

Abstract
Tumor development driven by inflammation is now an established phenomenon, but the role that complement plays remains uncertain. Recent evidence has suggested that various components of the complement (C) cascade may influence tumor development in disparate ways; however, little attention has been paid to that of the membrane attack complex (MAC). This is despite abundant evidence documenting the effects of this complex on cell behavior, including cell activation, protection from/induction of apoptosis, release of inflammatory cytokines, growth factors, and ECM components and regulators, and the triggering of the NLRP3 inflammasome. Here we present a novel approach to this issue by using global gene expression studies in conjunction with a systems biology analysis. Using network analysis of MAC-responsive expression changes, we demonstrate a cluster of co-regulated genes known to have impact in the extracellular space and on the supporting stroma and with well characterized tumor-promoting roles. Network analysis highlighted the central role for EGF receptor activation in mediating the observed responses to MAC exposure. Overall, the study sheds light on the mechanisms by which sublytic MAC causes tumor cell responses and exposes a gene expression signature that implicates MAC as a driver of tumor progression. These findings have implications for understanding of the roles of complement and the MAC in tumor development and progression, which in turn will inform future therapeutic strategies in cancer.
Keywords: cancer, complement, inflammation, systems biology, tumor metastasis
Introduction
Inflammation is now well established as a crucial contributor in the development and progression of tumors; indeed, it has been included among the second generation hallmarks of cancer (1). A key player in inflammatory responses is the complement (C)3 system, an innate immune effector with important roles in defense against infection. C provides recognition, early warning signals, and the initial fast response upon exposure to foreign organisms and has evolved to amplify the response to the initial signal (2). C comprises three activation pathways that converge on a common terminal pathway at the stage of C5 cleavage; release of a 74-amino acid peptide C5a, which has potent anaphylatoxic and chemotactic activities (3), leaves the large fragment, C5b, to form the nidus of a membrane-associated complex. Sequential recruitment of C6, C7, C8, and multiple C9 molecules creates a membrane-spanning pore-like cylindrical protein structure known as the membrane attack complex (MAC). The MAC can cause osmotic lysis of certain susceptible bacteria and of metabolically inert cells (4); however, lysis of self-cells is restricted by a combination of regulatory proteins, ion pumps, and MAC removal processes (5). Non-lytic MAC triggers numerous activation events in cells that likely contribute to the proinflammatory activity of C (6).
C has been strongly implicated as an effector in tumor clearance over the past 20 years, largely because of the success of monoclonal antibody (mAb)-based immunotherapies, many of which are designed to harness C as an effector to cause killing of tumor cells (7). In this context, the mAb triggers overwhelming C activation and tumor cell destruction; however, the role of C in tumor clearance in the absence of an activating mAb is much less clear. Indeed, it has been suggested that C activation has a tumor promoting role in many malignancies (8). C activation is known to occur on tumor cells both in vitro and in vivo in many malignancies, including breast (9), papillary thyroid (10, 11), colorectal (12), and ovarian (13) cancers. The best evidence implicating a C activation product as a promoter of tumor development was provided by the demonstration that locally generated C5a recruits myeloid-derived suppressor cells into the tumor where they suppress the anti-tumor activity of CD8+ T-cells (14, 15). Others have implicated C5a as a factor influencing the balance between tumor promotion and tumor clearance (16), whereas both C3a and C5a have been shown to cause proliferation in tumor cells, for example in neuroblastoma (17, 18). Studies in knock-out mice lacking C3 or C4 confirm important roles for C in tumorigenesis tumor growth being restricted in both C3 and C4 knockouts (14). Despite the large and growing body of evidence supporting a tumor promoting role of C activation (18–21), the role of MAC in tumor biology has been neglected.
Most tumors express and indeed often overexpress membrane bound C regulators CD55, CD59, and CD46 (22). As a consequence, although C is activated in the tumor microenvironment, activation will be restricted, and thus terminal pathway activation and MAC deposition may be insufficient to kill the tumor cell. Nevertheless, MAC deposition on tumor cells at a sublytic level may have a profound impact on the target, for example by causing an immediate increase in intracellular Ca2+ (23) and downstream activation of signaling cascades (24). The effects of sublytic MAC on cells in vitro include: release of inflammatory mediators such as reactive oxygen or nitrogen species, leukotrienes, arachidonic acid metabolites, and prostaglandins (5); the release of cytokines such as IL-1, TNF α, IL-8, IL-6, and MCP-1 (25); increased expression of adhesion molecules such as E-selectin, ICAM-1, VCAM-1, and ELAM-1 (26); release of growth factors such as basic FGF, PDGF, EGF, PlGF (placental growth factor), and RANTES (regulated on activation normal T cell expressed and secreted) (27, 28); secretion of extracellular matrix components such as collagen IV and fibronectin (29) and regulators such as MMP2 and MMP9 (30); increased cell proliferation (31); accelerated or inhibited apoptosis (32–34) and activation of the NLRP3 inflammasome (35). Given this catalogue of effects on cell function, it is likely that sublytic MAC will significantly influence tumor cell fate in vivo. Here we take a novel approach to addressing how MAC influences tumor cell fate by adopting an unbiased systems analysis of the effects of sublytic MAC on the patterns of gene expression in a tumor cell, and we identify key pathways implicated and discuss the impact that these might have on tumor survival.
Results
Sublytic C Attack and MAC inhibition on Tumor Cell Lines
The C5-binding protein Ornithodoros moubata C inhibitor (OmCI) has been extensively characterized and shown to specifically block formation of MAC in human and rodent plasma (36, 37). The dose of OmCI required to completely block MAC formation was titrated by assessing inhibition of hemolysis of antibody-sensitized sheep erythrocytes (ShEA) exposed to pooled normal human serum (pNHS), an assay where target hemolysis is absolutely dependent on MAC formation (Fig. 1A). At 10 μg/ml, OmCI caused complete inhibition of pNHS-induced hemolysis. This dose was used in all subsequent experiments. The sensitivity of each of the selected tumor cell lines, CT26 and B16, to pNHS-induced complement-directed cytoxicity was determined in a calcein release assay immediately before each experiment. Both CT26 and B16 cells were efficiently killed by pNHS without need for sensitization, calcein release correlating with dose of pNHS (Fig. 1B). The dose of pNHS causing <10% specific calcein release at 1 h was chosen as the maximum sublytic dose for the subsequent experiments. The 1 h time point was chosen based on our previous work showing that MAC killing of nucleated cell targets is an acute event and does not increase further with prolonged incubation (6). To identify MAC-specific effects, cells were also exposed to the same sublytic dose of pNHS but preincubated with the inhibitory dose of OmCI (10 μg/ml) to block MAC formation.
FIGURE 1.

Optimization of sublytic complement conditions. A, hemolytic activity testing lysis of ShEA by serum with or without 10 μg/ml OmCI (C5 blocker), titrated from 16% down to 0%. B, susceptibility of CT26 cells to C lysis; cells in a monolayer were loaded with calcein AM then exposed to serum for 1h at 37 °C. Lysis was calculated from the release of calcein into the supernatant and expressed as the percentage of the total entrapped calcein obtained by detergent lysis of the cells. Results are the means of four separate experiments ± S.E. C and D, expression analysis of OPN in CT26 cells in response to exposure to sublytic C for 1, 6, and 12 h. CT26 cells were exposed for 1 (C) and 6 and 12 (D) hours to 5% serum treated with or without a MAC-blocking dose of OmCI and OPN gene expression analyzed by qPCR. Expression was calculated as % of untreated control. Results are means of three determinations ± S.E. (*, p < 0.05; **, p < 0.01).
