCl3): δ 174.6, 158.9, 157.8, 132.3, 129.2, 113.6, 69.7, 55.0, 38.7, 35.1, 34.7, 28.3, 27.3, 25.5, 17.9, −5.1, −5.3; MS (ESI) m/z (%): 469.3 [(M+H)+, 100].
Synthesis of 40a,b
To a stirred solution of compound 39a,b (1.0 mmol) in dry toluene (3 mL) in a microwave vessel, TBAF (1M in THF, 1.0 mmol) was added. The reaction mixture was left stirring under microwave irradiation (initial setting at 90W) for 1 hour at 120 °C. The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C), 2:8].
1-(5-tert-Butyl-1,2,4-oxadiazol-3-yl)-5-(4-methoxyphenyl)pentan-1-ol (40a)
Yield 28%; colourless solid; m.p. 109- 111°C; 1H NMR (200 MHz, CDCl3): δ 7.09 (2H, d, J = 8.0 Hz), 6.81 (2H, d, J = 8.0 Hz), 4.82 (1H, t, J = 6.0 Hz), 3.78 (3H, s), 2.92 (1H, s), 2.57 (2H, t, J = 6.0 Hz), 2.02–1.87 (2H, m), 1.75–1.55 (4H, m), 1.43 (9H, s); 13C NMR (50 MHz, CDCl3): δ 186.4, 171.7, 157.5, 134.4, 129.1, 113.6, 66.6, 55.1, 35.3, 34.7, 33.0, 31.2, 28.3, 24.6; MS (ESI) m/z (%): 319.2 [(M+H)+, 100].
1-(5-tert-Butyl-1,2,4-oxadiazol-3-yl)-2-(4-methoxyphenethylthio)ethanol (40b)
Yield 25%; pale yellow oil; 1H NMR (200 MHz, CDCl3): δ 7.11 (2H, d, J = 8.6 Hz), 6.84 (2H, d, J = 8.6 Hz), 5.04–4.86 (1H, m), 3.79 (3H, s), 3.31–3.18 (1H, m), 3.11–3.01 (2H, m), 2.91–2.69 (4H, m), 1.43 (9H, s); 13C NMR (50 MHz, CDCl3): δ 186.8, 170.3, 158.2, 132.0, 129.4, 113.9, 65.4, 55.2, 37.9, 35.2, 34.2, 33.7, 28.3; MS (ESI) m/z (%): 337.2 [(M+H)+, 100].
Synthesis of 1,2,4-oxadiazoles 32, 33
To a stirred solution of compound 40a,b (1 mmol) in dry CH2Cl2 (10 mL), Dess-Martin periodinane was added (1.2 mmol, 509 mg) and the mixture was stirred for 1 h at room temperature. The organic solvent was evaporated under reduce pressure and Et2O (30 mL) was added. The organic phase was washed with saturated aqueous NaHCO3 (20 mL) containing Na2S2O3 (1.5 g, 9.5 mmol), H2O (20 mL), dried over Na2SO4, and the organic solvent was evaporated under reduced pressure. The residue was purified by column chromatography [EtOAc-petroleum ether (bp 40–60 °C), 2:8].
1-(5-tert-Butyl-1,2,4-oxadiazol-3-yl)-5-(4-methoxyphenyl)pentan-1-one (33)
Yield 88%; colourless oil; 1H NMR (200 MHz, CDCl3): δ = 7.08 (2H, d, J = 8.0 Hz), 6.81 (2H, d, J = 8.0 Hz), 3.77 (3H, s), 3.06 (2H, t, J = 6.0 Hz), 2.59 (2H, t, J = 6.0 Hz), 1.86–1.64 (4H, m), 1.47 (9H, s); 13C NMR (50 MHz, CDCl3): δ = 191.8, 187.8, 165.3, 157.6, 134.0, 129.2, 113.6, 55.2, 40.3, 34.6, 33.8, 30.9, 28.3, 23.0; HRMS (ESI) calcd for C18H24N2O3 [M+H]+, 317.1863, found, 317.1863.
