

Abstract
Nearly all members of the inwardly rectifying potassium (Kir) channel family share a cytoplasmic domain structure that serves as an unusual AP-1 clathrin adaptor-dependent Golgi export signal in one Kir channel, Kir2.1 (KCNJ2), raising the question whether Kir channels share a common Golgi export mechanism. Here we explore this idea, focusing on two structurally and functionally divergent Kir family members, Kir2.3 (KCNJ4) and Kir4.1/5.1 (KCNJ10/16), which have ∼50% amino identity. We found that Golgi export of both channels is blocked upon siRNA-mediated knockdown of the AP-1 γ subunit, as predicted for the common AP-1-dependent trafficking process. A comprehensive mutagenic analysis, guided by homology mapping in atomic resolution models of Kir2.1, Kir2.3, and Kir4.1/5.1, identified a common structure that serves as a recognition site for AP-1 binding and governs Golgi export. Larger than realized from previous studies with Kir2.1, the signal is created by a patch of residues distributed at the confluence of cytoplasmic N and C termini. The signal involves a stretch of hydrophobic residues from the C-terminal region that form a hydrophobic cleft, an adjacent cluster of basic residues within the N terminus, and a potential network of salt bridges that join the N- and C-terminal poles together. Because patch formation and AP-1 binding are dependent on proper folding of the cytoplasmic domains, the signal provides a common quality control mechanism at the Golgi for Kir channels. These findings identify a new proteostatic mechanism that couples protein folding of channels to forward trafficking in the secretory pathway.
Keywords: adaptor protein, clathrin, intracellular trafficking, potassium channel, protein trafficking (Golgi)
Introduction
The Golgi apparatus is the central biosynthetic sorting station in the cell, responsible for targeting newly synthesized membrane proteins to appropriate subcellular locales. In recent years, a new mechanistic understanding of Golgi export evolved from discoveries that some surface membrane-destined proteins can be selected for delivery to specific plasmalemma domains in signal and AP-1 clathrin adaptor-dependent manners (1–3) rather than by default, as once believed (4). The AP-1 clathrin adaptor belongs to a structurally diverse family of clathrin-associated sorting proteins (CLASPs) (5) that couple cargo selection to the formation of clathrin-coated vesicles at different membrane domains. AP-1 localizes to the trans-Golgi network (TGN)2 (6, 7), where it sorts specific subsets of membrane proteins into specialized tubulovesicular exit carriers. AP-1 was long believed to solely control trafficking between the Golgi and endocytic-lysosomal pathway (8), but recent studies revealed that AP-1 also selects subsets of membrane proteins for direct delivery to specialized surface membrane domains, including the basolateral membrane of polarized epithelial cells (3, 9), the myocyte sarcolemma and transverse tubule (10), and the somatodendritic membrane of neuronal cells (2, 11).
AP-1 is a heterotetrameric protein complex comprised of two large subunits, β1 and γ1, one medium subunit, μ1, and a small subunit, σ1 (12). The subunits assemble into a closed conformation until the complex associates with the TGN. Upon binding to phosphatidylinositol 4 phosphate (PIP4) (13) and the small GTPase Arf1 (14–16) at the TGN, AP-1 adopts an open conformation, which exposes clathrin binding sites on the hinge regions of the β1 and γ1 subunits (17–19) and cargo recognition sites on the μ1 and γ1σ1 subunits (2, 9, 11, 20). By simultaneously interacting with the TGN, clathrin, and specific proteins in the secretory pathway, AP-1 acts as a TGN-dependent scaffold, marking specific proteins as cargo for inclusion in clathrin-coated carriers for export to the cell surface.
Cargo selection is dictated by specific signals that serve as binding sites for the AP-1 adapter. Typically, AP-1 recognizes short peptide motifs that share a high degree of sequence similarity with tyrosine-based YXXΦ or dihydrophobically based [DE]XXXL[LI] endosomal sorting signals (X stands for any amino acid residue, and Φ stands for bulky hydrophobic residue) (21). YXXΦ signals bind to AP-1 through interaction with the μ subunit (2, 11), whereas dihydrophobic signals interact with the γ1σ1 subunits (22–24). These common types of Golgi sorting signals are often found in the cytoplasmic tail domains but can be located within more complex structures, particularly in polytypic integral membrane proteins. For example, in the chloride channel, ClC-2, Golgi export is dictated by a dihydrophobic signal that is embedded within the surface of its large cytosolic domain (9). In the inwardly rectifying potassium (Kir) channel, Kir2.1 (KCNJ2), the AP-1-dependent Golgi export signal is even more unusual and complex (10). Highly focused mutagenesis studies identified a patch structure within the tertiary structure of the Kir2.1 cytoplasmic domain that is necessary for AP-1 binding and Golgi export. The patch structure does not conform to the typical tyrosine or dihydrophobic trafficking signals. Although the binding site for AP-1 could potentially involve regions that were not screened, the finding indicated that AP-1 likely interacts with Kir2.1 through a completely novel signal.
Remarkably, residues in the Kir2.1 signal patch are highly conserved across the functionally diverse, 15-member Kir channel family, raising the major question of whether Kir channels share a common Golgi export mechanism. In this study, we address this question. A comprehensive, atomic structure-guided analysis of the Golgi export signal was performed in two functionally diverse, inwardly rectifying potassium channels, Kir2.3 (KCNJ4) and Kir4.1 (KCNJ10), which share only ∼50% overall sequence identity. We found that Kir2.3 and Kir4.1 have a common Golgi export signal that is larger than previously realized from studies with Kir2.1. The patch signal stretches across the cytoplasmic N and C termini to form a unique AP-1 binding site when the channels adopt an appropriate folding conformation.