Preliminary qPCR experiments were carried out on RNA harvested from CT26 cells used in the above experiments to validate targets for subsequent microarray expression analyses and determine optimum time points for RNA collection. Initially, RNA was harvested at 1 h and expression of osteopontin (OPN), a candidate gene chosen based on evidence from the literature (38), was measured by qPCR. Relative expression of OPN increased more in response to treatment with pNHS compared with OmCI-treated pNHS after 1 h exposure (Fig. 1C). To further refine the time points of exposure to sublytic MAC before RNA harvest, the experiment was repeated for 6- or 12-h time points, calculating the expression change in relation to untreated cells. OPN expression increased significantly at 6 h (p < 0.01) and 12 h (p < 0.05) in MAC-exposed cells compared with the OmCI control (Fig. 1D). For expression analysis, we chose to use 1 h as an initial time point to capture an immediate response and 12 h to capture sustained changes.
Global Gene Expression Analysis of Sublytic C
For microarray analysis cells were exposed to sublytic C attack and compared with a MAC-inhibited control as established in the optimization experiments described above: CT26 cells seeded at 1.6 × 103/mm2 were exposed to 5% pNHS with or without the addition of OmCI, then incubated for 1 or 12 h (4 replicates of each condition in wells of a 12-well plate) before harvest of RNA. RNA was also harvested from untreated control cells (four replicates) to establish a baseline. A total of 16 samples were used for microarray analysis, 3 from each of the 4 serum conditions and all 4 untreated controls. Quality control of microarray data was performed using principal components analysis (PCA), a method used to compress a high content dataset, enabling its description with a limited number of contributors to variation. PCA allows the effect of experimental parameters on the data to be observed and identifies data quality issues. Initial PCA showed a batch effect that was eliminated using the Partek batch remover tool to reveal the most important components (Fig. 2A). Data were plotted to explore the contributions of the top three components; PCA#1 and #3 were best correlated with time point and serum exposure and the presence of OmCI, respectively. Batch removal was not retained for downstream analysis because the robustness of the ANOVA model used rendered it unnecessary.
FIGURE 2.

Primary microarray data analysis. A, PCA plot of top three principle components. Three-dimensional plot showing the top three principle components of the microarray data as calculated using PCA. Contributing principle components (PC) are labeled on each axis alongside the calculated % contribution to overall variation. Each sample from the experiment is represented by a colored sphere; red = control, green = pNHS, blue = pNHS+OmCI. A centroid sphere shows how these samples are grouped according to their experimental conditions; black = control, pale blue = OmCI at 1 h, darker blue = OmCI at 12 h, light green = pNHS at 1 h, and darker green = pNHS at 12 h. B, scatter plot comparisons between samples exposed to pNHS and pNHS+OmCI at 1 (i) and 12 (ii) hours. Data are log2 transformed, median baseline-adjusted. Expression is presented as distribution around a median that represents equal gene expression in the two conditions. The parallel flanking lines represent gene expression changes of ±1.3-fold change; data points falling outside these lines are considered to be differentially expressed. Data points are colored according to their expression levels (median baseline-adjusted) upon exposure to pNHS; green = below median, red = above median.
To better visualize the impact of experimental conditions, scatter plots of median baseline-adjusted data were prepared to compare pNHS to pNHS + OmCI at 1 and 12 h (Fig. 2B, i and ii). This graphics representation revealed that, for most parameters, expression changes were more apparent 1 h after sublytic attack than at 12 h, indicating that in this system most of the changes were transient in nature. To measure significance of differential gene expression in cells exposed to pNHS with or without MAC blockade, a two-way ANOVA model was applied using the method of moments (39). Gene lists were prepared using the ANOVA-generated -fold change and p values to identify the most significantly differentially expressed (up or down) genes at each time point (Table 1). These show those genes that altered expression significantly (p < 0.05), filtered to include genes changed by a >2-fold at 1 h or a 1.5-fold at 12 h; different filters were chosen to reduce the disparity in number of differentially expressed genes at these time points. Five genes were up-regulated and 1 was down-regulated at 1 h post-exposure, whereas 2 genes were up-regulated 12 h post-exposure with none down-regulated. This difference in the number of genes differentially regulated between the time points supports the trends shown in the scatter plots, substantially more points falling outside the set confidence intervals at 1 h compared with 12 h (Fig. 2B).
TABLE 1.
Gene list of the top most significant expression changes when comparing pNHS with pNHS+OmCI at 1 and 12 h
List criteria: false discovery rate adjusted p value <0.05. Top: pNHS 1 h relative to pNHS+OmCI 1 h, up-regulated genes (-fold change >2) ranked by p value. Middle: pNHS 1 h relative to pNHS+OmCI 1 h, down-regulated genes (-fold change >2). Bottom: pNHS 12 h relative to pNHS+OmCI 12 h, up-regulated genes (-fold change >1.5) ranked by p value.
| Probeset ID | Symbol | Transcript | p value (pNHS 1 h vs. OmCI 1 h) | -Fold change (pNHS 1 h vs. OmCI 1 h) |
|---|---|---|---|---|
| ILMN_2623983 | Egr2 | ILMN_212209 | 4.57 × 10−11 | 2.08117 |
| ILMN_2600744 | Rgs16 | ILMN_209950 | 3.03 × 10−09 | 2.74759 |
| ILMN_2995794 | Itprip | ILMN_242056 | 6.49 × 10−08 | 2.16089 |
| ILMN_2662926 | Egr1 | ILMN_215729 | 9.47 × 10−08 | 2.37517 |
| ILMN_2757634 | Fam110c | ILMN_222981 | 1.89 × 10−07 | 2.07236 |
| ILMN_2834777 | Irf1 | ILMN_209850 | 5.15 × 10−10 | −2.19138 |
| Probeset ID | Symbol | Transcript | p value (pNHS 12 h vs. OmCI 12 h 12) | -Fold change (pNHS 12 h vs. OmCI 12 h) |
|---|---|---|---|---|
| ILMN_1260323 | Akr1c18 | ILMN_215518 | 4.16 × 10−07 | 1.61755 |
| ILMN_1225816 | Hbb-bh1 | ILMN_216390 | 1.99 × 10−07 | 1.58676 |
Identification of Secreted Effectors Induced by Sublytic C
To provide functional insight into the data a new gene list was prepared using less stringent thresholds for inclusion by applying the following filters to the ANOVA statistics: -fold change > 1 and an unadjusted p value cut-off <0.05 for both 1 h and 12 h comparisons. This latter filter selected for MAC-induced up-regulation events that were apparent at both 1 and 12 h time points, representing sustained changes. To understand the interactions between these genes the list was interrogated using MetaCore network building tools to automatically map genes to a representative component termed the network object (NO). The “shortest path” algorithm was selected, and canonical pathways were included for network building; to interpret the resulting network, nodes were arranged to identify the starting NOs and their overlapping connections. NOs not connected to the main network were removed, and those groups displaying little connectivity to the larger network were pruned. The network was then organized by cellular location from top to bottom (Fig. 3). The analysis revealed four key highly interrelated NOs, co-regulated by canonical pathways and with roles outside of the cell. These represented four genes; AREG (encoding amphiregulin), MMP3 (encoding matrix metalloproteinase-3; mmp-3), MMP13 (encoding matrix metalloproteinase-13; mmp-13), and CXCL1 (encoding chemokine (CXC motif) ligand 1; cxcl-1) (Table 2). Other highly connected NOs of note were epidermal growth factor receptor (EGFR) and the AP-1 complex.