1-(5-tert-Butyl-1,2,4-oxadiazol-3-yl)-2-(4-methoxyphenethylthio)ethanone (32)
Yield 85%; colorless oil; 1H NMR (200 MHz, CDCl3): δ 7.13 (2H, d, J = 8.6 Hz), 6.83 (2H, d, J = 8.6 Hz), 3.86–3.70 (5H, m), 2.94–2.70 (4H, m), 1.49 (9H, s); 13C NMR (50 MHz, CDCl3): δ 188.2, 184.4, 164.7, 158.2, 131.8, 129.5, 113.8, 55.2, 37.6, 34.4, 33.9, 33.8, 28.3; HRMS (ESI) calcd for C17H21N2NaO3S [M+Na]+, 357.1243, found, 357.1255.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant GM20501/40 (E. A. D) and by NIH, NSF, NBCR and HHMI (J.A.M). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. This research has been co-financed by the European Union (European Regional Development Fund-ERDF) and Greek national funds through the Operational Program “Competitiveness and Entrepreneurship” of the National Strategic Reference Framework (NSRF) - Research Funding Program: “Phospholipases A2 inhibitors: Developing a drug pipeline for the treatment of inflammatory neurological disorders” (G.K.)
Abbreviations used
- PLA2
Phospholipase A2
- cPLA2
cytosolic
- sPLA2
secreted
- iPLA2
calcium-independent
- PAF-AH
platelet-activating factor acetylhydrolase
- Lp-PLA2
lipoprotein-associated
- LPLA2
lysosomal
- AdPLA
adipose-PLA
- AA
arachidonic acid
- GVIA cPLA2
Group IVA cytosolic
- GVIA iPLA2
Group VIA calcium-independent
- GV sPLA2
Group V secreted
- COX
cyclooxygenase
- LOX
lipoxygenase
- DXMS
hydrogen/deuterium exchange mass spectrometry
- MD
molecular dynamics
- SAR
structure-activity relationship
- IFD
Induced Fit Docking
- XP GScore
extra-precision glide score
- XI(50)
mole fraction of the compound that is required for 50% inhibition of the enzyme
- TBDMSCN
tert-butyldimethylsilyl cyanide
- DCC
N,N’-dicyclocarbodiimide
- TBAF
tetrabutylammonium fluoride
- PAPC
1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- CGenFF
CHARMM General Force Field
- VMD
visual molecular dynamics
Footnotes
Additional figure and toppar stream files for compounds 2, 3, 21, 32 and 33 (PDF).
3D movies for MD simulation on enzyme-inhibitor complexes (mpg):
References
- 1.Dennis E, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15:511–523. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mouchlis VD, Dennis EA. Membrane and inhibitor interactions of intracellular phospholipases A2. Adv Biol Regul. 2015 doi: 10.1016/j.jbior.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kramer RM, Roberts EF, Manetta J, Putnam JE. The Ca2+-sensitive cytosolic phospholipase A2 is a 100-kDa protein in human monoblast U937 cells. J Biol Chem. 1991;266:5268–5272. [PubMed] [Google Scholar]
- 5.Sharp JD, White DL, Chiou XG, Goodson T, Gamboa GC, McClure D, Burgett S, Hoskins J, Skatrud PL, Sportsman JR. Molecular cloning and expression of human Ca2+-sensitive cytosolic phospholipase A2. J Biol Chem. 1991;266:14850–14853. [PubMed] [Google Scholar]
- 6.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 7.Dessen A, Tang J, Schmidt H, Stahl M, Clark J, Seehra J, Somers W. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999;97:349–360. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- 8.Buczynski M, Dumlao D, Dennis E. Thematic review series: proteomics. an integrated omics analysis of eicosanoid biology. J Lipid Res. 2009;50:1015–1038. doi: 10.1194/jlr.R900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackermann EJ, Kempner ES, Dennis EA. Ca2+-independent cytosolic phospholipase A2 from macrophage-like P388D1 cells. Isolation and characterization. J Biol Chem. 1994;269:9227–9233. [PubMed] [Google Scholar]
- 10.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 11.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem. 1998;273:207–214. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 12.Larsson Forsell PK, Kennedy BP, Claesson HE. The human calcium-independent phospholipase A2 gene multiple enzymes with distinct properties from a single gene. Eur J Biochem. 1999;262:575–585. doi: 10.1046/j.1432-1327.1999.00418.x. [DOI] [PubMed] [Google Scholar]
