

Abstract
Bacterial spores are the most resistant form of life known on Earth and represent a serious problem for (i) bioterrorism attack, (ii) horizontal transmission of microbial pathogens in the community, and (iii) persistence in patients and in a nosocomial environment. Stage II sporulation protein D (SpoIID) is a lytic transglycosylase (LT) essential for sporulation. The LT superfamily is a potential drug target because it is active in essential bacterial processes involving the peptidoglycan, which is unique to bacteria. However, the absence of structural information for the sporulation-specific LT enzymes has hindered mechanistic understanding of SpoIID. Here, we report the first crystal structures with and without ligands of the SpoIID family from two community relevant spore-forming pathogens, Bacillus anthracis and Clostridium difficile. The structures allow us to visualize the overall architecture, characterize the substrate recognition model, identify critical residues, and provide the structural basis for catalysis by this new family of enzymes.
Keywords: cell differentiation, cell surface enzyme, enzyme structure, peptidoglycan, sporulation, drug target, lytic transglycosylase, SpoIID
Introduction
Sporulation is an evolutionarily conserved process that allows some Gram-positive bacteria to differentiate into a dormant cell type called a spore that allows them to resist chemical agents such as antibiotics and disinfectants and physical agents such as radiation, boiling and drying (1). Sporulation is characterized by an asymmetric cell division, followed by migration of the mother cells' membrane around the nascent spore in a phagocyte-like process (2, 3). Engulfment of what will become the spore requires PGN2 degradation by the SpoIIM/SpoIIP/SpoIID molecular machine (4, 5). In this complex, SpoIIM serves as a scaffold upon which the SpoIID and SpoIIP enzymes assemble (1, 4, 6). SpoIIP is single-pass transmembrane protein with a large extracellular portion that has both amidase and endopeptidase activities. This enzyme cleaves the cross-links connecting the stem peptides of PGN and removes the stem peptides from the glycan chains (Fig. 1a). SpoIID is a lytic transglycosylase (LT), an enzyme that cleaves the glycan strands of PGN (Fig. 1b). It also has a single-pass transmembrane domain and a large extracellular portion that can bind PGN but can only cleave glycan strands after the stem peptides of PGN have been removed by SpoIIP. The requirement for a peptide-free glycan strand defines an ordered and sequential set of events during the engulfment process. SpoIID also functions to stimulate SpoIIP activity (4).
FIGURE 1.

Activities of SpoIID and SpoIIP on the PGN.
LTs represent a class of autolysin that cleaves the glycan chain of the PGN to facilitate biosynthesis, remodeling, degradation, and turnover of the bacterial cell wall (7). LTs differ from the other two classes of glycan strand-cleaving enzymes. Although muramidases hydrolyze the β-1,4 N-acetylmuramic acid (NAM)-N-acetylglucosamine (NAG) glycosidic bond and glucosaminidases hydrolyze the β-1,4 NAG-NAM glycosidic bond, LTs use the C-6 hydroxyl of NAM as a nucleophile in the cleavage reaction of the β-1,4 NAM-NAG glycosidic bond. Consequently, the product of LT action has a terminal 1,6-anhydro-NAM (Fig. 1b). The proposed intramolecular transglycosylation reaction requires the presence at the active site of a catalytic residue that has been identified as a glutamate in several enzymes (8).
Despite the low sequence homology observed among the LT superfamily, in most cases their catalytic domains include a region structurally similar to that of goose egg-white lysozyme (GEWL) (9). The LT superfamily has been subdivided into four families on the basis of conserved motifs (10). However, SpoIID does not show sequence homology with any of the characterized murein hydrolases and thus represents the founding member of a new LT family (1). Although several crystal structures are available for members of the 1–4 LT families, none have previously been determined for any members of the SpoIID family.
Experimental Procedures
Cloning, Protein Purification, Crystallization, Data Collection, and Phasing
The standard protocols from the Center for the Structural Genomics of Infectious Diseases were used for cloning, overexpression, and purification of CdSpoIID (YP_001086593.1) and Ba-SpoIID (AAP29172.1) (11, 12). Briefly, BaSpoIID (from residues 33 to 339) and CdSpoIID (from residues 28 to 354) were cloned into the pMCSG7 expression vector (bioinformatics.anl.gov). The mutants of CdSpoIID were generated by PCR and by using the polymerase incomplete primer extension technique (13). The list of the primers used is reported in supplemental Table S1. BL21 (DE3) Magic Escherichia coli strain (14) was grown in M9/seleniomethionine medium or TB at 37 °C up to an A600 of 1. At that point, the temperature was reduced to 25 °C, and protein overexpression was induced by 1 mm isopropyl 1-thio-β-d-galactopyranoside. After 16 h, cells were harvested by centrifugation, suspended in a buffer containing 10 mm Tris-HCl, pH 8.3, 500 mm NaCl, 10% glycerol, and 5 mm β-mercaptoethanol, and lysed by sonication. Proteins were purified by nickel-nitrilotriacetic acid affinity chromatography and eluted with a buffer containing 10 mm Tris-HCl, pH 8.3, 500 mm NaCl, 5 mm β-mercaptoethanol. Proteins were concentrated and stored at −80 °C in a buffer containing 0.5 m NaCl and 10 mm Tris-HCl, pH 7.2. The proteins were concentrated using Vivaspin centrifugal concentrators (GE Healthcare), and both its size and purity were checked by SDS-PAGE.