FIGURE 3.

Network analysis of overlap gene list. The list includes genes up-regulated by sublytic MAC at both time points. The network was generated in MetaCore using the following options: shortest path network-building algorithm with a maximum of two steps, inclusive of canonical pathway; this latter option allows sequences of interactions that occur frequently in the cell to be counted as single steps in the shortest path. The network describes the interconnected regulation of up-regulated genes and highlights four key downstream effector genes. The network is organized so that nodes are organized by the subcellular localization of their products, from extracellular to nuclear. Nodes present in the input list are in blue circles. Thick light blue lines highlight the various canonical pathways of signal transduction and transcription regulation. Seed nodes are circled in navy blue, lines represent interactions, either transcriptional regulation or protein-protein associations; red = inhibition and green = activation.
TABLE 2.
Gene list entities
Four key highly interrelated NOs, co-regulated by canonical pathways and with roles outside of the cell were identified; these represented four genes, AREG (encoding amphiregulin), MMP3 (encoding mmp-3), MMP13 (encoding mmp-13), and CXCL1 (encoding cxcl-1). These genes were sorted by p values at 1 h and 12 h, and -fold change display (pNHS relative to pNHS+OmCI).
| Probeset ID | Symbol | Transcript | p value (pNHS 1 h vs. OmCI 1 h) | -Fold change (pNHS 1 h vs. OmCI 1 h) | p value -fold change (pNHS 12 h vs. OmCI 12 h) | -Fold change (pNHS 12 h vs. OmCI 12 h) |
|---|---|---|---|---|---|---|
| ILMN_1238547 | Areg | ILMN_217903 | 2.31 × 10−06 | 1.43899 | 0.00185142 | 1.18086 |
| ILMN_2737685 | Mmp13 | ILMN_210384 | 1.06 × 10−06 | 1.52378 | 0.0100312 | 1.14512 |
| ILMN_2753809 | Mmp3 | ILMN_219123 | 9.89 × 10−05 | 1.23906 | 0.00367792 | 1.08968 |
| ILMN_2763245 | Cxcl1 | ILMN_223377 | 8.74 × 10−03 | 1.55636 | 0.0215031 | 1.45109 |
qPCR Validation of Identified Genes
Genes identified as differentially expressed by microarray were validated using qPCR to provide support for further bioinformatic and biochemical exploration of their relevance. The four genes identified above as secreted effectors during network analysis were selected (Table 2) together with four genes identified as showing the most significant differential expression (up or down at either time point) between MAC-exposed and MAC-inhibited pNHS-exposed CT26 cells when stringent thresholds were applied (Table 1). FAM110c (encoding family with sequence similarity 110, member C) and RGS16 (encoding regulator of G-protein signaling 16) were both increased at 1 h post-attack; IRF1 (encoding interferon regulatory factor 1) was decreased at 1 h; HBB-BH1 (encoding hemoglobin Z, β-like embryonic chain) was increased at 12 h. For each of these eight genes, qPCR was performed twice, first on cDNA prepared from RNA extracted for the microarray experiment and second on RNA from a fresh replication experiment. The qPCR expression data were replicated in these experiments and largely confirmed the expression patterns found in microarray for these same genes; data are presented together for comparison (Figs. 4 and 5). In a few instances, qPCR and microarray data did not completely replicate: CXCL1 showed the highest up-regulation at 1 h by microarray but at 12 h by qPCR; RGS16 peak up-regulation at 1 h in microarray was confirmed by qPCR using the same original RNA, but in RNA from the second experiment further up-regulation was seen at 12 h post exposure. Despite these minor differences, the data strongly correlated, confirming the capacity of the microarray to accurately detect expression changes.
FIGURE 4.

qPCR validation of statistically significant hits. Microarray = original microarray data, Primary Validation = RNA extracted in parallel with that used in microarray, and Secondary validation = RNA extracted in a fresh sublytic attack experiment. RNA was reverse-transcribed and FAM110C, RGS16, IRF1, and HBB-BH1 gene expression analyzed by qPCR and calculated as expression relative to housekeeping genes β-actin and Pol2ra using the ΔΔCt calculation then presented as % of untreated control. Results are the means of three determinations ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. MBL adj., median baseline-adjusted. Con, control.
FIGURE 5.

qPCR validation of network identified hits. Microarray = original microarray data, Primary Validation = RNA extracted in parallel with that used in microarray, and Secondary validation = RNA extracted in a fresh sublytic attack experiment. B16 validation = RNA extracted from fresh sublytic attack experiment using the B16 mouse myeloma cell line (MMP3 message was not significantly detected in this cell line). In all cases RNA was reverse-transcribed, and MMP3, MMP13, CXCL1, and AREG gene expression was analyzed by qPCR and calculated as expression relative to housekeeping genes β-actin and Pol2ra using the ΔΔCt calculation then presented as a % of untreated control. Results are the means of three determinations ± S.E. (*, p < 0.05; **, p < 0.01; ***, p < 0.001). MBL adj., median baseline-adjusted; Con, control.
To explore whether the observed expression changes were cell type-specific, RNA extracted from MAC-exposed and control B16 melanoma cells was analyzed by qPCR for expression of the four network identified hits, AREG, MMP3, MMP13, and CXCL1. Expression of MMP3 RNA was negligible in this cell type. Expression of RNA for both AREG and CXCL1 was markedly increased in MAC-exposed cells at both 1 and 12 h, replicating the results obtained in CT26 cells (Fig. 5). MMP13 RNA expression was low in B16 cells and not significantly different between MAC exposed and control cells.
Interconnectivity of Secreted Effectors and Regulatory Genes
In an effort to identify pathways and mechanisms by which MAC effected changes in expression of the identified genes, a model was developed that analyzed the combination of those genes identified as significantly differentially expressed under stringent statistics and those identified by network analysis as downstream secreted factors. Fig. 6 shows the shortest path network generated using the gene lists shown in Tables 1 and 2 as input. The main hub of the network contained 11 of the total 12 starting NOs; FAM110C, ITPRIP, and HBB-BH1 were unconnected and, therefore, hidden. The network shows that the starting 11 NOs are well connected with a central triangle containing EGR1, EGR2, and IRF1, suggesting they are key drivers of the gene expression response to MAC. NOs added by the algorithm included AP-1 transcription factor subunit c-JUN, several NFκB subunits, the glucocorticoid receptor-α, c-Myc, and the EGFR. AP-1 and NFκB are the only two transcription factor NOs connected to all four secreted NOs validated by qPCR.
FIGURE 6.

Network analysis of collated gene list. Network describing the interconnected regulation of the four key downstream effector genes as well as the eight statistically significant genes. The network was generated in MetaCore using the following options: shortest path network building algorithm with a maximum of two steps, excluding canonical pathways. The network is organized to show cellular localization from extracellular (top) to nucleus (bottom). From the list, ITPRIP, FAM110C, and HBB-BH1 are not represented on the network due to lack of connectivity. Seed nodes are circled in navy blue, and lines represent interactions either transcriptional regulation or protein-protein associations; red = inhibition, and green = activation.