Experimental Procedures
Plasmid Constructs and Antibodies
Modified mouse Kir channels, Kir2.3 (NM_008427) and Kir4.1 (NM_001039484), containing an HA tag at the external loop, were constructed as described previously (25). The pcDNA3.1 mammalian expression vector (Invitrogen) was used to express the cloned channel genes. The pGex5x-1 vector (GE Healthcare) was used to produce GST-Kir2.3 and Kir4.1 fusion proteins. Site-directed mutagenesis was carried out using a PCR-based strategy with the QuikChange site-directed mutagenesis kit (Stratagene, Agilent Technologies). All sequences were validated (Applied Biosystems, 3100). Primary antibodies included mouse anti-HA (Covance, catalog no. mms-101p), anti-GM130 (catalog no. 610822, BD Transduction Laboratories), and anti-γ-adaptin (catalog no. 610386, BD Transduction Laboratories) and rabbit anti-HA monoclonal (catalog no. 3724p, Upstate Cell Signaling Solutions) and α-tubulin (catalog no. 9099s, Upstate Cell Signaling Solutions). Secondary antibodies included HRP-conjugated goat anti-mouse or anti-rabbit (The Jackson Laboratory) and Alexa Fluor-conjugated goat anti-mouse or anti-rabbit antibodies (Molecular Probes). The specificity of the antibodies used in this study has been validated (10, 26).
Quantitative Chemiluminescence Detection of Surface Channel Proteins
Channel surface expression in COS-7 cells was quantified by chemiluminescence with a protocol adapted from previous studies (10, 26). Briefly, COS-7 cells at 60% to 70% confluence were transfected with external epitope-HA-tagged channel plasmids. After 40–48 h, cells were fixed (4% paraformaldehyde for 15 min) and blocked (PBS + 5% fetal bovine serum for 30 min) before incubation with mouse-anti-HA monoclonal antibody (25 μg/ml in PBS with 5% FBS) for 1 h at room temperature. After extensive washes with PBS (5 min, four times), cells were incubated with a secondary antibody (peroxidase-conjugated goat anti-mouse IgG) for 20 min. After extensive washes with PBS (5 min, four times), cells were scraped and resuspended in 500 μl of PBS. For each sample, 10 μl of resuspended cells was mixed with 100 μl of SuperSignal West Pico chemiluminescent substrate and incubated for 10 min prior to quantification. The chemiluminescence was quantified in relative light units by an analytic illuminometer (Berthold Detection Systems). Relative light unit values of mutant channels were normalized to the average of corresponding wild-type channels.
Immunocytochemistry and Fluorescence Microscopy
COS-7 cells transfected with external epitope-HA-tagged channel plasmids were fixed (4% paraformaldehyde for 15 min). To access cell surface and intracellularly localized channel proteins in the same cell, channel proteins on the cell surface were first labeled in non-permeabilized cells with mouse anti-HA monoclonal antibody (33 μg/ml) overnight at 4 °C and then stained with a secondary antibody (Alexa Fluor 488-conjugated goat anti-mouse IgG) at a saturating concentration (100 μg/ml). After extensive washes, cells were permeabilized (0.1% Triton X-100 in PBS), and intracellular channel proteins were labeled with rabbit anti-HA monoclonal antibody (0.5 μg/ml) and stained with a secondary antibody (Alexa Fluor 568-conjugated goat anti-rabbit IgG). Cells were examined using a ×40 objective lens by wide-field fluorescence microscopy (Olympus IX71). For co-localization analysis, cells were permeabilized. Channels and Golgi markers were labeled by antibodies raised in different hosts and stained with corresponding Alexa Fluor-conjugated secondary antibodies. Cells were examined using a ×40 oil lens and filters appropriate for fluorophores used by confocal microscopy (Zeiss LSM 510 Meta).
Protein Expression, Purification, and Pulldown Assays
GST fusion proteins of Kir channel cytoplasmic domains were constructed as before with Kir2.1 (10), following the approach pioneered by Nishida and MacKinnon (27) and Pegan et al. (28), who found that fusion of Kir cytoplasmic N and C termini appropriately fold into a structure that is identical to the cytoplasmic domain of the native Kir channels (Fig. 1A). Specifically, the Kir channel cytoplasmic N termini (amino acids 15–38 in Kir2.3, amino acids 24–47 in Kir4.1) were directly fused to the cytoplasmic C termini (amino acids 180–445 in Kir2.3, amino acids 175–379 in Kir4.1). GST was fused to the N termini of this construct. Proteins were produced in BL21-CodonPlus (DE3)-RIPL-competent cells (Agilent Technology), immobilized on glutathione beads, and washed with high-salt buffer (0.5 m NaCl, 25 mm HEPES, and 2.5 mm MgCl2 (pH 7.0)) to purify proteins to homogeneity. Proteins were suspended and stored in potassium acetate buffer (115 mm potassium acetate, 25 mm HEPES, and 2.5 mm MgCl2 (pH 7.0)). Clathrin adaptor-enriched rat brain extract was prepared as described previously (29). For the pulldown assay, purified GST fusion protein (2 μg) was incubated with the rat brain extract (300 μg) overnight and then washed (15 min, three times) with potassium acetate buffer supplemented with 5% Triton X-100 and 2 mm dithiothreitol at 4 °C. The specifically bound AP-1 clathrin adaptor complex was resolved on SDS-PAGE gels and visualized by Western blotting (Bio-Rad). Quantification of protein abundance was performed by scanning autofluorograms, followed by ImageJ analysis (National Institutes of Health).
FIGURE 1.