Sitting drop crystallization plates were set up at room temperature, and crystals were obtained for BaSpoIID at a concentration of 13.4 mg/ml in 0.15 m dl-malic acid, pH 7.0, 20% (w/v) PEG 3350, and for CdSpoIID at 7.0 mg/ml in 1.6 m tri-sodium citrate, pH 6.5, with elastase 1:100 (v/v) (Sigma, 50 units/ml) (11). Harvested crystals were transferred to the mother liquor before being frozen in liquid nitrogen. Diffraction data were collected at 100 K at the Life Sciences Collaborative Access Team at the Advance Photon Source, Argonne, IL (APS BEAMLINE 21-ID-G). All structural work was performed by the Structural Genomics of Infectious Diseases (15). Data were processed using HKL-2000 for indexing, integration, and scaling (16). A selenomethionine derivative of BaSpoIID was used to phase the structure by single-wavelength anomalous diffraction. The domains from this structure were used for molecular replacement to phase CdSpoIID. Structures were refined with Refmac (17). Models were displayed in Coot and manually corrected based on electron density maps (18). All structure figures were prepared using PyMOL Molecular Graphics System, Version 1.3 (Schrödinger, LLC). The topology diagrams (Fig. 2, a and d) were drawn according to the structural analysis generated with PDBsum (19). The visualization of the sequence conservation of the most 40 different members of Pfam08486 (the conserved domain of the SpoIID family) on the structure of CdSpoIID (Fig. 6e) was performed using the ConSurf server (20).
FIGURE 2.

Overall structure of BaSpoIID (top) and CdSpoIID (bottom). a and d, topology diagrams of BaSpoIID and CdSpoIID, respectively, with the highlighted α-helix rich α-domain in the slate box and the β-strand rich β-domain in the orange box. The three α-helices comprising the core of the α-domain are shown in white. b and e, ribbon diagrams of BaSpoIID and CdSpoIID, respectively, rainbow-colored from blue at the N terminus to red at the C terminus. e, two NAG3 molecules are represented as green sticks. c and f, surface representations of BaSpoIID and CdSpoIID, respectively, with the characteristic fist-like shape, with the α-helix rich α-domain (the hand) colored in light blue and the β-strand rich β-domain (the arm) in orange. f, the two NAG3 molecules are represented as red sticks in the main groove of the α-domain.
FIGURE 6.

CdSpoIID and BaSpoIID structural superimposition. a, superimposition of BaSpoIID (in slate ribbon) on CdSpoIID (in white ribbon). b, superimposition of BaSpoIID (slate ribbon) on CdSpoIID (white ribbon). c, zoom-in on the N-terminal β-sheet of BaSpoIID and CdSpoIID (colored as in A) with the extra β3-strand of CdSpoIID anchoring the turn-β-strand-turn motif of the zinc-binding site. d, phylogenetic tree of the SpoIID family based on the 40 most divergent members. The members that have conserved residues for the zinc coordination in CdSpoIID were identified and labeled with an open circle. The solid symbols show the position of CdSpoIID and BaSpoIID. e, CdSpoIID colored according to the sequence conservation of the most 40 different SpoIID family members. The most highly conserved residues are red, and the least conserved residues are blue.
Building of the (NAG-NAM)3 Model
The ligand-binding site of CdSpoIID was analyzed for multiprobe characterization using the server SiteComp to generate a model of the subsite −1 of the (NAG-NAM)3 glycan chain (21). Only the grid points with interaction energy below e−10 were included in the cluster. The result of this analysis was the molecular interaction field (MIF), which clearly showed the location for favorable interaction energy clusters in subsite −1, between the active site residues of CdSpoIID and the ligand. The MIF and the limited empty volume in subsite −1 of the CdSpoIID guided the modeling of the NAM moiety in Coot (18). In particular, to maintain the links with the NAG in subsites −2 and +1 by conserving the minimum energy conformation, the NAM in subsite −1 has (i) the C6 in axial position, (ii) the N-acetyl group, and (iii) the lactyl group modeled in the two lobes available in the MIF. Any alternative conformation of NAM in subsite −1 is not able to maintain the links with the NAG in subsites −2 and D. Additionally, the lactyl group in subsite −3 was modeled in a minimum energy conformation in accordance with the conformation of the pyranose ring of the NAG in subsite −3 of the NAG3-1 molecule in the crystal structure of CdSpoIID.
Sequence Alignment and Phylogenetic Analysis
The sequence alignment was generated with ESPript (22) and ClustalW (23). The maximum likelihood method was used to infer the evolutionary history of identified sequences in Mega 6.06 (24). Confidence limits of branch points were estimated by 103 bootstrap replications.
Enzymatic Assay
The activity of SpoIID from Bacillus subtilis (BsSpoIID) has been previously tested on PGN purified from the cell wall (i.e. sacculi) of E. coli (1). Sacculi were prepared from E. coli BL21DE3 as described previously (25). Briefly, 1 liter of cells was grown in LB until an A600 of 0.5. The growth was stopped by rapid cooling in an ice bath, and the cells were spun down (10 min; 10,000 × g; 4 °C) and boiled in 100 ml of 4% SDS with vigorous stirring for 30 min. Sacculi were then sedimented by ultracentrifugation (10 min; 10,000 × g; room temperature) and washed five times with double distilled H2O to remove SDS.
Isolated sacculi were suspended in 10 mm Tris-HCl buffer, 10 mm NaCl, 0.32 m imidazole and treated with 100 μg/ml amylase (Sigma) for 2 h at 37 °C, followed by incubation at 60 °C in the presence of 100 μl/ml of Pronase E (Sigma). The sacculi were boiled again with 4% SDS for 15 min and washed, as described above, until free of SDS. Sacculi were labeled with Remazol Brilliant Blue (RBB, Sigma) by incubating them with 20 mm RBB in 0.25 m NaOH overnight at 37 °C, as described previously (26). The preparation was neutralized with HCl, and the dye-labeled sacculi were pelleted by centrifugation (21,000 × g, 20 min, room temperature). The sacculi were then washed repeatedly with double distilled H2O until the supernatant was clear.
For the enzymatic reaction in Fig. 4, d and e, 600 μl of RBB-labeled PGN were incubated in buffer B1 (10 mm Hepes, pH 7.5, 50 mm NaCl) at 37 °C for 1 h with 100 μm CwlH, CdSpoIID, BaSpoIID or in combination. For Fig. 4e, the insoluble material of CwlH reaction was washed 10 times with 1 ml of B1 and 0.01% SDS and then 5 times with B1 alone to eliminate the SDS. 20 μl of CwlH-treated RBB-labeled PGN were incubated at 37 °C for 1 h with CdSpoIID mutants at 10 μm in buffer B1. Reactions were stopped by adding ½ reaction volume of 95% ethanol. Soluble cleavage products were separated by centrifugation (10 min, 10,000 × g) at room temperature.