Transcriptional Regulation Network
To interrogate the data further and explore transcription regulation patterns from a greater number of data points, a gene list was generated by applying a false discovery rate-adjusted p value cut-off of <0.05 to the ANOVA statistics for the pNHS versus pNHS+OmCI comparison. This was applied separately for 1-h and 12-h time points, and the two lists were combined to identify the most significant MAC-induced expression changes at either time point and regardless of direction. This new list was combined with that created to generate Fig. 3, and the entire gene list was uploaded to MetaCore. The list was used as the starting list for the Analyze Network (transcription factors) (AN(TF)) algorithm applied using the default settings.
The AN(TF) algorithm identifies transcription factors for which there are enriched numbers of targets in the starting list, then uses the list to find the shortest path back to a receptor for which there are ligands in the starting list, thereby creating networks for each transcription factor, ranked by significance, based on enrichment of starting NOs via calculated g-scores, z-scores, and p values. z-score indicates the saturation of starting NOs, the g-score is a modified z-score describing the number of canonical pathways used to build the network, and p value assesses the probability of the number of starting NOs falling on the generated network by chance accounting for the total number of NOs in the network and in the entire database (40).
The network with the highest g-score and z-score and smallest p value was selected: g- and z-scores = 187.12, p value = 7 × 10−211. To assist in interpretation, the network was organized by aligning the most connected NOs in the center and placing the remaining NOs by protein class and in context around these main hubs (Fig. 7). With c-Myc and CREB1 as the main controlling transcription factors, EGFR and TrkB are introduced as non-seed nodes to the network as putative receptor starting points with EGFR the most interacting of the two. Other important TF hubs include c-Jun, p53, ESR1, and Oct3/4. All four secreted effectors identified in microarray are present, and the NO with most direct connections with these is c-Jun. Other signaling molecules with high connectivity include AKT and ERK1/2. The network contains six of the eight validated genes. Overall the AN(TF) network includes 67 of the 118 candidate objects and provides evidence for a central role of EGFR activation by sublytic MAC.
FIGURE 7.

Network of transcriptional regulation and ligand receptor signaling. This network uses a greater number of genes, including all those identified as significantly changed by combining significantly differentially expressed genes with those significantly up-regulated at both time points. The network was generated in MetaCore using the following options: AN(TF)-building algorithm with “add ligands and TF targets” selected. The algorithm generates a list of possible networks, with scores based on the number of seed nodes to non-seed nodes and the presence of canonical pathway threads. The network with the highest score for these two factors was selected and manually organized, first showing the four validated genes and then the most highly connected objects regardless of functional type to their right. The remaining objects were sorted by function so that TFs, kinases, phosphatases, generic proteins, and binding proteins were from left to right. Ligands and receptors were placed to the far left. Seed nodes are circled in navy blue, the predicted receptor trigger is highlighted in green, the predicted controlling TF is highlighted in red, lines represent interactions either transcriptional regulation or protein-protein associations; red = inhibition and green = activation.
Discussion
The role of C as a tumor promoter has attracted a great deal of attention over the last few years because of evidence for significant C activation in diverse tumors. MAC is suspected to be influential given its published activating and proliferative effects on nucleated cells (5, 6, 14); however, signaling mechanisms underlying many of these effects remain ill defined. We took a novel approach to understanding the role of the MAC, taking advantage of an available terminal pathway inhibitor, global gene expression technology, and systems biology methodology. Sublytic conditions were optimized using pNHS as a C source and OmCI to block terminal pathway activation. CT26 colon carcinoma cells were selected as a model tumor cell, and MAC-specific gene expression changes were mapped by microarray, qPCR, and network analysis. These approaches revealed a gene expression pattern in tumor cells exposed to sublytic MAC, which could significantly impact cell survival and proliferation as well as reshape surrounding extracellular matrix. The key findings were replicated in an unrelated tumor cell line, B16 melanoma.
Statistical analysis of array data comparing MAC-exposed cells with controls confirmed a set of expression changes; genes involved in Ca2+ and G-protein signal transduction (ITPRIP, RGS16), early response transcription factors (EGR1, EGR2), and inflammatory responses (IRF1) were significantly altered. Network analysis to map the interactions of genes up-regulated at the 1-h and 12-h time points highlighted 4 further expression changes in genes encoding proteins with extracellular localization, AREG, CXCL1, MMP3, and MMP13 genes; these were co-regulated by putative canonical signaling cascades including PKC, PI3k/AKT, JNK, Erk1/2, and p38.
The product of the AREG gene is the amphiregulin protein), an EGF-like ligand capable of triggering erbB2 activation (41). CXCL1 ligand is a potent neutrophil chemoattractant, important in infection and signals via CXCR2, a G-protein-coupled receptor (42). MMP3 and MMP13 both code for matrix metalloproteinases (MMPs), which function in extracellular matrix regulation and remodeling; they are important during development, wound healing, proliferation, and inflammation (43).
Validation by qPCR confirmed that the expression changes highlighted in gene array and supported by statistics and network building were real and robust. Critically, expression changes for two of these genes, AREG and CXCL1, closely replicated in an unrelated tumor cell line, B16. MMP expression in this line was extremely low so changes could not be replicated.
Network generation using these and the remaining statistically significant changes found interactions between all identified genes apart from FAM110C and highlighted the central importance of IRF1, EGR1, and EGR2 in mediating the changes. EGFR is noteworthy in that it is placed in between two starting NOs, amphiregulin and Rgs16, the three connected in an extracellular to nuclear direction, supportive of EGFR activation. Rgs16 is further connected to Egr1, Egr2, and Irf1 via c-Myc. Egr1 protein is known to positively regulate EGR2 gene expression, whereas Egr2 protein negatively regulates EGR1 (44, 45); Egr1 protein is reported to inhibit IRF1 expression (46). AP-1 and NFκB were both highlighted as possible transcriptional regulators in the network. The AP-1 and NFκB complexes are known to regulate MMP3, MMP13, and CXCL1 gene expression in mouse and human cells (47–52).
Ap-1 (a heterodimer of c-fos and c-jun) and NFκB are known to be responsive to MAC (53) and have been cited as important regulators of the response to sublytic C (6). In particular, c-fos is up-regulated rapidly in MAC-exposed cells and is linked to Ca2+ flux and MAPK (particularly ERK) involvement (54). MMP3 and MMP13 up-regulation has been described in MAC-attacked chondrocytes in human disease and an experimental model of osteoarthritis (55). Each of the canonical signaling cascades identified in the network, PKC, PI3k/AKT, JNK, Erk1/2, and p38 have been reported to be activated in cells exposed to sublytic MAC (6).
The larger network generated using genes significantly changed either at 1 or 12 h exposure alongside those up-regulated at both time points provided greater insight into mechanisms responsible for observed gene expression changes in CT26 cells. The network placed EGFR central in the response, an assignment supported by the presence of this receptor in all three generated networks (Fig. 7). EGFR is a member of the erbB receptor-tyrosine kinase family, activated by ligand binding at the cell surface triggering phosphorylation of the intracellular tyrosine kinase domain (56). Activation of the EGFR system has also been described in response to cellular stressors such as UV, osmotic, and oxidative stress (57). MAC may cause analogous stress responses; indeed, there is evidence that it can induce expression of the EGF ligand and cause EGFR signaling activation without ligand binding (58). The response may involve Gαi protein activation independent of receptor, which is known to be activated by MAC (59). Indeed, our data showing an RGS16 expression response to MAC supports this assertion; RGS16 gene expression is induced as a feedback mechanism for G-protein signaling (60). MAC can transactivate several other receptor-tyrosine kinases, including fibroblast growth factor receptor-2 (FGFR2) and hepatocyte growth factor receptor (HGFR) (58). Potentiation of EGFR activation may come from MMP cleavage and release of EGF family ligands such as heparin-binding EGF-like growth factor (HB-EGF) at the cell surface, a pathway supported by our demonstration of AREG up-regulation (61).