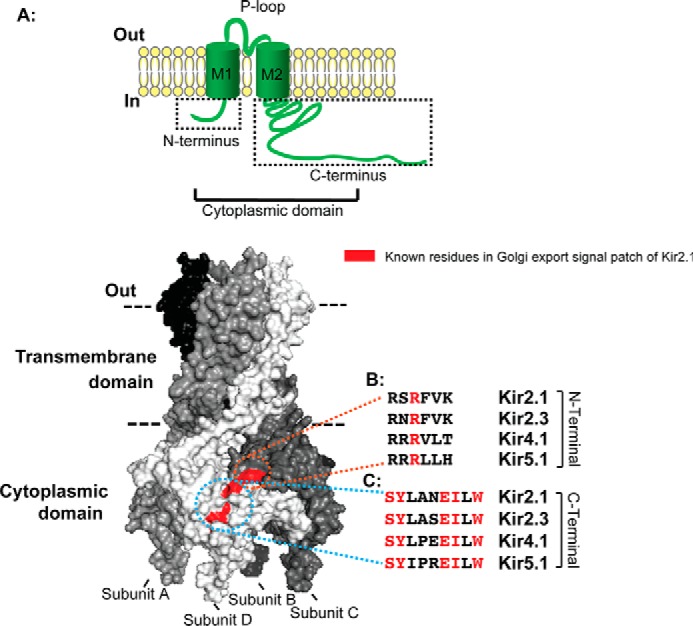
The Kir2.1 Golgi export signal patch is conserved in diverse Kir channels. A, schematic of a single Kir channel subunit. M1 and M2, transmembrane helix domains 1 and 2 with cytoplasmic N and C termini, which fold into a cytoplasmic domain. B and C, surface representation of a Kir2.1 tetramer model based on the crystal structure of chicken Kir2.2 (PDB code 3JYC) (47). The previously identified residues in the Golgi export signal patch of Kir2.1 with a surface-exposed side chain are marked in red. The Golgi export signal patch of Kir2.1 is embedded in the tertiary structure, minimally comprised of an arginine residue (R) from the N-terminal pole of the cytoplasmic domain of subunit C (B), juxtaposing a track of residues [SY]XXX[EI]X[W] from the C-terminal pole of the cytoplasmic domain of subunit D (C). Note that the known residues (red) of the Kir2.1 Golgi export signal patch are conserved in Kir2.1, 2.3, Kir4.1, and Kir5.1 (B and C).
RNA Interference
The double-stranded siRNA probes (siRNA-1, 5′-TAGCACAGGTTGCCACTAA-3′; siRNA-2, 5′-AGATTTGGATGTCTCAATA-3′; Dharmacon Inc.) corresponding to nucleotides 849–867 and1102–1120 of the human AP1γ-adaptin subunit mRNA, respectively, were used as before to specifically knock down AP-1 (10). These probes have no effect on the surface expression of influenza HA, a raft-associated transmembrane protein that leaves the Golgi through an AP-1-independent mechanism (10). COS-7 cells at 30–40% confluence were transfected with either a non-silencing scramble siRNA probe (Qiagen) or AP1γ-adaptin-targeted siRNA probes using X-tremeGene siRNA transfection reagent (Roche Diagnostics GmbH). After 24 h, the cells were co-transfected with corresponding siRNA probes and channel plasmids. After 40–48 h, cells were processed for immunoblot, immunocytochemistry, or surface antibody binding.
Quantitative Image Analysis
The degree of co-localized channels with Golgi markers were quantified and presented as Mander's coefficients (30). All analytical procedures were performed with Volocity 4.2 (Improvision). At least 30 cells from three separate transfections were analyzed and statistically compared.
Structure Models and Statistical Analysis
Structural homology models were generated based on available Protein Data Bank files (SWISS-MODEL) and visualized using PyMOL (Schrödinger, LLC). Data are presented as mean ± S.E. Statistical analysis was performed using GraphPad PRISM. Statistical significance was determined by unpaired t test when comparing two groups and by one-way randomized ANOVA followed by Tukey's post hoc test when comparing multiple groups or Dunnett's post hoc test when multiple test groups were compared with the control. p < 0.05 was considered significant.
Results
The Common “SY” Deletion Mutation Blocks Golgi Export of Diverse Kir Channels
To explore whether different members of the Kir channel family share a common Golgi export mechanism, we examined two conserved residues in Kir2.3 and Kir4.1, SY (Ser305-Tyr in Kir2.3, Ser299-Tyr in Kir4.1) that are positioned at the center of the Kir2.1 Golgi signal (Fig. 1, B and C). In Kir2.1, an Andersen-Tawil (Online Mendelian Inheritance in Man® (OMIM) no. 170390) disease-causing mutation that deletes SY (31), blocks Golgi export and prevents cell surface expression (10). Accordingly, we tested whether deletion of the SY sequence (ΔSY) in Kir2.3 and Kir4.1 causes a similar Golgi mistrafficking phenotype.
As measured by HA surface antibody binding and quantitative chemiluminescence of external HA epitope-tagged channels, deletion of the SY sequence blocked expression of Kir2.3 and Kir4.1 on the plasmalemma (Fig. 2A). Confocal microscopy of intact and permeabilized cells labeled with anti-HA antibodies revealed that both Kir2.3 and Kir4.1 ΔSY mutants largely accumulate in a perinuclear compartment (Fig. 2B). Co-localization with intracellular markers revealed that both Kir2.3 and 4.1 ΔSY mutants predominately reside in the Golgi (Fig. 3A). Quantitative co-localization analysis (30) revealed that ∼80% of the Kir2.3 and Kir4.1 ΔSY mutants co-localized with the cis-Golgi matrix protein GM130 (Fig. 3B). These observations are consistent with a shared Golgi export mechanism that is dependent on the conserved SY sequence in Kir2.1, Kir2.3, and Kir4.1.
FIGURE 2.
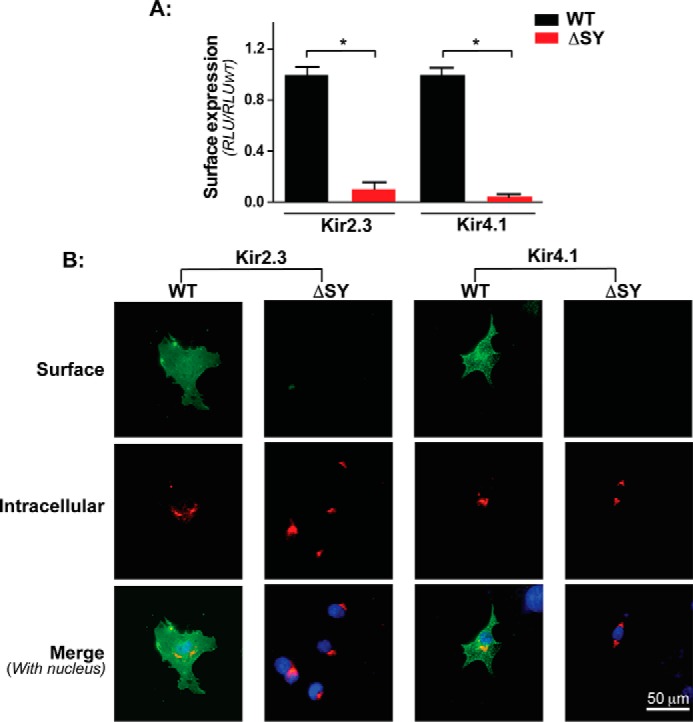
The common ΔSY mutation blocks channel surface expression. A, surface expression quantified by HA antibody binding and luminometry in COS7 cells transiently expressing external HA-tagged WT and ΔSY mutants of Kir2.3 and Kir4.1. *, p < 0.05 by unpaired t test. RLU, relative light unit. B, immunocytochemical analysis of external HA-tagged Kir2.3 and Kir4.1 channels in intact (green) and permeabilized (red) COS7 cells, counterstained with DAPI (blue) to show nuclei.