FIGURE 4.

Residues important in binding of the two NAG3, the zinc, and affecting the enzymatic activity CdSpoIID. a, zoom-in the binding site of CdSpoIID with the surface of the α-domain and ribbon of the β-domain. The surfaces of the residues interacting with NAG3-1 and NAG3-2 are colored in orange. b, white ribbon of CdSpoIID and the residues interacting with the ligands represented as orange sticks. The NAG3-1 and NAG3-2 are represented as dark gray sticks, and the electron density map (omit map), countered at 2σ level, is shown in green. The black dashed lines indicate possible hydrogen bonds between the atoms. c, turn-β-strand-turn motif with the residues coordinating the binding of the zinc atom (slate) drawn as orange sticks. d, PGN degradation activity of BaSpoIID andCdSpoIID. RBB dye release assay for BaSpoIID and CdSpoIID alone or in combination with N-acetylmuramoyl-l-alanine amidase CwlH from B. anthracis. Data represent the mean of three independent experiments and include the standard deviation. e, RBB-dye release assay of the CdSpoIID and CdSpoIID mutants on the RBB-dye PGN-labeled pretreated with CwlH. Data represent the mean and the standard deviation from three independent experiments.
Conservation of the Structural Motif in SpoIID Family
Structural multialignment was performed using PROMALs3D on a set of representative structures from 14 different LTs available in the Protein Data Bank (Table 2) (27).
TABLE 2.
Structures from the LT family available in the Protein Data Bank
The Protein Data Bank codes in bold were used as representative structures in each subfamily to perform the structural multialignment.
| Family | Subfamily | Founder member | Structure (PDB code) | Proteins |
|---|---|---|---|---|
| Family 1 | A | Slt70 (E. coli) | 1QSA, 1QTE, 1SLY | Slt70 (E. coli) |
| 4F55 | SleB (B. cereus) | |||
| 4FET | SleB (B. anthracis) | |||
| B | YfhD (E. coli) | |||
| C | MltC (E. coli) | 4C5F, 4CFO, 4CFP, 4CHX | MltC (E. coli) | |
| D | MltD (E. coli) | 1E01, 1E0G | MltD (E. coli) | |
| E | EmtA (E. coli) | 3T36, 4HJV, 4HJY, 4HJZ, 2Y8P | EmtA/MltE (E. coli) | |
| Family 2 | MltA (E. coli) | 2PNW | MltA (Agrobacterium tumefaciens) | |
| 2PI8, 2PIC, 2PJJ, 2AEO, 2GAE | MltA (E. coli) | |||
| 2G5D, 2G6G | MltA (Neisseria gonorrhoeae) | |||
| Family 3 | MltB/Slt35(E. coli) | 1D0L, 1QDR, 1QUS, 1QUT, 1D0K, 1D0M, 1QDT, 1LTM | MltB/Slt35 (E. coli) | |
| 4ANR | SltB1 (Pseudomona aeruginosa) | |||
| 4P11, 4P0G, 4OZ9, 4OYV, 4OXV, 4OWD | MltF (P. aeruginosa) | |||
| Family 4 | Phage λ | 1AM7, 1D9U, 3D3D | R (phage λ) | |
| 3BKH, 3BKV | gp144 (phage φKZ) | |||
| Family 5 SpoIID-Like | A | SpoIID (B. subtilis) | 4RWR | SpoIID (B. anthracis) |
| B | SpoIID (C. difficile) | 5I1T | SpoIID (C. difficile) |
Results
Overall Structures of SpoIID from Bacillus anthracis and Clostridium difficile
To better understand the mechanism of SpoIID, we initiated structural studies on SpoIID from B. anthracis (BaSpoIID). This protein was successfully expressed, purified, and crystallized. Single-wavelength anomalous diffraction with a selenomethionine-substituted sample of BaSpoIID provided initial experimental phases. The final model (Fig. 2, a–c) was refined at 2.10 Å resolution (Table 1). BaSpoIID has a fist-like shape, which includes an α-helix rich α-domain (“the hand”), and a β-strand rich β-domain (“the arm”). A deep groove runs across the palm of the α-domain. The α-domain includes seven α-helices, with α2 spanning the α-domain under the transverse groove. Two clusters of α-helices (α1-α3-α5 and α9-α10) and three small β-sheets (β1-β2, β4-β5-β14, and β6–β13) were assembled around α2 and participate in the formation of the main groove. A β-strand rich β-domain is organized in two β-sheets (β7-β12-β11 and β8-β9-β10) connected by two α-helices (α7 and α8) (Fig. 2, a–c).
TABLE 1.
Data collection and refinement statistics for crystal structures of BaSpoIID and CdSpoIID
| BaSpoIID (PDB code 4RWR) | CdSpoIID (PDB code 5I1T) | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P3121 |
| Cell dimensions | ||
| a, b, c (Å) | 46.62, 144.48, 47.08 | 98.08, 98.08, 108.33 |
| α, β, γ (°) | 90.00, 115.36, 90 | 90.00, 90.00, 120.00 |
| Resolution (Å) | 30.00-2.10 (2.14-2.10)a | 30.00-2.60 (2.64-2.60) |
| Rsym or Rmerge | 0.105 (0.623) | 0.067 (0.608) |
| I/σI | 12.4 (2.2) | 30.8 (4.1) |
| Completeness (%) | 100.0 (100.0) | 99.9 (100.0) |
| Redundancy | 4.3 (4.3) | 8.8 (9.3) |
| Refinement | ||
| Resolution (Å) | 27.53-2.10 (2.15-2.10) | 29.08-2.60 (2.67-2.60) |
| No. of reflections | 31,083 (2,402) | 17,967 (1,362) |
| Rwork/Rfree | 0.188/0.243 (0.246/0.302) | 0.155/0.196 (0.200/0.308) |
| No. of atoms | ||
| Protein | 4,203 | 2,314 |
| Ligand/ion | 0/0 | 82/3 |
| Water | 290 | 148 |
| B-Factors | ||
| Protein | 39.5 | 68.6 |
| Ligand/ion | 97.5/68.4 | |
| Water | 36.6 | 65.6 |
| r.m.s.d. | ||
| Bond lengths (Å) | 0.009 | 0.011 |
| Bond angles (°) | 1.44 | 1.48 |
a Values in parentheses are for highest resolution shell.
