

Abstract
Cytoplasmic protein aggregates are one of the pathological hallmarks of neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Several RNA-binding proteins have been identified as components of inclusion bodies. Developmentally regulated RNA-binding protein 1 (Drb1)/RNA-binding motif protein 45 is an RNA-binding protein that was recently described as a component in ALS- and FTLD-related inclusion bodies. However, the molecular mechanism underlying cytoplasmic Drb1 aggregation remains unclear. Here, using an in vitro cellular model, we demonstrated that Drb1 co-localizes with cytoplasmic aggregates mediated by TAR DNA-binding protein 43, a major component of ALS and FTLD-related inclusion bodies. We also defined the domains involved in the subcellular localization of Drb1 to clarify the role of Drb1 in the formation of cytoplasmic aggregates in ALS and FTLD. Drb1 predominantly localized in the nucleus via a classical nuclear localization signal in its carboxyl terminus and is a shuttling protein between the nucleus and cytoplasm. Furthermore, we identify a double leucine motif serving as a nuclear export signal. The Drb1 mutant, presenting mutations in both nuclear localization signal and nuclear export signal, is prone to aggregate in the cytoplasm. The mutant Drb1-induced cytoplasmic aggregates not only recruit TAR DNA-binding protein 43 but also decrease the mitochondrial membrane potential. Taken together, these results indicate that perturbation of Drb1 nuclear-cytoplasmic trafficking induces toxic cytoplasmic aggregates, suggesting that mislocalization of Drb1 is involved in the cause of cytotoxicity in neuronal cells.
Keywords: ALS (Lou Gehrig disease); neurodegenerative disease; protein aggregation; RNA binding protein; TAR DNA-binding protein 43 (TDP-43, TARDBP); cytoplasmic inclusion body; frontotemporal lobar degeneration (FTLD); nuclear-cytoplasmic trafficking
Introduction
ALS,2 one of the most devastating and untreatable neurodegenerative diseases, affects motor neurons selectively with relentless progression and causes respiratory failure within 2–5 years from onset. Although 5–10% of ALS cases are familial, ∼90% of cases are sporadic with unknown etiology (1). In contrast, frontotemporal lobar degeneration (FTLD) is a group of neurodegenerative diseases that affect frontal and temporal cortices with characteristic cognitive defects such as personality and behavioral changes as well as progressive deterioration of language skills (2). A subgroup of patients presents overlapped clinical characteristics of ALS and FTLD. Cytoplasmic inclusion bodies in neuronal cells are a common pathological hallmark in ALS and FTLD, implying that a common etiological pathway exists in ALS and FTLD pathogenesis (1, 3). TAR DNA-binding protein 43 (TDP-43), an RNA-binding protein, was the first protein identified in cytoplasmic inclusion bodies in most sporadic ALS and several FTLD cases (4, 5). Since then, other RNA-binding proteins, such as fused in sarcoma/translated in liposarcoma (FUS/TLS), and heterogeneous nuclear ribonucleoproteins were also identified as components of cytoplasmic inclusion bodies. Because disease-associated mutations in the TDP-43 or FUS/TLS gene were identified in several familial ALS and FTLD cases (6–9), TDP-43 and FUS/TLS are supposed to play a crucial role in ALS and FTLD pathogenesis.
Common features in TDP-43 pathology are its mislocalization from the nucleus to the cytoplasm, the formation of cytoplasmic aggregates, and protein modifications such as ubiquitination, phosphorylation, and truncation (4). TDP-43 contains two RNA recognition motifs (RRMs) and plays important roles in RNA metabolism such as RNA processing, exporting, and stabilizing and translational regulation between the nucleus and cytoplasm (10). Under normal conditions, TDP-43 predominantly localizes in the nucleus and rapidly shuttles between the nucleus and cytoplasm (11). Nuclear-cytoplasmic trafficking is mediated by a nuclear localization signal (NLS) upstream of the first RRM and a nuclear export signal (NES) in the second RRM (11). Either disease-linked or artificial mutations in TDP-43 NLS are known to induce protein redistribution in the cytoplasm of cultured cells (12–14). In addition to mislocalization, protein misfolding is thought to be essential for the formation of cytoplasmic inclusion bodies (15). Most mutations causing familial ALS are recognized in the glycine-rich domain, a prion-like domain at the carboxyl-terminal region of TDP-43 that mediates protein-protein interaction (16–18).