Increased expression of AREG, CXCL1, MMP3, and MMP13 is described at mRNA and protein levels in a number of human cancers such as breast, colorectal, ovarian, and pancreatic (62–69). This increased expression of the four effector genes often correlates with tumor development and aggressivity and can be predictive of patient prognosis (70–72). Their activities promote cell proliferation, activation, and motility through various mechanisms. AREG (amphiregulin) contributes to tumorigenesis via its function as a growth factor, the development of autocrine or juxtacrine loops that promote cell proliferation and survival, and increased cell motility (63, 71), and CXCL1 acts through recruitment of myeloid-derived suppressor cells to the tumor microenvironment where they support tumor growth and metastasis and suppress the local immune response (70). MMP family members contribute through their role in regulating the extracellular matrix (73), promoting angiogenesis, tumor invasion, and metastasis (72). In addition they cleave and activate molecules in the extracellular matrix that promote proliferation, motility, and induce alterations in adhesion (74). Increased expression of MMPs is associated with poor tumor differentiation, increased invasiveness, poor prognosis, increased likelihood of metastasis, and shorter survival time (72). In particular, MMP3 promotes tumor progression by releasing/activating E-cadherin, L-selectin, HB-EGF, and TNFα and is described as a central mediator of mammary tumorigenesis (69). MMP3 is reported to induce a stable epithelial to mesenchymal transition, a process that is closely linked to tumor development (75). MMP13 contribution to tumor promotion is mainly through its pro-angiogenic activity, increasing vascular density in tumors (76). Together, alterations in the expression of these four genes represent a powerful influence on tumor development. Induction of expression of these genes in response to MAC may, therefore, indicate a tumor-promoting role.
A role for C as a tumor promoting system has recently gained mainstream recognition (19). The work presented here represents a novel approach to uncover this relationship using global expression data and systems biology analysis to explore both the mechanisms and the characteristics of such a response. The approach has provided evidence to suggest that MAC deposition, which does not result in cell lysis, is a potent tumor cell activator leading to significant changes in gene expression in several critical and interlinked pathways. These data fit well with published piecemeal studies in diverse cell types. The work not only sheds light on the signaling cascades and responsive transcription factor systems that respond to MAC but also reveals a downstream gene expression response to MAC that will alter tumor behavior through induction of proliferative, migratory, and survival pathways. Interestingly, a central role for the EGFR system was identified, although it was not clear whether this was activated directly by MAC or indirectly after MAC exposure. Overall, this work provides additional evidence implicating sublytic MAC in tumor cell activation and has implications not only for our understanding of the tumor promoting effects of C but also for new approaches to cancer therapy.
Experimental Procedures
Materials
Pooled pNHS was obtained from whole blood collected from consenting volunteers. Blood was placed in 20-ml glass vials and allowed to clot. Serum was separated by centrifugation, pooled, 0.22-μm-filtered, and stored in aliquots at −80 °C. Sheep erythrocytes (ShEs) in Alsever's solution were purchased from TCS Biosciences (Buckingham, UK). Complement fixation diluent (CFD) was from Oxoid (Basingstoke, UK). Anti-ShE antiserum (Amboceptor) was from Siemens (Forchheim, Germany). CT26 mouse colon carcinoma and B16 mouse melanoma cell lines were from American Type Culture Collection (Manassas VA). RPMI 1640 medium (RPMI), fetal bovine serum (FBS), and additives and calcein-AM were obtained from Invitrogen. Complete medium comprised RPMI 1640 with 5% heat-inactivated FBS. All other chemicals were from Sigma.
Preparation of ShEA
ShEs were washed into CFD and resuspended at 4% (v/v) in CFD (10 ml total volume) at 37 °C. Amboceptor, diluted 1:2000 in 10 ml of CFD, was mixed with the ShE suspension and incubated for 30 min at 37 °C. The resultant antibody-sensitized sheep erythrocytes were washed and diluted to 2% (v/v) in CFD.
Hemolytic Assay
Hemolytic activity in pNHS was used as a measure of MAC formation, assessed by incubating (37 °C for 60 min) triplicate serum dilutions in CFD with an equal volume of ShEA in wells of a 96-well plate. No serum (CFD alone) and 100% lysis (CFD containing 0.1% Triton-X-100) controls in triplicate were included. Plates were spun, supernatant was transferred to a flat-bottomed 96-well plate, and absorbance measured at 410 nm using a FLUOstar OPTIMA plate reader (BMG Labtech, Aylesbury, UK). Percentage hemolysis was calculated using the following equation where FI indicates fluorescence intensity,
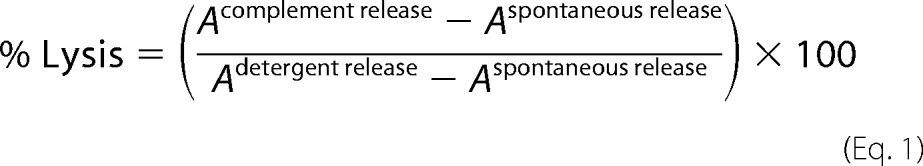 |
To titrate the effect of the C5 inhibitor OmCI (a gift from Dr. Miles Nunn) on MAC formation and hemolytic activity, aliquots of pNHS were preincubated with different doses of OmCI before measurement of hemolysis as above.
Complement-directed Cytoxicity Assay
We chose the well-described calcein release assay to measure tumor cell killing. The cell-permeant calcein AM is taken into cells and trapped by de-esterification to calcein; release of calcein from the cells then correlates with lytic cell death. CT26 cells or B16 cells were grown as monolayers in complete medium to 80% confluence in 75-cm2 tissue culture flasks then washed in saline and harvested by incubation in 10 mm EDTA in PBS (30 min). Harvested cells were washed in RPMI, diluted to a density of 5 × 105 cells per ml in complete medium, aliquoted at 100 μl/well into flat-bottomed 96-well plates, and incubated for 16 h at 37 °C in 5% CO2. Adherent cells were washed, and 100 μl of complete medium containing 2 μg/ml calcein AM was dispensed into each well. Plates were incubated for 1 h at 37 °C in 5% CO2. Calcein-loaded cell monolayers were washed twice in RPMI, then pNHS dilutions (0–40% in 100 μl of RPMI) were dispensed directly into wells and incubated for a further 1 h at 37 °C in 5% CO2. Supernatants were transferred to fresh 96-well plates, and fluorescence was measured (excitation, 485 nm; emission, 520 nm) in a Fluostar Optima plate reader. The remaining cells were lysed by the addition of 100 μl of 0.2% Triton-X-100 in RPMI per well, and released fluorescence was measured as above. Percentage lysis was calculated using the equation FI = fluorescence intensity,
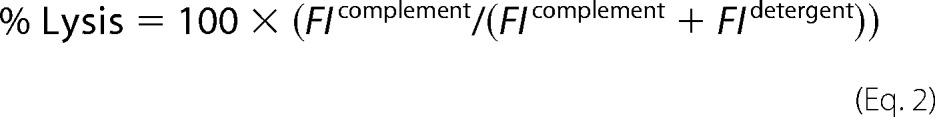 |
Titrating Sublytic C Attack
CT26 or B16 cell monolayers were washed with saline then incubated (37 °C, 5% CO2, 1 or 12 h) with pNHS at a dilution previously titrated to give <10% lysis at 1 h, a time point when maximum lytic killing has been reached, either in the presence or absence of a dose of OmCI that completely inhibited hemolytic activity (10 μg/ml). Monolayers were washed in RPMI, and RNA was harvested using the Genelute Mammalian Total RNA Miniprep kit (Sigma).