SpoIIDs exhibit considerable sequence divergence, and we decided it would be useful to compare divergent SpoIID structures. The SpoIID from C. difficile (CdSpoIID) shares only 34.8% sequence identity with BaSpoIID and was selected for subsequent studies (Fig. 3). CdSpoIID was successfully expressed, purified, and crystallized. The structure was determined to 2.6 Å resolution by the molecular replacement method using the structure of BaSpoIID as a search model (Fig. 2, d–f) (Table 1). CdSpoIID has the same structural organization of BaSpoIID with a α-helix rich α-domain forming the hand and a β-strand rich β-domain, forming the arm (Fig. 2f).
FIGURE 3.

Sequence conservation for SpoIID from C. difficile, B. anthracis, and B. subtilis and key residues. The sequences for CdSpoIID, BaSPoIID, and BsSpoIID are aligned and numbered according to the secondary structure of CdSpoIID. The sequences are colored according to the conservation. As indicated in the legend, the residues of CdSpoIID that make interactions with the two NAG3 molecules and the zinc atom are indicated with orange and blue bars, respectively. The residues that have been tested by alanine substitution in CdSpoIID (this work) and BsSpoIID (1, 4) are indicated with black bars. The red bars indicate the mutations with a phenotype.
Well diffracting CdSpoIID crystals only grew in the presence of N,N′,N″-tri-acetyl-chitotriose (NAG3) molecules, which mimic the glycan chains of the PGN. In the structure, a NAG3 molecule (NAG3-1) occupies subsites −4, −3, and −2 of the substrate-binding site, whereas a second NAG3 molecule (NAG3-2) occupies subsites +1, +2, and +3 (Fig. 4a). CdSpoIID makes direct interactions with sugars spanning positions −4 to +1 (Fig. 4b). The glycosidic torsion angles of the NAG3 molecules adopt an intermediate configuration between the rotation of NAG-NAM (pentapeptide) observed for the PGN with three NAG-NAM (pentapeptide) disaccharides per turn (28) and the rotation of α-chitin (NAG polymer) in which the planes of the saccharide rings are oriented at 180° to each other (29). This indicates that the SpoIID interactions modify the sugar orientations in the glycan chains.
The CdSpoIID crystal structure also revealed a new zinc-binding site made up of a turn-β-strand-turn motif that contributes two cysteines (Cys-40 and Cys-146) and two histidines (His-134 and His-145) to the coordination of the zinc (Fig. 4c). This binding site has no counterpart in the BaSpoIID.
Substrate Recognition Model and Critical Residues for the Enzymatic Activity of CdSpoIID
We next sought to leverage our structural insight to clarify SpoIID function. First, we confirmed that both CdSpoIID and BaSpoIID require peptide-free glycan chains as substrate for their hydrolase activities (1, 4). In fact, no blue release was detected in the supernatant for reactions containing BaSpoIID and CdSpoIID when incubated with RBB-labeled PGN. Consequently, the RBB-labeled PGN was incubated with the CwlH enzyme in combination with CdSpoIID or BaSpoIID confirming the requirement for CdSpoIID and BaSpoIID for stem-free glycan chains as substrate (Fig. 4d). Furthermore, based on a structural analysis, we generated 14 CdSpoIID single mutants (in each case mutated to alanine) and tested their enzymatic activity (Fig. 4e). The Glu-101 to Ala substitution inactivated the enzyme. This result, complemented by the structural data and by the fact that the corresponding Glu-88 in the BsSpoIID has been previously proposed to be the catalytic residue (1, 4), confirms that Glu-101 of CdSpoIID is the essential catalytic residue for the reaction. Interestingly, the single mutations of His-145, Cys-146, or Cys-140, three of the residues involved in the zinc coordination, also inactivated the enzyme, highlighting the importance of this structural motif for CdSpoIID activity. Besides the mutations of Glu-101 and the residues involved in the zinc binding coordination, other mutations affect the in vitro activity of CdSpoIID. We identified Tyr-194 as another key residue. Its hydroxyl group is positioned at 3.4 Å from the carboxylic group of the catalytic glutamate Glu-101 in CdSpoIID. This position is occupied by tyrosines or phenylalanines in all the enzymes in the SpoIID family. Consequently, besides the hydroxyl group of the tyrosine, the aromatic ring that the two residues share seems to be particularly important in the active site. The Tyr-194 of CdSpoIID, which corresponds to Phe-176 in BaSpoIID, makes hydrophobic interactions with the acetyl group of NAG at subsite −2. The Tyr-213 substitution to alanine also impacts the activity of the enzyme. This residue makes hydrophobic interactions with the NAG3-1 in the subsite −4 (Fig. 4b). Furthermore, we observed a reduction in the catalytic activity of mutants S196A and Q324A, which interact with the acetyl group of the NAG3-2 subsite +1 and NAG3-1 subsite −2, respectively. Gln-324 seems to have also a structural importance due to the interaction that occurs between its amide group and the carbonyl of Gln-114 from the α2-helix (Fig. 4b). The side chain amide group of Gln-324 is also at 3 Å from a side chain oxygen of Glu-101. His-318 is highly conserved in the family, and its side chain interacts with the carbonyl of the acetyl group of NAG3 at subsite −2. His-318 in CdSpoIID corresponds to His-297 in BsSpoIID, and its alanine substitution completely abrogates spore formation in B. subtilis (1, 4).