We previously identified developmentally regulated RNA-binding protein 1 (Drb1), also known as RNA-binding motif protein 45 (RBM45), and suggested its critical role in neural development (19). The gene encoding Drb1 is highly conserved evolutionarily, and its ortholog exists in many species, including fruit fly, zebrafish, frog, chicken, mouse, and human. In the rat, Drb1 is highly expressed in the early embryonic brain, and its expression gradually tapers during the development. Drb1 contains three or four RRM-type RNA-binding domains and is suggested to bind to the GACGAC motif in a large analysis of the sequence specificity of RNA-binding proteins (20). Drb1 was recently identified as a protein that is statistically elevated in the cerebrospinal fluid of patients with sporadic and familial ALS. Additionally, cytoplasmic co-aggregation of Drb1 with TDP-43 was observed in patients with ALS and FTLD (21, 22). Using a yeast two-hybrid system, it was determined that the carboxyl-terminal region of TDP-43 interacts with Drb1 (23). Drb1 is also redistributed from the nucleus to the cytoplasm by oxidative stresses such as H2O2 and paraquat and modulates the antioxidant response via cytoplasmic interaction with Kelch-like ECH-associated protein 1 (KEAP1), an inhibitor of the antioxidant response transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) (24). Despite these findings in relation to ALS pathology, the mechanism underlying the formation of Drb1 cytoplasmic aggregation remains unclear.
In this study, to unveil the role of Drb1 in ALS/FTLD pathology, we investigated the molecular mechanism of co-localization of Drb1 with TDP-43 in cytoplasmic aggregates in a cellular model. First, we elucidated the subcellular localization of Drb1 and identified its NLS and NES. We then evaluated Drb1 subcellular distribution when its NLS and/or NES were abrogated. Surprisingly, mutations in both NLS and NES but not in a single domain of Drb1 tended to induce TDP-43-positive cytoplasmic aggregates with cytotoxicity. Our findings indicate that a novel mechanism related to Drb1 subcellular localization causes neuronal toxicity.
Experimental Procedures
Plasmid Construction
The plasmids used in this study were produced in the pcDNA3 (Invitrogen), pEF-Bos-T7 (25), pEGFP (Clontech, Tokyo, Japan), and pGEX-6p-1 (GE Healthcare) vectors, and each was fused with EGFP, Clover (Addgene plasmid 40259), a T7 tag, a myc tag, or GST at the N terminus and with mRuby2 (Addgene plasmid 40260) at the C terminus. EGFP, Clover, and mRuby2 were engineered from GFP and red fluorescent protein, respectively (26). To produce the construct for T7-tag-Drb1-WT, full-length Drb1 cDNA, amino acids 2–474, was amplified by PCR using a pair of primers (Drb-BamF, 5′-ATGGATCCTGGACGAAGCTGGCAGCTC-3′ (forward) and Drb-BamR, 5′-ATGGATCCTCAGTAAGTTCTTTGCCGTTTG-3′ (reverse); underlined text represents BamHI sites) from a HeLa cDNA library and inserted into the BamHI site of the pEF-Bos-T7 vector. pcDNA3-Clover-T7-tag-Drb1-WT was generated by PCR using pEF-Bos-T7-tag-Drb1-WT as a template with a pair of primers (T7-XbaF, 5′-AGGTCTAGAATGGCCAGCATGACTGGTGG-3′ (forward) and T7-XbaR, 5′-AAATCTAGAGGTGGGGACCCTCACTCTAG-3′ (reverse); underlined text represents XbaI sites), and the amplified fragment was inserted into the XbaI site of pcDNA3-Clover. A deletion mutant of Clover-T7-tag-Drb1-2–400 was generated by PCR using pcDNA3-Clover-T7-tag-Drb1-WT as a template with a pair of primers (T7-XbaF (forward) and 5′-CGGTCTAGACTAAGGATGAGGATTAAACACAATA-3′ (reverse); the underline represents the XbaI site) to insert the mutated cDNA into the XbaI site of pcDNA3-Clover. The other deletion mutants of