Global Gene Expression Analysis
RNA concentration and quality were measured using the Agilent 2100 Bioanalyzer (Agilent Technologies, Stockport, UK), and global gene expression analyses performed on the Illumina Microarray platform (Illumina, Saffron Walden, UK; Cardiff University Central Biotechnology Services). Amplification of material to generate cRNA and labeling was carried out according to manufacturer's instructions. Hybridization experiments were performed using the mouse ref8v2 BeadChips (2 × 8 samples) and analyzed using the iScan Reader and Control Software (Illumina). GenomeStudio Expression Module software (lllumina) was used to convert signal intensity data into expression data. Data were normalized using the quantile method and log-transformed (77). PCA and primary statistical analysis were performed using PartekGenomics Suite version 6.6 (build 6.13.0213, Partek Inc., Chesterfield, MO), and graphic representations were obtained using GeneSpring 12.0 GX (Agilent Technologies). Pathway analysis was performed using MetaCore software (Thomson Reuters, London, UK).
qPCR Analysis of mRNA
To validate differences in the relative expression of genes of interest implicated from gene expression analysis, extracted RNA samples (1 μg) were reverse-transcribed using TaqMan Reverse Transcription Reagents (Applied Biosystems, Paisley, UK) following the manufacturer's instructions; resulting cDNA was stored at −80 °C. For qPCR, sufficient cDNA for triplicate reactions of each primer pair diluted in ultrapure H2O, was mixed with 1× SYBR Green Jump Start Readymix (Sigma) according to manufacturer's instructions. Reaction mixes were aliquoted into 48-well white PCR plates, sealed with optical flat 8-cap strips (Bio-Rad), and placed in a MiniOpticon Real-Time PCR System (Bio-Rad) controlled using the Opticon Monitor 3.1 software. Thermocycling was adjusted from the manufacturer's protocol (65 °C annealing temperature and 40 cycles) to take account of relative expression, assessed using the ΔΔCt method where calculated Ct was the cycle number at which fluorescence crossed a threshold level selected as the point where PCR expansion was linear in all samples. The mean Ct values for the housekeeping genes β-actin and Polr2A were assessed, ΔΔCt was calculated for each sample, and results are expressed as a percentage of the control.
Author Contributions
B. P. M. and T. R. H. conceived the idea for the project. L. D. T. conducted most of the experiments and analyzed the results. R. A. W. performed confirmatory experiments with additional cell lines. B. P. M. and T. R. H. contributed to experimental design and data analysis at all stages. B. P. M., T. R. H., and L. D. T. contributed to writing of the manuscript.
Acknowledgments
We thank the members of the Complement Biology Group for support and input.
This work was supported by a Cardiff University School of Medicine Ph.D. studentship (to L. D. T.). The authors declare that they have no conflicts of interest relevant to the contents of this article.
- C
- complement
- MAC
- membrane attack complex; extracellular matrix
- ShE
- sheep erythrocytes
- ShEA
- antibody-sensitized sheep erythrocytes
- CFD
- complement fixation diluent
- pNHS
- pooled normal human serum
- AN(TF)
- analyze network (transcription factor)
- NO
- network object
- EGFR
- EGF receptor
- MMP
- matrix metalloproteinase
- PCA
- principal components analysis
- ANOVA
- analysis of variance
- OmCI
- O. moubata C inhibitor
- qPCR
- quantitative PCR.
References
- 1. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Walport M. J. (2001) Complement: first of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 3. Monk P. N., Scola A. M., Madala P., and Fairlie D. P. (2007) Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 152, 429–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Müller-Eberhard H. J. (1986) The membrane attack complex of complement. Annu. Rev. Immunol. 4, 503–528 [DOI] [PubMed] [Google Scholar]
- 5. Morgan B. P. (1989) Complement membrane attack on nucleated cells: resistance, recovery, and non-lethal effects. Biochem. J. 264, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cole D. S., and Morgan B. P. (2003) Beyond lysis: how complement influences cell fate. Clin. Sci. 104, 455–466 [DOI] [PubMed] [Google Scholar]
- 7. Taylor R. P., and Lindorfer M. A. (2014) The role of complement in mAb-based therapies of cancer. Methods 65, 18–27 [DOI] [PubMed] [Google Scholar]
- 8. Pio R., Ajona D., and Lambris J. D. (2013) Complement inhibition in cancer therapy. Semin. Immunol. 25, 54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niculescu F., Rus H. G., Retegan M., and Vlaicu R. (1992) Persistent complement activation on tumor cells in breast cancer. Am. J. Pathol. 140, 1039–1043 [PMC free article] [PubMed] [Google Scholar]
- 10. Lucas S. D., Karlsson-Parra A., Nilsson B., Grimelius L., Akerström G., Rastad J., and Juhlin C. (1996) Tumor-specific deposition of immunoglobulin g and complement in papillary thyroid carcinoma. Hum. Pathol. 27, 1329–1335 [DOI] [PubMed] [Google Scholar]
- 11. Yamakawa M., Yamada K., Tsuge T., Ohrui H., Ogata T., Dobashi M., and Imai Y. (1994) Protection of thyroid-cancer cells by complement-regulatory factors. Cancer 73, 2808–2817 [DOI] [PubMed] [Google Scholar]
- 12. Ytting H., Jensenius J. C., Christensen I. J., Thiel S., and Nielsen H. J. (2004) Increased activity of the mannan-binding lectin complement activation pathway in patients with colorectal cancer. Scand. J. Gastroenterol. 39, 674–679 [DOI] [PubMed] [Google Scholar]
- 13. Bjørge L., Hakulinen J., Vintermyr O. K., Jarva H., Jensen T. S., Iversen O. E., and Meri S. (2005) Ascitic complement system in ovarian cancer. Br. J. Cancer 92, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Markiewski M. M., DeAngelis R. A., Benencia F., Ricklin-Lichtsteiner S. K., Koutoulaki A., Gerard C., Coukos G., and Lambris J. D. (2008) Modulation of the antitumor immune response by complement. Nat. Immunol. 9, 1225–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corrales L., Ajona D., Rafail S., Lasarte J. J., Riezu-Boj J. I., Lambris J. D., Rouzaut A., Pajares M. J., Montuenga L. M., and Pio R. (2012) Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 189, 4674–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gunn L., Ding C., Liu M., Ma Y., Qi C., Cai Y., Hu X., Aggarwal D., Zhang H. G., and Yan J. (2012) Opposing roles for complement component C5A in tumor progression and the tumor microenvironment. J. Immunol. 189, 2985–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Barr S. A., Caguioa J., Gruol D., Perkins G., Ember J. A., Hugli T., and Cooper N. R. (2001) Neuronal expression of a functional receptor for the C5a complement activation fragment. J. Immunol. 166, 4154–4162 [DOI] [PubMed] [Google Scholar]
- 18. Rutkowski M. J., Sughrue M. E., Kane A. J., Mills S. A., and Parsa A. T. (2010) Cancer and the complement cascade. Mol. Cancer Res. 8, 1453–1465 [DOI] [PubMed] [Google Scholar]