FIGURE 3.
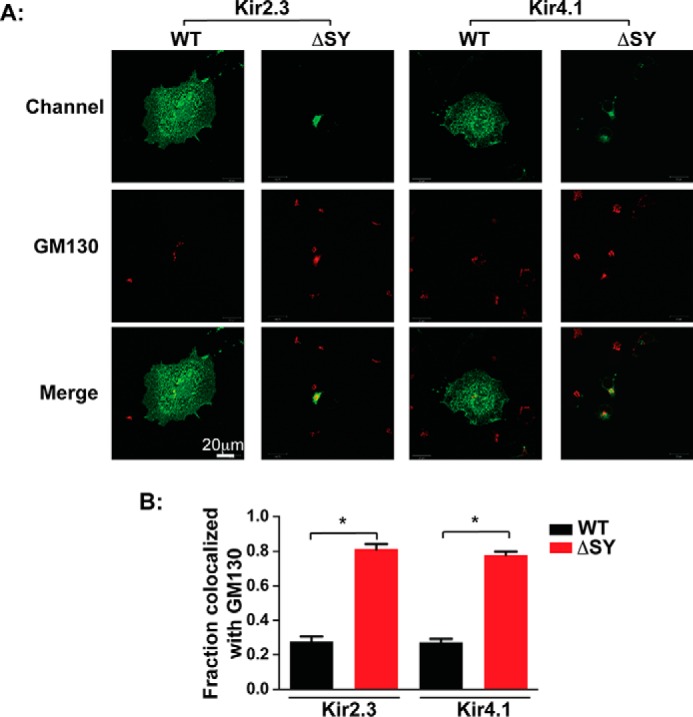
Kir2.3 and Kir4.1 bearing the ΔSY mutation accumulate in the Golgi. A, co-localization of Kir2.3 and Kir4.1 bearing the ΔSY mutation (green) with the Golgi marker GM130 (red) in permeabilized COS7 cells. B, quantification of co-localization. The fraction of co-localized channel is presented as Mander's coefficient (n = 30 cells from three separate transfections; *, p < 0.05 by one-way randomized ANOVA followed by Tukey's post hoc test).
Diverse Kir Channels Share Common Sequence Determinants for Golgi Export
Previously, the Golgi export signal in Kir2.1 was defined from a highly focused mutagenic screen as a patch of six residues at the confluence of cytoplasmic N and C termini, extending an ∼15-Å distance from SY (10). These residues are conserved in Kir2.3 and Kir4.1, suggesting that they may form part of a common recognition site for AP-1 binding. Because the footprint of AP-1 is potentially twice as large as the region originally screened in Kir2.1, we performed a more comprehensive analysis of the Golgi export signal. Homology mapping in atomic resolution models of Kir2.1, Kir2.3, and Kir4.1 identified 14 additional conserved residues that either have surface-exposed side chains or contribute to potential hydrophobic binding pockets within an ∼30-Å radius of Tyr315 (Fig. 5A), and these were selected for analysis in Kir2.3 (Fig. 4, A and C) and Kir4.1 (Fig. 4, B and D). Because Kir4.1 naturally forms heterotetrameric channels with Kir5.1 (32, 33) that are more efficiently expressed on the cell surface than Kir4.1 alone, we screened for the determinants of a Golgi export signal in the heterotetramer of Kir4.1/5.1 (Fig. 4, B and D). For these studies, an external HA tag was placed in Kir4.1, and heterotetrameric channels bearing mutations at comparable positions in both Kir4.1 and Kir5.1 subunits were examined. Alanine replacement mutations that block channel expression at the cell surface and cause the channel to accumulate in the Golgi were considered to be determinants of the Golgi export signal (Fig. 4, A–D).
FIGURE 5.

The Golgi export signal patch in Kir2.3 and Kir4.1. A, surface representation of a Kir2.1 tetramer model. Tyr315 is marked (orange) to show the center of the Golgi export signal patch in Kir2.1. Yellow, known surface-exposed residues in the Golgi export signal patch of Kir2.1; blue, additional candidates with a surface-exposed side chain for Golgi export sequence determinants (∼30-Å radius). B and C, surface representation of the entire cytoplasmic domain of Kir2.3 and Kir4.1 tetramer models. D and E, ribbon display of two subunits against a surface rendering of the other two subunits for Kir2.3 and Kir4.1. Relevant residues are color-coded.
FIGURE 4.

Golgi export sequence determinants of Kir2.3 and Kir4.1 elucidated by structure-guided mutagenesis. Residue candidates for the Golgi export sequence determinants were subjected to alanine replacement mutagenesis. A and B, cell surface expression of external HA-tagged Kir2.3 and Kir4.1 channels as quantified by surface HA antibody binding (n = 3). RLU, relative light unit. C and D, quantification of Kir2.3 and Kir4.1 co-localization with the Golgi marker GM130 (n = 30 cells from three individual transfections, the fraction of co-localized channel is presented as Mander's coefficient). Red bars, Golgi export sequence determinants; orange text, previously identified residues in the Kir 2.1 Golgi export signal. *, p < 0.05 by one-way randomized ANOVA followed by Dunnett's post hoc test.