Clover-T7-tag-Drb1-, 401–474, 2–300, 301–474, and 301–400 were generated by the following two-step subcloning. First, the deletion mutants of pEF-Bos-T7-Drb1 were generated by PCR using pcDNA3-Clover-T7-tag-Drb1-WT as a template with the following primer pairs to insert the mutated cDNA into the BamHI site of pEF-Bos-T7: 401–474, 5′-CGAGGATCCCTTTACCTTTAGACGTATTAGA-3′ (forward) and Drb-BamR (reverse); 2–300, Drb-BamF (forward) and 5′-ATGGATCCTCATGCATAAATAGCTGATGC-3′ (reverse); 301–474, 5′-GCAGGATCCAAAAATACAAATTACATGGATT-3′ (forward) and Drb-BamR (reverse); and 301–400, 5′-GCAGGATCCAAAAATACAAATTACATGGATT-3′ (forward) and 5′-CGGGGATCCTAAGGATGAGGATTAAACACAATA-3′ (reverse); the underlines represent BamHI sites. Then each of the deletion mutants of pcDNA3-Clover-T7-tag-Drb1 was generated by PCR using each of the deletion mutants of pEF-Bos-T7-Drb1 as a template with the pair of primers T7-XbaF and T7-XbaR to insert each of the mutated cDNA into the XbaI site of the pcDNA3-Clover construct. To create the substitution mutants of Clover-T7-tag-Drb1, including mtNLSc1 (K454A and R456A) and mtLL (LL329,330AA), the overlap extension PCR method (27) was performed using pcDNA3-Clover-T7-tag-Drb1-WT and its deletion mutants as templates with the following two sets of primers: For mtNLSc1, first, T7-XbaF (forward) and 5′-CTGCCAGCATAACTGCAAGTGCCACCCCATTCAGAATCTTTC-3′ (reverse) and second, 5′-CTGAATGGGGTGGCACTTGCAGTTATGCTGGCAGATTCGCC-3′ (forward) and T7-XbaR (reverse) and for mtLL; first, T7-XbaF (forward) and 5′-TGCCATTTTTCTAGCGGCATCTGTTGCATTACTTCCATC-3′ (reverse) and second, 5′-TAATGCAACAGATGCCGCTAGAAAAATGGCAACAC-3′ (forward) and T7-XbaR (reverse), where boldface letters represent mutation sites. Substitution mutations of mtNLSc2 (K469A, R470A, and R472A) and R470G were generated by single PCR with the following primer pairs (mtNLSc2, 5′-TAGGGATCCTCAGTAAGTTGCTTGCGCTGCGTTAGATTCTTCTCTTGGCG-3′ (forward) and Drb-BamR (reverse); R470G, 5′-AAGGATCCTCAGTAAGTTCTTTGCCCTTTGTTAGATTCTTCTC-3′ (forward) and Drb-BamR (reverse); underlined text represents BamHI sites and boldface letters indicate mutation sites) to insert the mutated cDNA into pEF-Bos-T7 constructs. Then each of the substitution mutants of pcDNA3-Clover-T7-tag-Drb1 was generated from each of the substitution mutants of pEF-Bos-T7-tag-Drb1 with the same method as the deletion mutants of pcDNA3-Clover-T7-tag-Drb1. The mtNLSc1+c2 construct was generated by additional NLSc1 mutagenesis for the NLSc2 construct. To generate pGEX-6p-1-Drb1-WT, the Drb1-WT cDNA fragment was amplified from pEF-Bos-T7-tag-Drb1-WT using a set of primers (5′-CGGGATCCGACGAAGCTGGCAGCTCTGCGAGC-3′ (forward) and 5′-CCGCTCGAGTCAGTAAGTTCTTTGCCGTTTGTTAGA-3′ (reverse); underlined text represents BamHI and XhoI sites, respectively). The amplified fragment was inserted into the BamHI/XhoI site of pGEX-6p-1. The pcDNA3-TDP-43 and pEGFP-TDP-43 expression plasmids and its mutants were provided by Dr. T. Nonaka (described in Ref. 12). pcDNA3-TDP-43-WT-mRuby2 was generated by PCR using pcDNA3-TDP-43 as a template with a pair of primers (5′-TAAGGATCCGTCTGAATATATTCGGGTAAC-3′ (forward) and 5′-ATCGGATCCATTCCCCAGCCAGAAGAC-3′ (reverse); underlined text indicates BamHI sites) to insert the TDP-43 fragments into the BamHI site of pcDNA3-mRuby2. pcDNA3-Clover-Myc-tag-NPc-NLS was generated by PCR using pcDNA3-Myc-tag-NPc-NLS-REV-NES (a gift from Dr. T. Fujiwara (28)) as a template with a pair of primers (5′-AAGTCTAGAATGGGAGAGCAGAAACTGATC-3′ (forward) and 5′-CACTCTAGATCACACTTTGCGTTTCTTTTTTGG-3′ (reverse); underlined text indicates XbaI sites) to insert the mutated cDNA into the XbaI site of pcDNA3-Clover. All constructs were confirmed by DNA sequencing.