Additionally, a number of BsSpoIID mutations that reduce PGN degradation activity and/or sporulation efficiency have previously been reported (Fig. 3). These residues are very conserved in all the members of the SpoIID family and correspond in CdSpoIID to Arg-119, Thr-202, Lys-217, Asp-224, Met-322, Tyr-344, and Tyr-345 (1, 4). All of these residues are situated in the α-domain and have a structural role (Fig. 5). In fact, the side chains of Thr-202, Asp-224, Tyr-344, and Tyr-345 make several hydrogen bonds with the side chain or the main chain atoms of several other residues. Met-322 is buried in the middle of the α-domain and is involved in several hydrophobic interactions and influences the position of the side chains of many residues that are directly exposed in the substrate-binding groove. Arg-119 is located under the α2-helix and seems to be important for the relative orientation of the α2- and α5-helices of the catalytic core domain of the enzyme by making hydrogen bonds with the main chain of Val-175 and the side chain atoms of Thr-178 at the end of the α5-helix. The main chain of Gln-181 in the β4 β-strand is also at hydrogen bonding distance. Lys-217 is surface-exposed and is located in the flexible loop β6-η1 of the SpoIID. In CdSpoIID, the residue forms a hydrogen bond with the main chain of Tyr-213.
FIGURE 5.
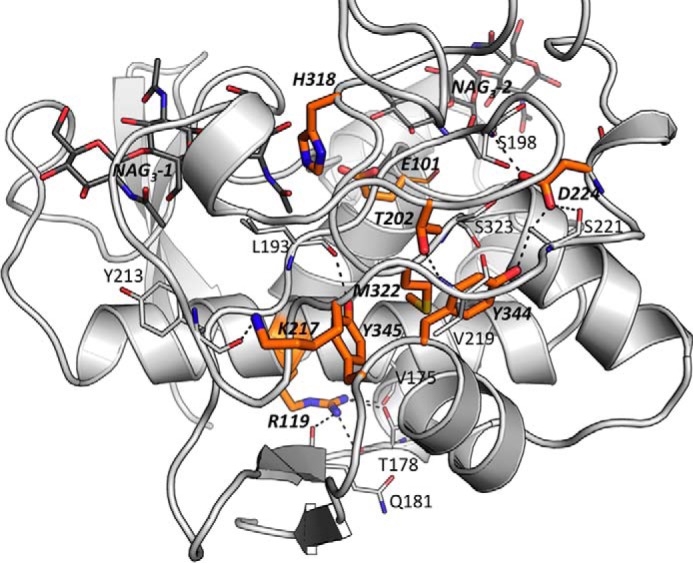
Correspondent positions of the important residues for BsSpoIID's activity in CdSpoIID. View of the outer face of CdSpoIID α-domain, represented as white ribbons with the orange sticks of the residues that correspond to the mutation in BsSpoIID affecting the B. subtilis sporulation. The black dashes indicate the hydrogen bonds occurring with the other residues shown as white sticks.
Structural Homology of SpoIID-LT Family
Overall, CdSpoIID and BsSpoIID exhibit high structural homology (Fig. 6a) with an r.m.s.d. of 1.49 Å (superimposition of 198 Cα atoms). The major differences include the extra α6-helix in CdSpoIID (Fig. 6b), the zinc-binding site (Fig. 6c), the conformation of the helix α7-strand β8-connecting loop in the β-domain, and an insertion in the BaSpoIID α3-helix that results in two distinct helices (α3 and α4) in CdSpoIID.
Regarding the differences in the secondary structure elements between the two molecules, besides the sequence divergence, a major modification that may be induced by the binding of the NAG3 ligands in CdSpoIID is the reorganization of the loop Ser-205/Tyr-213 (Ala-187/Tyr-195 in BaSpoIID) with consequent formation of the extra α6 α-helix (Fig. 6b). Many interactions occur between several residues of this region with the NAG3-1 molecule in CdSpoIID. In CdSpoIID the major interactions with NAG3-1 occur though Phe-209 and Tyr-213, corresponding in BaSpoIID to the position of Trp-191 and Tyr-195, respectively. Although Phe-209 in CdSpoIID is interacting with NAG in subsite −3 of NAG3-1, Trp-191 is involved in hydrophobic interactions with the other molecule of BaSpoIID in the crystal lattice.
The clearest and seemingly most meaningful difference between the two SpoIIDs concerns the zinc-binding site (Fig. 6c). The N-terminal β-sheet in CdSpoIID contains longer β1 and β2 β-strands and a unique β-strand, β3, which structurally anchors the zinc-binding site (Fig. 6c). Furthermore, the four residues involved in the coordination of the zinc cation are not conserved in BaSpoIID or BsSpoIID, suggesting a divergent evolution for CdSpoIID at this site.
Because BaSpoIID and CdSpoIID differ with respect to the zinc-binding site, we examined the prevalence of the zinc-binding motif. BaSpoIID and CdSpoIID both belong to the sequence domain family Pfam08486 (E-value 1.09e−36 and E-value 2.27e−34, respectively). Pfam08486 is common and is found in 5,183 genes from 4,210 species. The lowest sequence homology in the family is about 28%. A phylogenetic tree was built from 40 divergent members of the SpoIID family (Fig. 6d). Our analysis revealed that the zinc-binding motif is present in ∼39% of these sequences and is spread across clades 2–4. Within these clades, the zinc-binding motif is not strictly conserved, as shown in the sequence conservation of the SpoIID family (Fig. 6e).
To identify proteins more distantly related to SpoIID, we performed searches of protein structure databases to identify structural homologs of CdSpoIID and BaSpoIID. Several servers (such as Dali, CACH, ProFunc, and Salign) were searched but failed to identify significant similarity with any other protein. Despite this, a multiple structural alignment was performed with a set of representative structures from 14 different LTs available in the Protein Data Bank (Table 2). The analysis identified a shared structural motif with the catalytic domain of SleB from Bacillus cereus (BcSleB) or B. anthracis (BaSleB). The motif corresponds to α1, α2, and α5 α-helices in the SpoIID enzymes and to the core region encompassing helices α1, α2, and α5 in SleB (30). This is a very conserved structural motif that resembles that occurring in the GEWL (Fig. 7a) (30). The superimpositions of the core domains highlight the conservation of the relative orientations of the three helices that position the essential catalytic residue, despite the low sequence homology (Fig. 7, b and c).