The analysis revealed a conserved Golgi export patch in Kir2.3 and Kir4.1/5.1 that is larger and more complex than previously realized from studies with Kir2.1. In addition to the conserved residues in the Kir2.1 Golgi export patch that were found to be necessary for Golgi export in Kir2.3 and Kir4.1/5.1, the comprehensive mutagenic screen revealed other Golgi export determinants, expanding potential recognition sites for AP-1 binding (Fig. 5, B–E). As observed in Kir2.1, the Kir2.3 and Kir4.1/5.1 patch signal is formed in part by a common cluster of hydrophobic residues that extend diagonally from the equator of the cytoplasmic domain toward the apex of Kir2.3 and Kir4.1/5.1 (Ile311-X-Trp313 in Kir 2.3, Ile305-X-Trp307 in Kir 4.1). This structure joins newly identified residues (Leu222-Ile in Kir2.3, Leu217-Leu in Kir4.1) that extend from the base of the C terminus to create a previously unrecognized inverted V-shaped hydrophobic cleft. The patch signal also involves a cluster of basic residues in the N terminus (Arg20 and Lys23-Lys in Kir2.3, Arg27-Arg-Arg and Lys33 in Kir4.1) that juxtapose the hydrophobic cleft. The screen revealed a potential salt bridge network that brings N- and C-terminal elements of the signal together. As recognized previously, an arginine-glutamic acid pair (Arg20-Glu310 in Kir2.3, Arg29-Glu304 in Kir4.1) is at the center of the network. More complex than previously appreciated, other potential salt bridges may be involved. In particular, a neighboring N-terminal lysine (Lys24 in Kir2.3, Lys33 in Kir4.1) likely interacts with the C-terminal glutamic acid (Glu310 in Kir2.3, Glu304 in Kir4.1), and a nearby C-terminal glutamic acid (Glu284 in Kir2.3, Glu278 in Kir4.1) may interact with the N-terminal arginines. In Kir 2.3, Arg20 is required for Golgi export and may interact with Glu284. Although mutation of the equivalent residue in Kir 4.1 (Arg29) did not alter Golgi trafficking, any one of the neighboring arginines [Arg-Arg-Arg29], which are collectively required for Golgi export, may support a salt bridge with Glu278 when Arg29 is mutated.
Although the Golgi export signal patch is highly conserved between Kir2.1, Kir2.3, and Kir 4.1/5.1, a few channel-specific elements were identified. For example, an arginine residue (Arg209 in Kir2.3, Arg204 in Kir4.1) in the Golgi export patch of Kir2.3 is dispensable in Kir4.1. In fact, the R204A mutation in Kir4.1 actually increased Kir4.1 expression at the cell surface. The other difference between Kir2.3 and Kir4.1/5.1 relates to the requirements of two phenylalanine residues (Phe186/Phe283 in Kir2.3, Phe181/Phe277 in Kir4.1) for Kir2.3 Golgi export. The comparable residues in Kir4.1 are also required for surface expression (Fig. 4B), but we could not detect an accumulation of Kir4.1 at the Golgi when these residues were mutated to alanine (Fig. 4D).
Importantly, the common patch signal does not include typical AP-1 binding motifs. A tyrosine in a potential YXXΦ motif was reported to be necessary for Golgi export of Kir2.1 (34), although the requirements for the terminal hydrophobic residue were not studied. Our evaluation of the comparable YXXΦ sequence in Kir2.3 revealed that the comparable Φ residue (Leu236) is not necessary for Golgi export (Fig. 4, A and C). Furthermore, Kir4.1/5.1 channels lack the YXXΦ sequence and exit the Golgi in an AP-1-dependent manner (see below). Taken together, our findings indicate that export of Kir channels from the Golgi is driven by a common patch structure that has no resemblance to known trafficking signals.
The Common Kir Signal Patch Is a Binding Site for AP-1
To explore whether the shared Golgi export determinants in Kir2.3 and Kir4.1 form a recognition site for AP-1 clathrin adaptor interaction, protein-protein interaction studies were performed. Bacterial GST fusion proteins of the entire Kir2.3 and Kir4.1 cytoplasmic domains were immobilized on glutathione beads, incubated with a cytoplasmic lysate enriched with the AP-1 clathrin adaptor (29), and washed extensively, and bound material was subjected to Western blotting analysis with antibodies to the AP-1 γ subunit. As shown in Fig. 6, the WT GST-Kir2.3 and Kir4.1 cytoplasmic domains (Fig. 6A) interact with the AP-1 clathrin adaptor, whereas the ΔSY mutation abolished this interaction, consistent with a common signal-dependent interaction (Fig. 6, B and C). To test whether the interaction of AP-1 requires the patch of residues that are necessary for Golgi export, a comprehensive mutagenesis analysis was performed. As predicted for the formation of an AP-1-dependent Golgi trafficking signal, alanine replacement mutations of residues that are required for Golgi export (Lys33, Leu218, Glu278 and Glu304, Ile305, and Trp307 in Kir4.1) blocked the interaction with the AP-1 clathrin adaptor (Fig. 6, E and F). Thus, the shared Golgi export determinants collectively form the AP-1 binding site and define an AP-1-dependent trafficking signal.
FIGURE 6.

Common Golgi export sequence determinants in Kir channels are required for AP-1 interaction. A, GST and GST fusion proteins of the entire Kir2.3 and 4.1 cytoplasmic domain (WT and ΔSY mutants) following gel electrophoresis and Coomassie Brilliant Blue staining. MW, molecular weight. B, ΔSY mutations abolished AP-1 γ subunit binding. AP-1 clathrin adaptors bound to the Kir2.3 and Kir4.1 cytoplasmic domains were detected by immunoblot (IB) with an AP-1 γ subunit-specific antibody. ΔSY mutations abolished AP-1 γ subunit binding. C and D, quantification of relative AP-1 binding to Kir2.3 and Kir4.1 (n = 3; *, p < 0.05 by unpaired t test). E, mutations of the common Golgi export sequence determinants in Kir4.1 disrupt AP-1 γ subunit interaction. F, quantification of relative AP-1 binding to Kir4.1 (n = 3; *, p < 0.05 by one-way randomized ANOVA followed by Dunnett's post hoc test).