Cell Culture and Transfection
HeLa cells and NIH3T3 cells were cultured in high-glucose DMEM (Wako, Tokyo, Japan) supplemented with 10% (v/v) fetal bovine serum (Japan Bio Serum) and 1% (v/v) penicillin/streptomycin (Invitrogen) in 5% CO2 at 37 °C. SH-SY5Y cells were cultured in DMEM supplemented with 15% (v/v) FBS and 1% (w/v) penicillin/streptomycin. HeLa cells and SH-SY5Y cells were transfected using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Cells were transfected for 24 h unless otherwise indicated. For stable transfection of HeLa or SH-SY5Y cells, pcDNA3-Clover-T7-Drb1 or pcDNA3-Clover-NPc-NLS was co-transfected with pBluescriptII-SK+ to adjust DNA amounts. The transfected cells were typically split 1:10 into selection medium 2 days post-transfection. Geneticin (Gibco) was added to the medium at a final concentration of 500 μg/ml for the selection and the maintenance of stably expressing cells.
Immunofluorescence Imaging Analysis
Cells cultured in poly-l-lysine-coated 35-mm glass bottom dishes (Matsunami Glass, Osaka, Japan) were fixed with 4% (w/v) paraformaldehyde at room temperature for 15 min, followed by permeabilization with ice-cold 100% methanol for 10 min, and then washed with PBS three times at room temperature. Hoechst 33258 (1:2000, Dojindo, Tokyo, Japan) was used for nuclear staining. For immunofluorescence staining, fixed cells were treated with blocking solution (PBS with 5% (v/v) horse serum (Sigma) and 0.02% (w/v) sodium azide) for 45 min at room temperature and then incubated with the primary antibody diluted in PBS at 4 °C overnight. After three washes with PBS, the cells were incubated at room temperature for 2 h with the secondary antibody in blocking solution and then washed three times with PBS. Fluorescence images were obtained using a confocal laser-scanning microscope (TCS-SP5, Leica, Tokyo, Japan) and processed using ImageJ (National Institutes of Health, Bethesda, MD).
Co-immunoprecipitation
Cells were washed once with ice-cold PBS and lysed in immunoprecipitation (IP) buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1% (v/v) Nonidet P-40, protease inhibitor mixture (Nacalai, Kyoto, Japan), phosphatase inhibitor mixture (Sigma), and Benzonase nuclease (Novagen, Madison, WI)). The lysates were sheared by passing through 22- and 27-gauge needles five times and incubated at 4 °C for 30 min. Cell debris was removed by centrifugation at 20,400 × g for 20 min at 4 °C. The supernatant was incubated with the antibody overnight at 4 °C, followed by incubation with protein A or G magnetic beads (Invitrogen) pretreated with PBS with 5% (w/v) skim milk. After washing the beads with IP buffer three times, the beads were boiled with 2 × sample buffer containing 10% (v/v) β-mercaptoethanol for 5 min at 90 °C. The extracted proteins were separated on 12% (w/v) SDS-polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Millipore, Tokyo, Japan) using a semidry blotting apparatus. The membranes were analyzed by immunoblot assay using a rabbit anti-GFP antibody (1:3000, TP401, Torrey Pines Biolabs, San Diego, CA), a mouse anti-T7 antibody (1:5000, 69522-3, Novagen), a goat anti-T7 antibody (1:1000, NB600-371, Novus Biologicals, Littleton, CO), or a mouse anti-TDP-43 antibody (1:10,000, H00023435-M01, 2E2-D3 clone, raised for the 1–260 amino acids of human TDP-43, Abnova, Taipei, Taiwan). Horseradish peroxidase-conjugated (anti-rabbit, NA9340V, and anti-mouse, NA931V, GE Healthcare) secondary antibodies were used. Positive bands on the blots were visualized using ECL chemiluminescence reagent (GE Healthcare).