FIGURE 7.

Conservation of the structural motifs of SpoIID. a, structural conservation of the α1α2α5 core of the α-domain of SpoIID with other proteins. b, superimposition of the core domains from CdSpoIID (gray), BaSPoIID (orange), BaSleB (purple), EcYgeG (cyan), EcSlt70 (yellow), and GEWL (red). c, sequence alignment based on structural superimposition of the three helices of the core domain from CdSpoIID, BaSleB, BcSleB EcYgeG, EcSlt70, and GEWL and the corresponding position of the catalytic glutamic acid in motif I.
The superimposition of α1α2α5 of CdSpoIID with α1α2α5 of BcSleB and α1α2α5 of BaSleB gives a r.m.s.d. of 1.20 and 1.22 Å on 39 Cα, respectively, with a local sequence homology of ∼12%. SleB is a germination-specific LT responsible for the degradation of the cortex PGN, an essential step that allows full spore core rehydration and resumption of metabolism to initiate germination (30, 31). In the SpoIID family, the identified catalytic Glu-101 and Glu-87 for CdSpoIID and BaSpoIID, respectively, corresponds to the glutamate of BcSleB-BaSleB in motif I, at the C terminus of the α1-helix, and the Ser of motif I of the LTs' classification is replaced by a methionine. This Glu-Met motif is conserved across the SpoIID family (Fig. 7c).
Despite the different topological arrangements, the α1α2α5 core helices of SpoIIDs can be superimposed on the helices α2α5α1 of the catalytic domain of Slt70 from E. coli with a r.m.s.d. of 1.26 Å for 36 Cα, and the α1-helix of Slt70 is in the position of α5 of SpoIID and SleB (Fig. 7, a–c) (30, 31).
Among the three different lysozymes, hen egg-white lysozyme (HEWL), T4 lysozyme (T4L), and GEWL, the best superimposition of the core helices α1α2α5 of SpoIIDs occurs with the α2α5α1-helices of GEWL (r.m.s.d. of 2.54 Å for 40 Cα, sequence identity 7.7%) (Fig. 7, a–c). Several structures in complex with the glycan chain are available for this superfamily. We tried to superimpose the NAG moieties occupying the analogous subsites −4, −3, −2, and −1 of the available structures of HEWL, T4L, and GEWL on the binding site of SpoIID, but without success. However, some similar patterns of interactions between the lysozyme superfamily and the substrates are found in CdSpoIIDs (32). In the HEWL, GEWL, and T4 lysozyme, the most distinctive interactions occur for the saccharide bound in the subsite −2 (33, 34). One important network of interactions that is conserved throughout SpoIID, HEWL, and GEWL is the interaction of the side chains of His-318, His-101, and Trp-63, respectively, with the sugars in subsites −2. Furthermore, the hydrophobic interaction described for Trp-62 of the HEWL with the pyranose ring in −3, which has no apparent analog in either GEWL or T4L, is similar to the interaction occurring at the same subsite in CdSpoIID via Phe-209 (32). This residue corresponds to a tryptophan in BaSpoIID and BsSpoIID.
Structural Model for the Catalytic Events of SpoIID
Finally, we built a model for the mechanistic events catalyzed by SpoIID. The two NAG3 molecules in the CdSpoIID structure are aligned on the same axis, and the distance between the O1 of NAG in subsite −2 and the O4 of NAG in subsite +1 is 6.1 Å (Fig. 8a), appropriate for accommodating a NAM residue at subsite −1 in a glycan chain of the PGN. At subsite −1, we identified the MIF, the location for favorable interaction energy clusters, between the active site residues of CdSpoIID and the ligand (Fig. 8a) (21). The structure of CdSpoIID in complex with the two NAG3 and our model of the NAM moiety in subsite −1 provide support for several of the proposed steps of the mechanism of reaction for LTs. When we fit the NAM in the MIF at subsite −1, the saccharide can adopt a “boat” conformation (Fig. 8b), which it must to form the product. Indeed, a steric distortion of the sugar in subsite −1 has been previously proposed in the mechanisms of action of HEWL, T4L, and Slt70 (35, 36). The model suggests that the binding site favors the distortion of the NAM moiety bound in subsite −1 at the intermediate state (Fig. 8, c and d), conferring the higher energy conformation to the pyranose ring. The boat conformation can also be stabilized by the interaction of the lactic acid group to Arg-290 (∼3 Å), a residue that is conserved throughout the SpoIID family.
FIGURE 8.

Subsite −1 of SpoIID and the enzyme's catalytic mechanism. a, MIF (21) in lime green calculated by the analysis of the biochemical properties of the binding site identifies the location for favorable interaction clusters in the subsite −1 of CdSpoIID (white surface) and the (NAG-NAM)3 model. b, rotated views of (NAG-NAM)3 with NAM at subsite −1 fitted in the MIF (lime green). c, zoom-in of the subsite −1 of the superimposed CdSpoIID (white ribbon and orange sticks) and BaSpoIID (slate ribbon in transparence and slate sticks). (NAG-NAM)3 showed as dark gray sticks. The black dashes indicate hydrogen bond distances between atoms. d, mechanism of the reaction. In the first step, the catalytic glutamic acid donates its proton to the oxygen of the β1–4 bond, with the consequent formation of the oxocarbonium ion and release of the leaving group from the subsite +1 in the active site. In second step, the catalytic glutamate extracts the proton from the hydroxyl group at C6 of the NAM, allowing the intramolecular nucleophilic attack on C1 to form the 1–6-anhydro product (7).