Kir 2.3 and Kir4.1 Are Exported from the Golgi in an AP-1-dependent Manner
To test whether the AP-1 clathrin adaptor is required for the export of Kir2.3 and Kir4.1 from the Golgi, trafficking of Kir2.3 and Kir4.1 was examined following siRNA-mediated knockdown of the AP-1 γ subunit. Two different γ subunit siRNA probes, together with a non-silencing scramble siRNA probe, were used. As revealed by immunoblotting, they both reduce γ subunit protein abundance without suppressing other proteins, such as α-tubulin (Fig. 7A), or altering the Golgi structure (see the characteristic localization of GM130; Fig. 7, D and E). Quantification of surface channel expression revealed a significant reduction of the Kir2.3 and Kir4.1 channels at the cell surface (Fig. 7, B and C). As evidenced by co-localization analysis with the Golgi matrix protein GM130 (35), ablation of the AP-1 γ subunit led to the accumulation of Kir2.3 and Kir4.1 in the Golgi (Fig. 7, D and E). Quantitative analysis further revealed that knockdown of the AP1 γ subunit significantly increased the fraction of the channel that co-localizes with GM130, identical to the Golgi-mistrafficking phenotype of mutant channels lacking the Golgi export signal (Fig. 7, F and G). Taken together, these findings reveal that Kir channels contain a common Golgi export patch signal in the cytoplasmic domain that serves as a recognition site for AP-1 clathrin adaptor binding.
FIGURE 7.

Golgi export of Kir channels requires the AP-1 clathrin adaptor. A, Western blots of AP-1, γ subunit and alpha-tubulin in COS-7 cells treated with two different AP-1 γ siRNA probes (AP1 γ-1 and AP1 γ-2) as well as scramble siRNA as the negative control. MW, molecular weight. B and C, AP-1 γ knockdown by each probe reduced the surface expression of Kir2.3 and Kir4.1 as measured by surface antibody binding (n = 6; *, p < 0.05 by one-way randomized ANOVA followed by Dunnett's post hoc test). RLU, relative light unit. D and E, cellular localization of HA-tagged Kir2.3 and Kir4.1 (green) and the Golgi marker GM130 (red) in permeabilized COS-7 cells after transfection with the indicated RNAi probes. Arrowheads, cells where channel co-localized with GM130. F and G, quantification of Kir2.3 and Kir4.1 co-localization with the Golgi marker GM130, presented as the fraction of co-localized channel with GM130, in COS-7 cells transfected with the indicated RNAi probes (n = 20 cells from three individual transfections; *, p < 0.05 by one-way randomized ANOVA followed by Dunnett's post hoc test).
Discussion
Our comprehensive analysis of Golgi export in divergent members of the Kir channel family that do not share canonical signaling motifs revealed a novel AP-1-dependent Golgi export signal. Unlike short peptide trafficking motifs, a patch of residues within the cytoplasmic domain, at the interface between the N and C termini, forms the AP-1 binding signal in Kir 2.1, Kir 2.3, and Kir4.1/5.1. Remarkably, most Kir channels share a similar patch structure, raising the possibility that this unusual AP-1-dependent Golgi export mechanism is shared among many members of the channel family.
Conserved Golgi Export Signal in Kir Channels
We found that the signal patch is more complex than previously revealed from our previous studies with Kir 2.1 (10) (Fig. 5). The patch signal has two chemically distinct poles. A cluster of shared residues extends ∼20 Å from the equator of the cytoplasmic domain toward the apex, forming an inverted V-shaped hydrophobic cleft with a neighboring pair of hydrophobic residues that extend from the base of the cytoplasmic domain. The other pole involves an adjacent cluster of basic residues that are brought into juxtaposition with the hydrophobic cleft by salt bridging. It is possible that the salt-bridging residues in the patch structure control proper folding, whereas the others form the recognition site for AP-1 interaction.
The unique arrangement of the patch signal residues at the confluence of cytoplasmic domains implies that the signal is a conformational structure, sharing some similarities with the “conformational epitopes” identified previously in the SNARE proteins Sec22 and VAMP8 (36, 37). Indeed, the vesicle-sorting signals of SNARE proteins are embedded in the tertiary structure and are conformation-dependent. Incorporation of Sec22 into coat protein complex II (COPII)-coated vesicles relies on a transport signal that is exposed when the protein aquires an unassembled closed conformation (36). Similarly, the retrieval of VAMP8 from the plasmalemma into the clathrin-coated vesicles depends on a folded epitope that is exposed only when the protein is not assembled into the SNARE complex (37). In the Kir channels, conformation-dependent formation of the Golgi export signal provides a mechanism to ensure that only properly folded channels exit the Golgi, offering an additional quality control checkpoint beyond the well known control processes in the endoplasmic reticulum (38).
The Shared Golgi Export Patch as an AP-1 clathrin Adaptor Recognition Site
Our protein-protein interaction studies identify the signal patch as the recognition site for AP-1 clathrin adaptor binding. Further studies will be required to identify the complementary binding site for the channel on the AP-1 complex. Two distinct binding pockets for the canonical trafficking signals have been described. The site for the [Y]XX[Φ] signal, which resides in the AP-1 μ1 subunit (39), is comprised of two hydrophobic pockets that separately interact with the tyrosine and Φ bulky hydrophobic residue. The dihydrophobic [DE]XXXL[LI] motif-binding site resides in the AP-1 γ1σ1 subunits (24, 40). In σ1, a hydrophobic pocket serves as the docking site for the L[LI] part of the signal, whereas a cluster of basic residues at the boundary of γ and σ1 bind the acidic residue (Asp/Glu) resides. Our previous studies with Kir2.1 revealed that the signal patch binds to the AP-1 γ1σ1 hemicomplex rather than μ1 or β subunits (10). Given that the signal patch has no resemblance to typical dihydrophobic trafficking signals, it is logical to suggest that the corresponding binding site on AP-1 γ1σ1 for Kir channels is distinct. The AP-1 γ1σ1 complex has several structures that are complementary to the patch signal. It will be interesting to test whether they form the binding site.