GST Pulldown Assay
35S-labeled TDP-43 (predicted size, 43 kDa) was generated by in vitro translation using the TnT T7 Quick for PCR DNA-coupled transcription/translation system (Promega, Tokyo, Japan) as described in the instructions of the manufacturer. Template DNA was generated from the pcDNA3-TDP-43 plasmid by PCR using a set of primers (forward, 5′-GGCAGTACATCTACGTATTAGTCATCGC-3′; reverse, 5′-ATCGCAGTGATTCCTAGTCGACTGAGTCATCTACATTCCCCAGCCAGAAG-3′). One hundred ng of the template was used for the standard 50-μl reaction in the presence of 20 μCi of EasyTag L-[35S]methionine (PerkinElmer Japan Co., Ltd., Kanagawa, Japan). The reaction was incubated for 60 min at 30 °C, followed by Benzonase nuclease treatment for 20 min. Recombinant GST-fused Drb1 (predicted size, 79 kDa) and GST alone were produced in C41(DE3) (Lucigen) and XL-2 bacterial cells, respectively, and purified using glutathione-Sepharose 4B beads (GE Healthcare) after treatment with Benzonase nuclease. GST recombinant protein (∼5 μg) was rebound to glutathione beads in 200 μl of buffer D (20 mm HEPES-KOH (pH 8.0), 100 mm KCl, 5% (v/v) glycerol, 0.2 mm EDTA, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Nacalai)). After combination with 10 μl of 35S-labeled TDP-43, the reaction was incubated for 1 h at 4 °C with rotation. Glutathione beads were washed six times with buffer D. Pulldown proteins were eluted in 2× sample buffer containing 100 mm DTT by boiling at 80 °C for 3.5 min. The eluted proteins were subjected to 12% (w/v) SDS-polyacrylamide gel electrophoresis and visualized by staining using EZblue gel stain reagent (Sigma). After gel-drying and exposure to the imaging plate (Fujifilm, Tokyo, Japan), 35S-labeled protein was detected by a Typhoon 9410 scanner (GE Healthcare).
Heterokaryon Assay
Interspecies heterokaryon assays were performed as described previously (29) using hemagglutinating virus of Japan envelope, an inactivated Sendai virus extract (Ishihara Sangyo Co., Tokyo, Japan). Murine NIH3T3 cells plated in poly-l-lysine-coated 35-mm glass bottom dishes and Clover-fused protein stably expressing human HeLa or SH-SY5Y cells were pretreated with 50 μg/ml cycloheximide (CHX) to inhibit protein synthesis for 4 h at 37 °C. After an additional 30 min-treatment with 100 μg/ml CHX, HeLa or SH-SY5Y cells (2 × 105 cells) were collected in tubes, mixed with 4 μl hemagglutinating virus of Japan envelope and 25 μl PBS and placed on ice for 5 min. After addition of 200 μl PBS, the human cell suspension with hemagglutinating virus of Japan envelope was plated on the PBS-rinsed NIH3T3 cell dishes. The cell mixture was incubated for 20 min at 37 °C, and then fresh medium containing 50 μg/ml CHX was added to the dish. After 2 h of incubation, the cells were fixed as described above and stained with Hoechst 33258 to verify the murine or human nuclei. Phalloidin (Life Technologies), staining actin filaments, was used to verify cell fusion. The cells were observed with a confocal laser-scanning microscope (Leica) and images were captured using LAS AF software. The regions of murine nuclei and cytoplasm in each heterokaryon cell were set using the drawing tool, and the average Clover intensity values of each region were measured with LAS AF software. The average intensity values of each murine nucleus were then corrected with those of the cytoplasm in the same heterokaryon cell and plotted on a box plot graph. The cutoff value to classify cells with shuttling of Drb1 or cells without shuttling was defined as the maximum value of the negative control protein, and the percentages of shuttling cells in non-neuronal or neuronal cells were evaluated.