- 13.Ramanadham S, Ali T, Ashley JW, Bone RN, Hancock WD, Lei X. Calcium-independent phospholipases A2 (iPLA2s) and their roles in biological processes and diseases. J Lipid Res. 2015;56:1643–1668. doi: 10.1194/jlr.R058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, Iwata T, Yagi M, Hasturk H, Van Dyke TE. Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J Immunol. 2010;184:1507–1515. doi: 10.4049/jimmunol.0901219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, Ren MC. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. PNAS. 2009;106:2337–2341. doi: 10.1073/pnas.0811224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Dennis EA. Expression and characterization of human group V phospholipase A2. Biochim Biophys Acta. 1998;1394:57–64. doi: 10.1016/s0005-2760(98)00098-8. [DOI] [PubMed] [Google Scholar]
- 17.Shirai Y, Balsinde J, Dennis EA. Localization and functional interrelationships among cytosolic group IV, secreted group V, and Ca2+-independent group VI phospholipase A2s in P388D 1 macrophages using GFP/RFP constructs. Biochim Biophys Acta. 2005;1735:119–129. doi: 10.1016/j.bbalip.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Masuda S, Murakami M, Komiyama K, Ishihara M, Ishikawa Y, Ishii T, Kudo I. Various secretory phospholipase A2 enzymes are expressed in rheumatoid arthritis and augment prostaglandin production in cultured synovial cells. FEBS J. 2005;272:655–672. doi: 10.1111/j.1742-4658.2004.04489.x. [DOI] [PubMed] [Google Scholar]
- 19.Magrioti V, Kokotos G. Phospholipase A2 inhibitors for the treatment of inflammatory diseases: a patent review (2010 - present) Expert Opin Ther Pat. 2013;23:333–344. doi: 10.1517/13543776.2013.754425. [DOI] [PubMed] [Google Scholar]
- 20.Ong WY, Farooqui T, Kokotos G, Farooqui AA. Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci. 2015;6:814–831. doi: 10.1021/acschemneuro.5b00073. [DOI] [PubMed] [Google Scholar]
- 21.Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker SF, Dennis EA, Kokotos G. Structure-activity relationship of 2-oxoamide inhibition of group IVA cytosolic phospholipase A2 and group V secreted phospholipase A2. J Med Chem. 2007;50:4222–4235. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 22.Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. Differential inhibition of group IVA and group VIA phospholipases A2 by 2-oxoamides. J Med Chem. 2006;49:2821–2828. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonopoulou G, Barbayianni E, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. Structure-activity relationships of natural and non-natural amino acid-based amide and 2-oxoamide inhibitors of human phospholipase A2 enzymes. Bioorg Med Chem. 2008;16:10257–10269. doi: 10.1016/j.bmc.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokotos G, Hsu YH, Burke J, Baskakis C, Kokotos C, Magrioti V, Dennis E. Potent and selective fluoroketone inhibitors of group VIA calcium-independent phospholipase A2. J Med Chem. 2010;53:3602–3610. doi: 10.1021/jm901872v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magrioti V, Nikolaou A, Smyrniotou A, Shah I, Constantinou-Kokotou V, Dennis E, Kokotos G. New potent and selective polyfluoroalkyl ketone inhibitors of GVIA calcium-independent phospholipase A2. Bioorg Med Chem. 2013;21:5823–5829. doi: 10.1016/j.bmc.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baskakis C, Magrioti V, Cotton N, Stephens D, Constantinou-Kokotou V, Dennis EA, Kokotos G. Synthesis of polyfluoro ketones for selective inhibition of human phospholipase A2 enzymes. J Med Chem. 2008;51:8027–8037. doi: 10.1021/jm800649q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Mosior M, Johnson C, Chen Y, Dennis E. Group-specific assays that distinguish between the four major types of mammalian phospholipase A2. Anal Biochem. 1999;269:278–288. doi: 10.1006/abio.1999.4053. [DOI] [PubMed] [Google Scholar]