- 19. Pio R., Corrales L., and Lambris J. D. (2014) The role of complement in tumor growth. in Tumor Microenvironment and Cellular Stress: Signaling, Metabolism, Imaging, and Therapeutic Targets (Koumenis C., Hammond E., and Giaccia A. eds.), pp 229–262, Springer-Verlag New York Inc., New York [Google Scholar]
- 20. Markiewski M. M., and Lambris J. D. (2009) Unwelcome Complement. Cancer Res. 69, 6367–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Markiewski M. M., and Lambris J. D. (2009) Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 30, 286–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fishelson Z., Donin N., Zell S., Schultz S., and Kirschfink M. (2003) Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 40, 109–123 [DOI] [PubMed] [Google Scholar]
- 23. Campbell A. K., Daw R. A., and Luzio J. P. (1979) Rapid increase in intracellular free Ca2+ induced by antibody plus complement. FEBS Lett. 107, 55–60 [DOI] [PubMed] [Google Scholar]
- 24. Lo T. N., and Boyle M. D. (1979) Relationship between the intracellular cyclic adenosine 3′:5′-monophosphate level of tumor cells and their sensitivity to killing by antibody and complement. Cancer Res. 39, 3156–3162 [PubMed] [Google Scholar]
- 25. Hänsch G. M., Seitz M., and Betz M. (1987) Effect of the late complement components-C5b-9 on human monocytes: release of prostanoids, oxygen radicals, and of a factor inducing cell-proliferation. Int. Arch. Allergy Appl. Immunol. 82, 317–320 [DOI] [PubMed] [Google Scholar]
- 26. Kilgore K. S., Shen J. P., Miller B. F., Ward P. A., and Warren J. S. (1995) Enhancement by the complement membrane attack complex of tumor necrosis factor-α-induced endothelial-cell expression of E-selectin and ICAM-1. J. Immunol. 155, 1434–1441 [PubMed] [Google Scholar]
- 27. Benzaquen L. R., Nicholson-Weller A., and Halperin J. A. (1994) Terminal complement proteins C5b-9 release basic fibroblast growth-factor and platelet-derived growth-factor from endothelial-cells. J. Exp. Med. 179, 985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fosbrink M., Niculescu F., Rus V., Shin M. L., and Rus H. (2006) C5b-9-induced endothelial cell proliferation and migration are dependent on Akt inactivation of forkhead transcription factor FOXO1. J. Biol. Chem. 281, 19009–19018 [DOI] [PubMed] [Google Scholar]
- 29. Wagner C., Braunger M., Beer M., Rother K., and Hänsch G. M. (1994) Induction of matrix protein-synthesis in human glomerular mesangial cells by the terminal complement complex. Exp. Nephrol. 2, 51–56 [PubMed] [Google Scholar]
- 30. Lueck K., Wasmuth S., Williams J., Hughes T. R., Morgan B. P., Lommatzsch A., Greenwood J., Moss S. E., and Pauleikhoff D. (2011) Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye 25, 1074–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Halperin J. A., Taratuska A., and Nicholson-Weller A. (1993) Terminal complement complex C5b-9 stimulates mitogenesis in 3T3 cells. J. Clin. Invest. 91, 1974–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rus H. G., Niculescu F., and Shin M. L. (1996) Sublytic complement attack induces cell cycle oligodendrocytes: S phase induction is dependent on c-jun activation. J. Immunol. 156, 4892–4900 [PubMed] [Google Scholar]
- 33. Hila S., Soane L., and Koski C. L. (2001) Sublytic C5b-9-stimulated Schwann cell survival through PI 3-kinase-mediated phosphorylation of BAD. Glia 36, 58–67 [DOI] [PubMed] [Google Scholar]
- 34. Liu L., Qiu W., Wang H., Li Y., Zhou J., Xia M., Shan K., Pang R., Zhou Y., Zhao D., and Wang Y. (2012) Sublytic C5b-9 Complexes induce apoptosis of glomerular mesangial cells in rats with Thy-1 nephritis through role of interferon regulatory factor-1-dependent caspase 8 activation. J. Biol. Chem. 287, 16410–16423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Triantafilou K., Hughes T. R., Triantafilou M., and Morgan B. P. (2013) The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913 [DOI] [PubMed] [Google Scholar]
- 36. Nunn M. A., Sharma A., Paesen G. C., Adamson S., Lissina O., Willis A. C., and Nuttall P. A. (2005) Complement inhibitor of C5 activation from the soft tick Ornithodoros moubata. J. Immunol. 174, 2084–2091 [DOI] [PubMed] [Google Scholar]
- 37. Hepburn N. J., Williams A. S., Nunn M. A., Chamberlain-Banoub J. C., Hamer J., Morgan B. P., and Harris C. L. (2007) In vivo characterization and therapeutic efficacy of a C5-specific inhibitor from the soft tick Ornithodoros moubata. J. Biol. Chem. 282, 8292–8299 [DOI] [PubMed] [Google Scholar]
- 38. Wai P. Y., Mi Z., Guo H., Sarraf-Yazdi S., Gao C., Wei J., Marroquin C. E., Clary B., and Kuo P. C. (2005) Osteopontin silencing by small interfering RNA suppresses in vitro and in vivo CT26 murine colon adenocarcinoma metastasis. Carcinogenesis 26, 741–751 [DOI] [PubMed] [Google Scholar]
- 39. Eisenhart C. (1947) The assumptions underlying the analysis of variance. Biometrics 3, 1–21 [PubMed] [Google Scholar]
- 40. Bessarabova M., Ishkin A., JeBailey L., Nikolskaya T., and Nikolsky Y. (2012) Knowledge-based analysis of proteomics data. BMC Bioinformatics 13, S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson G. R., Kannan B., Shoyab M., and Stromberg K. (1993) Amphiregulin induces tyrosine phosphorylation of the epidermal growth-factor receptor and p185(ERBB2): evidence that amphiregulin acts exclusively through the epidermal growth-factor receptor at the surface of human epithelial cells. J. Biol. Chem. 268, 2924–2931 [PubMed] [Google Scholar]
- 42. Chintakuntlawar A. V., and Chodosh J. (2009) Chemokine CXCL1/KC and its receptor CXCR2 are responsible for neutrophil chemotaxis in adenoviral keratitis. J. Interferon Cytokine Res. 29, 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nagase H., Visse R., and Murphy G. (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 69, 562–573 [DOI] [PubMed] [Google Scholar]
- 44. Kumbrink J., Kirsch K. H., and Johnson J. P. (2010) EGR1, EGR2, and EGR3 activate the expression of their coregulator NAB2 establishing a negative feedback loop in cells of neuroectodermal and epithelial origin. J. Cell. Biochem. 111, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fang F. (2011) The early growth response gene Egr2 (alias Krox20) is a novel transcriptional target of transforming growth factor-β that is up-regulated in systemic sclerosis and mediates profibrotic responses. Am. J. Pathol. 178, 2077–2090; Correction (2011) Am. J. Pathol.179, 537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. James A. B., Conway A. M., and Morris B. J. (2005) Genomic profiling of the neuronal target genes of the plasticity-related transcription factor-Zif268. J. Neurochem. 95, 796–810 [DOI] [PubMed] [Google Scholar]
- 47. Ahmad R., Sylvester J., and Zafarullah M. (2007) MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibits interleukin-1-induced matrix metalloproteinase-13 gene expression and promoter activity by impairing MAP kinase activation. Cell. Signal. 19, 2549–2557 [DOI] [PubMed] [Google Scholar]