The carboxyl group of the catalytic Glu-101, which protonates the oxygen of the β-1,4 NAM-NAG glycosidic bond in the first step of the reaction, is 2.8 Å from the O4 of the NAG3-2 (Fig. 8, c and d). In the model we observe that the position of the amide and the carbonyl of the acetamido group of NAM may favor the protonation of the O1–4 by providing an extra stabilization of the intermediate oxocarbonium ion. The position of the 2-acetamido group may also be stabilized by Tyr-316, which is within hydrogen bonding distance. Repositioning of Tyr-143 under the pyranose ring may additionally participate in this process. Interaction with Ser-196 may stabilize the acetamido group of NAM at subsite −1 upon the release of the leaving group.
Consistent with the proposal that Glu-101 deprotonates the NAM C6 hydroxyl group in the second step of the reaction, O6 is ∼3 Å from the carboxylate group of Glu-101 (Fig. 8, c and d). The intramolecular nucleophilic attack of O6 to C1 to form the 1–6-anhydro ring-containing product may be favored by Tyr-194 inducing the rotation of the C5–C6 bond moving O6 under the pyranose ring. Tyr-194 (Phe-176 in BaSpoIID) is held in place by a web of interactions occurring between Gln-114, Gln-324, and Glu-101 (corresponding to Gln-100, Gln-299, and Glu-87 in BaSpoIID). The aromatic ring in this position is conserved in all members of the SpoIID family, and its mutation to alanine in CdSpoIID inactivates the enzyme, suggesting that may not only be important for substrate binding but also for catalysis.
Discussion
LTs are key enzymes for many fundamental microbial processes by acting in PGN synthesis, remodeling, degradation, and recycling (7). They have been defined as space-making enzymes by cleaving the linkage in the glycan strands of the PGN sacculus. Because they act on a structure unique to bacteria, LTs are very interesting targets for structure-based design of a new class of antibiotics different from penicillin and related β-lactams. Interestingly, inhibition of LTs, i.e. with bulgecin A, increases the sensitivity of bacteria toward β-lactam antibiotics, and a combined therapeutic approach has been proposed (9). Consistent with this is the observation that 1,6-anhydro-MurNAc is used as an inducer of expression of the chromosomally encoded β-lactamase in some Gram-negative bacteria, e.g. Citrobacter freundii and Enterobacter cloacae (37).
We confirmed that the glutamic acid of the newly identified motif “EM” at the end of the α1-helix of SpoIID enzymes is the catalytic residue of the reaction. Furthermore, other biochemical features in the active site are important to coordinate the intramolecular reaction of the NAM. The aromatic ring of Tyr-194 of CdSpoIID and Phe-176 in BaSpoIID make hydrophobic interactions with the acetyl group of NAG at subsite −2 and are essential for the reaction. This position is occupied by tyrosines or phenylalanines in all the enzymes in the SpoIID family.
The turn-β-strand-turn motif of the zinc coordination site has a fundamental structural function to order the binding site in CdSpoIID. The alanine substitution of the tested residues involved in zinc coordination inactivates the enzyme. The turn-β-strand-turn motif occurs in a large group of SpoIID family members and identifies a new key feature in a subfamily of SpoIID-like LTs.
The structure of CdSpoIID in complex with the two NAG3 molecules allowed development of a model of the complex with the (NAG-NAM)3 substrate, which suggests that enzyme interactions contribute to accommodating the boat form of the NAM at subsite −1 and the 2-acetamido group in a position that should favor the approach of the carbonyl oxygen atom to the C1 atom and the oxazoline intermediate.
In conclusion, the crystal structures of BaSpoIID and CdSpoIID increase understanding of the catalytic function of the SpoIID enzymes and the LT family. They represent an important milestone toward the development of new drugs that could prevent spore formation and thus reduce transmission and recurrence of infections caused by important spore-forming pathogens.
Author Contributions
S. N. conducted the experiments, analyzed the results, and prepared the paper. G. M. was responsible for crystallographic structure determination. L. S. S. and I. D. were responsible for purity and crystallization. E. S. coordinated the project. W. F. A. conceived the idea for the project and prepare the paper with S. N.
Supplementary Material
Acknowledgments
X-ray data collection was done at the LS-CAT beamlines 21ID-G (3ROH) and 21ID-F (4Q7G) at the Advanced Photon Source Science User Facility operated for the United States Department of Energy (DOE), supported by the United States Department of Energy under Contract Number DE-AC02-06CH11357.
This work was supported by National Institutes of Health Contracts HHSN272200700058C and HHSN272201200026C from NIAID (to Structural Genomics of Infectious Diseases). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1.
The atomic coordinates and structure factors (code 5I1T) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PGN
- peptidoglycan
- LT
- lytic transglycosylase
- SpoIID
- stage II sporulation protein D
- GEWL
- goose egg-white lysozyme
- RBB
- Remazol Brilliant Blue
- NAM
- N-acetylmuramic acid
- NAG
- N-acetylglucosamine
- r.m.s.d.
- root mean square deviation
- MIF
- molecular interaction field
- HEWL
- hen egg-white lysozyme.