- 28.Mouchlis VD, Bucher D, McCammon JA, Dennis EA. Membranes serve as allosteric activators of phospholipase A2, enabling it to extract, bind, and hydrolyze phospholipid substrates. PNAS. 2015;112:E516–E525. doi: 10.1073/pnas.1424651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bucher D, Hsu YH, Mouchlis VD, Dennis EA, McCammon JA. Insertion of the Ca2+-independent phospholipase A2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput Biol. 2013;9:e1003156. doi: 10.1371/journal.pcbi.1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu YH, Bucher D, Cao J, Li S, Yang SW, Kokotos G, Woods V, McCammon J, Dennis E. Fluoroketone inhibition of Ca2+-independent phospholipase A2 through pinding pocket association defined by hydrogen/deuterium exchange and molecular dynamics. J Am Chem Soc. 2013;135:1330–1337. doi: 10.1021/ja306490g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burke J, Babakhani A, Gorfe A, Kokotos G, Li S, Woods V, McCammon J, Dennis E. Location of inhibitors bound to group IVA phospholipase A2 determined by molecular dynamics and deuterium exchange mass spectrometry. J Am Chem Soc. 2009;131:8083–8091. doi: 10.1021/ja900098y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Protein preparation wizard 2014–4; Epik version 2.4. Schrödinger, LLC; New York, NY: 2014. Impact version 5.9, Schrödinger, LLC, New York, NY, 2014; Prime version 3.2, Schrödinger, LLC, New York, NY, 2014. 2014. [Google Scholar]
- 34.Snyder DW, Bach NJ, Dillard RD, Draheim SE, Carlson DG, Fox N, Roehm NW, Armstrong CT, Chang CH, Hartley LW. Pharmacology of LY315920/S-5920,[[3-(Aminooxoacetyl)-2-ethyl-1-(phenylmethyl)-1H-indol-4-yl] oxy] acetate, a Potent and Selective Secretory Phospholipase A2 Inhibitor: A New Class of Anti-Inflammatory Drugs, SPI. J Pharmacol Exp Ther. 1999;288:1117–1124. [PubMed] [Google Scholar]
- 35.Carman G, Deems R, Dennis E. Lipid signaling enzymes and surface dilution kinetics. J Biol Chem. 1995;270:18711–18714. doi: 10.1074/jbc.270.32.18711. [DOI] [PubMed] [Google Scholar]
- 36.Douglas BK, Hélène D, John RF, Jürgen B. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 37.Hammock BD, Abdel-Aal YAI, Mullin CA, Hanzlik TN, Roe RM. Substituted thiotrifluoropropanones as potent selective inhibitors of juvenile hormone esterase. Pestic Biochem Physiol. 1984;22:209–223. [Google Scholar]
- 38.Ashour MBA, Hammock BD. Substituted trifluoroketones as potent, selective inhibitors of mammalian carboxylesterases. Biochem Pharmacol. 1987;36:1869–1879. doi: 10.1016/0006-2952(87)90483-7. [DOI] [PubMed] [Google Scholar]
- 39.Holmeide AK, Skattebol L. Syntheses of some polyunsaturated trifluoromethyl ketones as potential phospholipase A2 inhibitors. J Chem Soc, Perkin Trans 1. 2000:2271–2276. [Google Scholar]
- 40.Huwiler A, Feuerherm AJ, Sakem B, Pastukhov O, Filipenko I, Nguyen T, Johansen B. The ω3-polyunsaturated fatty acid derivatives AVX001 and AVX002 directly inhibit cytosolic phospholipase A2 and suppress PGE2 formation in mesangial cells. Br J Pharmacol. 2012;167:1691–1701. doi: 10.1111/j.1476-5381.2012.02114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musmade DS, Pattan SR, Yalgatti MS. Oxadiazole a nucleus with versatile biological behaviour. IJPC. 2015;5:11–20. [Google Scholar]
- 42.Maryanoff BE, Costanzo MJ. Inhibitors of proteases and amide hydrolases that employ an alpha-ketoheterocycle as a key enabling functionality. Bioorg Med Chem. 2008;16:1562–1595. doi: 10.1016/j.bmc.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Beno BR, Yeung KS, Bartberger MD, Pennington LD, Meanwell NA. A survey of the role of noncovalent sulfur interactions in drug design. J Med Chem. 2015;58:4383–4438. doi: 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- 44.Induced fit docking protocol 2014. Vol. 2014 Schrödinger, LLC; New York, NY: 2014. Glide version 6.5, Prime version 3.8. [Google Scholar]
- 45.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 46.Maestro, version 9.9. Vol. 2014 Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 47.LigPrep, version 3.2. Vol. 2014 Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 48.Glide, version 6.5. Vol. 2014 Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 49.Prime, version 3.8. Vol. 2014 Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 50.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 51.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AC. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2009;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RWC. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.