- 48. Fleischmann A., Hafezi F., Elliott C., Remé C. E., Rüther U., and Wagner E. F. (2000) Fra-1 replaces c-Fos-dependent functions in mice. Genes Dev. 14, 2695–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shen F., Hu Z., Goswami J., and Gaffen S. L. (2006) Identification of common transcriptional regulatory elements in interleukin-17 target genes. J. Biol. Chem. 281, 24138–24148 [DOI] [PubMed] [Google Scholar]
- 50. Yee J., Kuncio G. S., Bhandari B., Shihab F. S., and Neilson E. G. (1997) Identification of promoter activity and differential expression of transcripts encoding the murine stromelysin-1 gene in renal cells. Kidney Int. 52, 120–129 [DOI] [PubMed] [Google Scholar]
- 51. Zenz R., Eferl R., Scheinecker C., Redlich K., Smolen J., Schonthaler H. B., Kenner L., Tschachler E., and Wagner E. F. (2008) Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 10, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liacini A., Sylvester J., Li W. Q., and Zafarullah M. (2002) Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1), and nuclear factor κB (NF-κB) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 21, 251–262 [DOI] [PubMed] [Google Scholar]
- 53. Viedt C., Hänsch G. M., Brandes R. P., Kübler W., and Kreuzer J. (2000) The terminal complement complex C5b-9 stimulates interleukin-6 production in human smooth muscle cells through activation of transcription factors NF-κB and AP-1. FASEB J. 14, 2370–2372 [DOI] [PubMed] [Google Scholar]
- 54. Badea T. D., Park J. H., Soane L., Niculescu T., Niculescu F., Rus H., and Shin M. L. (2003) Sublytic terminal complement attack induces c-fos transcriptional activation in myotubes. J. Neuroimmunol. 142, 58–66 [DOI] [PubMed] [Google Scholar]
- 55. Wang Q., Rozelle A. L., Lepus C. M., Scanzello C. R., Song J. J., Larsen D. M., Crish J. F., Bebek G., Ritter S. Y., Lindstrom T. M., Hwang I., Wong H. H., Punzi L., Encarnacion A., Shamloo M., et al. (2011) Identification of a central role for complement in osteoarthritis. Nat. Med. 17, 1674–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Olayioye M. A., Neve R. M., Lane H. A., and Hynes N. E. (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carpenter G. (1999) Employment of the epidermal growth factor receptor in growth factor-independent signaling pathways. J. Cell Biol. 146, 697–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cybulsky A. V., Takano T., Papillon J., and McTavish A. J. (1999) Complement C5b-9 induces receptor-tyrosine kinase transactivation in glomerular epithelial cells. Am. J. Pathol. 155, 1701–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Niculescu F., Rus H., and Shin M. L. (1994) Receptor-independent activation of guanine-nucleotide-binding regulatory proteins by terminal complement complexes. J. Biol. Chem. 269, 4417–4423 [PubMed] [Google Scholar]
- 60. Beadling C., Druey K. M., Richter G., Kehrl J. H., and Smith K. A. (1999) Regulators of G protein signaling exhibit distinct patterns of gene expression and target G protein specificity in human lymphocytes. J. Immunol. 162, 2677–2682 [PubMed] [Google Scholar]
- 61. Bolitho C., Hahn M. A., Baxter R. C., and Marsh D. J. (2010) The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr. Relat. Cancer 17, 929–940 [DOI] [PubMed] [Google Scholar]
- 62. D'Antonio A., Losito S., Pignata S., Grassi M., Perrone F., De Luca A., Tambaro R., Bianco C., Gullick W. J., Johnson G. R., Iaffaioli V. R., Salomon D. S., and Normanno N. (2002) Transforming growth factor α, amphiregulin and cripto-1 are frequently expressed in advanced human ovarian carcinomas. Int. J. Oncol. 21, 941–948 [PubMed] [Google Scholar]
- 63. Johnson G. R., Saeki T., Gordon A. W., Shoyab M., Salomon D. S., and Stromberg K. (1992) Autocrine action of amphiregulin in a colon carcinoma cell line and immunocytochemical localization of amphiregulin in human colon. J. Cell Biol. 118, 741–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. LeJeune S., Leek R., Horak E., Plowman G., Greenall M., and Harris A. L. (1993) Amphiregulin, epidermal growth-factor receptor, and estrogen-receptor expression in human primary breast-cancer. Cancer Res. 53, 3597–3602 [PubMed] [Google Scholar]
- 65. Rubie C., Frick V. O., Wagner M., Schuld J., Gräber S., Brittner B., Bohle R. M., and Schilling M. K. (2008) ELR plus CXC chemokine expression in benign and malignant colorectal conditions. BMC Cancer 8, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang G., Rosen D. G., Zhang Z., Bast R. C. Jr., Mills G. B., Colacino J. A., Mercado-Uribe I., and Liu J. (2006) The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 16472–16477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huang M.-Y., Chang H.-J., Chung F.-Y., Yang M.-J., Yang Y.-H., Wang J.-Y., and Lin S.-R. (2010) MMP13 is a potential prognostic marker for colorectal cancer. Oncol. Rep. 24, 1241–1247 [PubMed] [Google Scholar]
- 68. Morgia G., Falsaperla M., Malaponte G., Madonia M., Indelicato M., Travali S., and Mazzarino M. (2005) Matrix metalloproteinases as diagnostic (MMP-13) and prognostic (MMP-2, MMP-9) markers of prostate cancer. Urol. Res. 33, 44–50 [DOI] [PubMed] [Google Scholar]
- 69. Sternlicht M. D., Lochter A., Sympson C. J., Huey B., Rougier J. P., Gray J. W., Pinkel D., Bissell M. J., and Werb Z. (1999) The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Acharyya S., Oskarsson T., Vanharanta S., Malladi S., Kim J., Morris P. G., Manova-Todorova K., Leversha M., Hogg N., Seshan V. E., Norton L., Brogi E., and Massagué J. (2012) A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Castillo J., Erroba E., Perugorría M. J., Santamaría M., Lee D. C., Prieto J., Avila M. A., and Berasain C. (2006) Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. 66, 6129–6138 [DOI] [PubMed] [Google Scholar]
- 72. Deryugina E. I., and Quigley J. P. (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 25, 9–34 [DOI] [PubMed] [Google Scholar]
- 73. Egeblad M., and Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 74. Lukashev M. E., and Werb Z. (1998) ECM signalling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 8, 437–441 [DOI] [PubMed] [Google Scholar]
- 75. Lochter A., Galosy S., Muschler J., Freedman N., Werb Z., and Bissell M. J. (1997) Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 139, 1861–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kudo Y., Iizuka S., Yoshida M., Tsunematsu T., Kondo T., Subarnbhesaj A., Deraz E. M., Siriwardena S. B., Tahara H., Ishimaru N., Ogawa I., and Takata T. (2012) Matrix Metalloproteinase-13 (MMP-13) Directly and indirectly promotes tumor angiogenesis. J. Biol. Chem. 287, 38716–38728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bolstad B. M., Irizarry R. A., Astrand M., and Speed T. P. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 [DOI] [PubMed] [Google Scholar]