References
- 1. Morlot C., Uehara T., Marquis K. A., Bernhardt T. G., and Rudner D. Z. (2010) A highly coordinated cell wall degradation machine governs spore morphogenesis in Bacillus subtilis. Genes Dev. 24, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Errington J. (2003) Regulation of endospore formation in Bacillus subtilis. Nat. Rev. Microbiol. 1, 117–126 [DOI] [PubMed] [Google Scholar]
- 3. Hilbert D. W., and Piggot P. J. (2004) Compartmentalization of gene expression during Bacillus subtilis spore formation. Microbiol. Mol. Biol. Rev. 68, 234–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gutierrez J., Smith R., and Pogliano K. (2010) SpoIID-mediated peptidoglycan degradation is required throughout engulfment during Bacillus subtilis sporulation. J. Bacteriol. 192, 3174–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abanes-De Mello A., Sun Y. L., Aung S., and Pogliano K. (2002) A cytoskeleton-like role for the bacterial cell wall during engulfment of the Bacillus subtilis forespore. Genes Dev. 16, 3253–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chastanet A., and Losick R. (2007) Engulfment during sporulation in Bacillus subtilis is governed by a multi-protein complex containing tandemly acting autolysins. Mol. Microbiol. 64, 139–152 [DOI] [PubMed] [Google Scholar]
- 7. Scheurwater E., Reid C. W., and Clarke A. J. (2008) Lytic transglycosylases: bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 40, 586–591 [DOI] [PubMed] [Google Scholar]
- 8. Thunnissen A. M., Dijkstra A. J., Kalk K. H., Rozeboom H. J., Engel H., Keck W., and Dijkstra B. W. (1994) Doughnut-shaped structure of a bacterial muramidase revealed by x-ray crystallography. Nature 367, 750–753 [DOI] [PubMed] [Google Scholar]
- 9. Thunnissen A. M., Isaacs N. W., and Dijkstra B. W. (1995) The catalytic domain of a bacterial lytic transglycosylase defines a novel class of lysozymes. Proteins 22, 245–258 [DOI] [PubMed] [Google Scholar]
- 10. Blackburn N. T., and Clarke A. J. (2001) Identification of four families of peptidoglycan lytic transglycosylases. J. Mol. Evol. 52, 78–84 [DOI] [PubMed] [Google Scholar]
- 11. Anderson W. F. (2009) Structural genomics and drug discovery for infectious diseases. Infect. Disord. Drug Targets 9, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim Y., Bigelow L., Borovilos M., Dementieva I., Duggan E., Eschenfeldt W., Hatzos C., Joachimiak G., Li H., Maltseva N., Mulligan R., Quartey P., Sather A., Stols L., Volkart L., Wu R., et al. (2008) Chapter 3. High-throughput protein purification for x-ray crystallography and NMR. Adv. Protein Chem. Struct. Biol. 75, 85–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klock H. E., and Lesley S. A. (2009) The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 498, 91–103 [DOI] [PubMed] [Google Scholar]
- 14. Wu N., Christendat D., Dharamsi A., and Pai E. F. (2000) Purification, crystallization and preliminary x-ray study of orotidine 5′-monophosphate decarboxylase. Acta Crystallogr. D Biol. Crystallogr. 56, 912–914 [DOI] [PubMed] [Google Scholar]
- 15. Anderson W. F. (2009) Structural genomics and drug discovery for infectious diseases. Infect. Disord. Drug Targets 9, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otwinowski Z, Wladek M. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, p. 307–326 [DOI] [PubMed] [Google Scholar]
- 17. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 19. de Beer T. A., Berka K., Thornton J. M., and Laskowski R. A. (2014) PDBsum additions. Nucleic Acids Res. 42, D292–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashkenazy H., Erez E., Martz E., Pupko T., and Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin Y., Yoo S., and Sanchez R. (2012) SiteComp: a server for ligand binding site analysis in protein structures. Bioinformatics 28, 1172–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 24. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., and Kumar S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harz H., Burgdorf K., and Höltje J. V. (1990) Isolation and separation of the glycan strands from murein of Escherichia coli by reversed-phase high-performance liquid chromatography. Anal. Biochem. 190, 120–128 [DOI] [PubMed] [Google Scholar]
- 26. Zhou R., Chen S., and Recsei P. (1988) A dye release assay for determination of lysostaphin activity. Anal. Biochem. 171, 141–144 [DOI] [PubMed] [Google Scholar]
- 27. Pei J., Kim B. H., and Grishin N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meroueh S. O., Bencze K. Z., Hesek D., Lee M., Fisher J. F., Stemmler T. L., and Mobashery S. (2006) Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 103, 4404–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minke R., and Blackwell J. (1978) The structure of α-chitin. J. Mol. Biol. 120, 167–181 [DOI] [PubMed] [Google Scholar]
- 30. Jing X., Robinson H. R., Heffron J. D., Popham D. L., and Schubot F. D. (2012) The catalytic domain of the germination-specific lytic transglycosylase SleB from Bacillus anthracis displays a unique active site topology. Proteins 80, 2469–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y., Jin K., Setlow B., Setlow P., and Hao B. (2012) Crystal structure of the catalytic domain of the Bacillus cereus SleB protein, important in cortex peptidoglycan degradation during spore germination. J. Bacteriol. 194, 4537–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weaver L. H., Grütter M. G., and Matthews B. W. (1995) The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the “goose-type” lysozymes lack a catalytic aspartate residue. J. Mol. Biol. 245, 54–68 [DOI] [PubMed] [Google Scholar]
- 33. Anderson W. F., Grütter M. G., Remington S. J., Weaver L. H., and Matthews B. W. (1981) Crystallographic determination of the mode of binding of oligosaccharides to T4 bacteriophage lysozyme: implications for the mechanism of catalysis. J. Mol. Biol. 147, 523–543 [DOI] [PubMed] [Google Scholar]
- 34. Matthews B. W., Remington S. J., Grütter M. G., and Anderson W. F. (1981) Relation between hen egg white lysozyme and bacteriophage T4 lysozyme: evolutionary implications. J. Mol. Biol. 147, 545–558 [DOI] [PubMed] [Google Scholar]
- 35. Thunnissen A. M., Rozeboom H. J., Kalk K. H., and Dijkstra B. W. (1995) Structure of the 70-kDa soluble lytic transglycosylase complexed with bulgecin A. Implications for the enzymatic mechanism. Biochemistry 34, 12729–12737 [DOI] [PubMed] [Google Scholar]
- 36. Hadfield A. T., Harvey D. J., Archer D. B., MacKenzie D. A., Jeenes D. J., Radford S. E., Lowe G., Dobson C. M., and Johnson L. N. (1994) Crystal structure of the mutant D52S hen egg white lysozyme with an oligosaccharide product. J. Mol. Biol. 243, 856–872 [DOI] [PubMed] [Google Scholar]
- 37. Jacobs C., Huang L. J., Bartowsky E., Normark S., and Park J. T. (1994) Bacterial cell wall recycling provides cytosolic muropeptides as effectors for β-lactamase induction. EMBO J. 13, 4684–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.