Complexity of Golgi Export in Kir Channels
It remains to be determined how widely the Golgi export mechanism is shared across the Kir channel family. Although all Kir channels, except for Kir7.1 (KCNJ13), share the Golgi export patch identified here, it seems likely that the constitutive Golgi export mechanism might be different for some Kir channels. The Kir1.1 (KCNJ1) channel, for example, requires phosphorylation of a residue that is near the patch signal for trafficking in the biosynthetic pathway (41–43), suggesting that formation of the Golgi export signal might be regulated. For KATP channels it is unlikely that the signal patch is used at all. Formation of the fully functional KATP channels at the cell surface requires the assembly of the pore-forming Kir subunits Kir6.1 (KCNJ8)/Kir6.2 (KCNJ11) with the accessory subunits sulfonylurea receptor Sur1/Sur2 (44). The arrangement of subunits predicts that the Golgi export signal patch in Kir6.1 or Kir6.2 would be masked by the Sur subunits, suggesting that KATP channels exit the Golgi by another mechanism. It is conceivable that binding of 14-3-3 proteins and phosphorylation, which are required for KATP trafficking from the endoplasmic reticulum to the Golgi (45, 46), govern the export of these channels from the Golgi.
In summary, here we uncovered a shared AP-1 clathrin adaptor trafficking mechanism that controls the export of several Kir channel proteins from the Golgi. We found that a highly conserved patch of residues that are embedded within the tertiary structure of all but one member of the Kir channel family serves as a unique binding site for AP-1. We propose that the shared conformational sorting signal provides a proteostatic mechanism for most Kir channels, ensuring that only properly folded channels exit the Golgi for delivery to the cell surface membrane.
Author Contributions
P. A. W. and X. L. conceived and coordinated the study, designed the experiments, and wrote the paper. X. L., B. O., and B. K. performed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful for excellent service provided by confocal microscopy and Biopolymer/genomics core facility at the University of Maryland School of Medicine, technical advice regarding generating the recombinant proteins provided by Dr. Donghui Ma at Origene, and proofreading of the final manuscript by Richard Coleman and P. Richard Grimm.
This study was supported in part by funds from the NIDDK, National Institutes of Health (DK063049, DK054231, and DK063049 to P. A. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TGN
- trans-Golgi network
- Kir
- inwardly rectifying potassium channel
- ANOVA
- analysis of variance.
References
- 1. Deborde S., Perret E., Gravotta D., Deora A., Salvarezza S., Schreiner R., and Rodriguez-Boulan E. (2008) Clathrin is a key regulator of basolateral polarity. Nature 452, 719–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Farías G. G., Cuitino L., Guo X., Ren X., Jarnik M., Mattera R., and Bonifacino J. S. (2012) Signal-mediated, AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron 75, 810–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gravotta D., Carvajal-Gonzalez J. M., Mattera R., Deborde S., Banfelder J. R., Bonifacino J. S., and Rodriguez-Boulan E. (2012) The clathrin adaptor AP-1A mediates basolateral polarity. Dev. Cell 22, 811–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Traub L. M., and Kornfeld S. (1997) The trans-Golgi network: a late secretory sorting station. Curr. Opin. Cell Biol. 9, 527–533 [DOI] [PubMed] [Google Scholar]
- 5. Traub L. M., and Bonifacino J. S. (2013) Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 5, a016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fölsch H., Pypaert M., Schu P., and Mellman I. (2001) Distribution and function of AP-1 clathrin adaptor complexes in polarized epithelial cells. J. Cell Biol. 152, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fölsch H., Pypaert M., Maday S., Pelletier L., and Mellman I. (2003) The AP-1A and AP-1B clathrin adaptor complexes define biochemically and functionally distinct membrane domains. J. Cell Biol. 163, 351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonifacino J. S., and Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 9. de la Fuente-Ortega E., Gravotta D., Perez Bay A., Benedicto I., Carvajal-Gonzalez J. M., Lehmann G. L., Lagos C. F., and Rodríguez-Boulan E. (2015) Basolateral sorting of chloride channel 2 is mediated by interactions between a dileucine motif and the clathrin adaptor AP-1. Mol. Biol. Cell 26, 1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma D., Taneja T. K., Hagen B. M., Kim B. Y., Ortega B., Lederer W. J., and Welling P. A. (2011) Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell 145, 1102–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mattera R., Farías G. G., Mardones G. A., and Bonifacino J. S. (2014) Co-assembly of viral envelope glycoproteins regulates their polarized sorting in neurons. PLoS Pathog. 10, e1004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boehm M., and Bonifacino J. S. (2001) Adaptins: the final recount. Mol. Biol. Cell 12, 2907–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y. J., Wang J., Sun H. Q., Martinez M., Sun Y. X., Macia E., Kirchhausen T., Albanesi J. P., Roth M. G., and Yin H. L. (2003) Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114, 299–310 [DOI] [PubMed] [Google Scholar]
- 14. Ren X., Farías G. G., Canagarajah B. J., Bonifacino J. S., and Hurley J. H. (2013) Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stamnes M. A., and Rothman J. E. (1993) The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 73, 999–1005 [DOI] [PubMed] [Google Scholar]
- 16. Traub L. M., Ostrom J. A., and Kornfeld S. (1993) Biochemical dissection of AP-1 recruitment onto Golgi membranes. J. Cell Biol. 123, 561–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doray B., and Kornfeld S. (2001) Gamma subunit of the AP-1 adaptor complex binds clathrin: implications for cooperative binding in coated vesicle assembly. Mol. Biol. Cell 12, 1925–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. ter Haar E., Harrison S. C., and Kirchhausen T. (2000) Peptide-in-groove interactions link target proteins to the β-propeller of clathrin. Proc. Natl. Acad. Sci. U.S.A. 97, 1096–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shih W., Gallusser A., and Kirchhausen T. (1995) A clathrin-binding site in the hinge of the β 2 chain of mammalian AP-2 complexes. J. Biol. Chem. 270, 31083–31090 [DOI] [PubMed] [Google Scholar]
- 20. Jain S., Farías G. G., and Bonifacino J. S. (2015) Polarized sorting of the copper transporter ATP7B in neurons mediated by recognition of a dileucine signal by AP-1. Mol. Biol. Cell 26, 218–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonifacino J. S. (2014) Adaptor proteins involved in polarized sorting. J. Cell Biol. 204, 7–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janvier K., Kato Y., Boehm M., Rose J. R., Martina J. A., Kim B. Y., Venkatesan S., and Bonifacino J. S. (2003) Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 γ-sigma1 and AP-3 δ-σ3 hemicomplexes. J. Cell Biol. 163, 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doray B., Lee I., Knisely J., Bu G., and Kornfeld S. (2007) The γ/σ1 and α/σ2 hemicomplexes of clathrin adaptors AP-1 and AP-2 harbor the dileucine recognition site. Mol. Biol. Cell 18, 1887–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mattera R., Boehm M., Chaudhuri R., Prabhu Y., and Bonifacino J. S. (2011) Conservation and diversification of dileucine signal recognition by adaptor protein (AP) complex variants. J. Biol. Chem. 286, 2022–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma D., Zerangue N., Lin Y. F., Collins A., Yu M., Jan Y. N., and Jan L. Y. (2001) Role of ER export signals in controlling surface potassium channel numbers. Science 291, 316–319 [DOI] [PubMed] [Google Scholar]
- 26. Ma D., Tang X. D., Rogers T. B., and Welling P. A. (2007) An Andersen-Tawil syndrome mutation in Kir2.1 (V302M) alters the G-loop cytoplasmic K+ conduction pathway. J. Biol. Chem. 282, 5781–5789 [DOI] [PubMed] [Google Scholar]
- 27. Nishida M., and MacKinnon R. (2002) Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111, 957–965 [DOI] [PubMed] [Google Scholar]
- 28. Pegan S., Arrabit C., Zhou W., Kwiatkowski W., Collins A., Slesinger P. A., and Choe S. (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8, 279–287 [DOI] [PubMed] [Google Scholar]
- 29. Le Borgne R., Griffiths G., and Hoflack B. (1996) Mannose 6-phosphate receptors and ADP-ribosylation factors cooperate for high affinity interaction of the AP-1 Golgi assembly proteins with membranes. J. Biol. Chem. 271, 2162–2170 [DOI] [PubMed] [Google Scholar]
- 30. Manders E. M. M., Verbeek F. J., and Aten J. A. (1993) Measurement of colocalization of objects in dual-color confocal images. J. Microsc-Oxford 169, 375–382 [DOI] [PubMed] [Google Scholar]
- 31. Plaster N. M., Tawil R., Tristani-Firouzi M., Canún S., Bendahhou S., Tsunoda A., Donaldson M. R., Iannaccone S. T., Brunt E., Barohn R., Clark J., Deymeer F., George A. L. Jr., Fish F. A., Hahn A., et al. (2001) Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 105, 511–519 [DOI] [PubMed] [Google Scholar]
- 32. Zhang C., Wang L., Zhang J., Su X. T., Lin D. H., Scholl U. I., Giebisch G., Lifton R. P., and Wang W. H. (2014) KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc. Natl. Acad. Sci. U.S.A. 111, 11864–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lourdel S., Paulais M., Cluzeaud F., Bens M., Tanemoto M., Kurachi Y., Vandewalle A., and Teulon J. (2002) An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J. Physiol. 538, 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hofherr A., Fakler B., and Klöcker N. (2005) Selective Golgi export of Kir2.1 controls the stoichiometry of functional Kir2.x channel heteromers. J. Cell Sci. 118, 1935–1943 [DOI] [PubMed] [Google Scholar]
- 35. Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T. E., and Warren G. (1995) Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131, 1715–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mancias J. D., and Goldberg J. (2007) The transport signal on Sec22 for packaging into COPII-coated vesicles is a conformational epitope. Mol. Cell 26, 403–414 [DOI] [PubMed] [Google Scholar]
- 37. Miller S. E., Sahlender D. A., Graham S. C., Höning S., Robinson M. S., Peden A. A., and Owen D. J. (2011) The molecular basis for the endocytosis of small R-SNAREs by the clathrin adaptor CALM. Cell 147, 1118–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ellgaard L., and Helenius A. (2003) Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191 [DOI] [PubMed] [Google Scholar]
- 39. Owen D. J., and Evans P. R. (1998) A structural explanation for the recognition of tyrosine-based endocytotic signals. Science 282, 1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kelly B. T., McCoy A. J., Späte K., Miller S. E., Evans P. R., Höning S., and Owen D. J. (2008) A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 456, 976–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Connell A. D., Leng Q., Dong K., MacGregor G. G., Giebisch G., and Hebert S. C. (2005) Phosphorylation-regulated endoplasmic reticulum retention signal in the renal outer-medullary K+ channel (ROMK). Proc. Natl. Acad. Sci. U.S.A. 102, 9954–9959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoo D., Kim B. Y., Campo C., Nance L., King A., Maouyo D., and Welling P. A. (2003) Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J. Biol. Chem. 278, 23066–23075 [DOI] [PubMed] [Google Scholar]
- 43. Yoo D., Fang L., Mason A., Kim B. Y., and Welling P. A. (2005) A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J. Biol. Chem. 280, 35281–35289 [DOI] [PubMed] [Google Scholar]
- 44. Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 [DOI] [PubMed] [Google Scholar]
- 45. Arakel E. C., Brandenburg S., Uchida K., Zhang H., Lin Y. W., Kohl T., Schrul B., Sulkin M. S., Efimov I. R., Nichols C. G., Lehnart S. E., and Schwappach B. (2014) Tuning the electrical properties of the heart by differential trafficking of KATP ion channel complexes. J. Cell Sci. 127, 2106–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zerangue N., Schwappach B., Jan Y. N., and Jan L. Y. (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22, 537–548 [DOI] [PubMed] [Google Scholar]
- 47. Tao X., Avalos J. L., Chen J., and MacKinnon R. (2009) Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 326, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]