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential (Δψm) was evaluated using tetramethylrhodamine ethyl ester (TMRE, Life Technologies) according to the instructions of the manufacturer. HeLa cells overexpressing Clover or Clover-Drb1 fusion protein (WT, R470G, or mtLL/R470G) were plated in poly-l-lysine-coated 35-mm glass bottom dishes, and the following assay was performed. At 24 h post-transfection, cells were treated with 100 ng/ml TMRE in culture medium containing 1 μg/ml Hoechst 33342 (Dojindo) for 20 min and rinsed twice with PBS. After filling the dish with prewarmed phenol red-free Opti-MEM, cells were observed by using a confocal laser-scanning microscope (Leica), and the images were captured with LAS AF software. To quantify Δψm, the same treatment was performed for cells plated in 24-well dishes (Sumitomo Bakelite, Tokyo, Japan), and the images were captured by IN Cell Analyzer 1000 (GE Healthcare). All Clover-expressing cells were selected to evaluate Δψm values using IN Cell Developer Toolbox software (GE Healthcare). Cells expressing Clover-Drb1-mtLL/R470G were classified into two groups: cells with cytoplasmic aggregates and cells without cytoplasmic aggregates. Average Δψm intensity values per unit area of each cell were measured, and the box plot graph was drawn. More than 250 cells were counted in each group.
Statistical Analysis
Drawing of box plot graphs and Fisher's exact test, Mann-Whitney U test, Kruskal-Wallis rank-sum test, and Steel-Dwass test were performed using Easy R statistical software v. 1.29 (30). In box plot graphs, the bottom of each box was the 25th percentile (Q1), the top of each box was the 75th percentile (Q3), and the line in the middle was the median value. Interquartile range (IQR = Q1 − Q3) is shown as a box. The top and bottom whiskers are defined as 1.5 × IQR + Q3 or 1.5 × IQR − Q1. The outliers from the whiskers are depicted as dots.
Results
Drb1 Co-localizes with TDP-43 Mutant-induced Cytoplasmic Aggregates
To elucidate the mechanism underlying the co-localization of Drb1 into TDP-43-positive cytoplasmic inclusions in ALS pathology, we established a cellular model of HeLa cells to form disease-like cytoplasmic aggregates using EGFP-fused TDP-43 (Fig. 1A). An artificial deletion mutant of TDP-43, TDP-mt, forms cytoplasmic aggregates in human neuroblastoma SH-SY5Y cells possessing similar biochemical features as those of ALS motor neurons (12). EGFP-TDP-43-WT dominantly localized in the nucleus of HeLa cells, whereas EGFP-TDP-mt localized in both the nucleus and cytoplasm with some cytoplasmic aggregates (Fig. 1B, a–f), similar to those in SH-SY5Y cells (12). In HeLa cells, T7-tagged Drb1 (Fig. 1A) dominantly localized in the nucleus with no cytoplasmic aggregates (Fig. 1B, g–i). When cells were co-transfected with T7-Drb1-WT and EGFP-TDP-mt, T7-Drb1-WT co-localized with TDP-mt-induced cytoplasmic aggregates and presented a partial nucleoplasmic co-localization (Fig. 1B, j–l). To confirm the ability of Drb1 to associate with TDP-43 in HeLa cells, co-immunoprecipitation experiments were performed using nuclease-treated whole lysates extracted from cells transiently expressing T7-Drb1-WT co-transfected with EGFP, EGFP-TDP-43-WT, or EGFP-TDP-mt. Immunoprecipitation with rabbit polyclonal anti-GFP antibody showed that both EGFP-TDP-43-WT and -mt could precipitate T7-Drb1-WT whereas lysates expressing only EGFP as a control did not (Fig. 1C). Conversely, immunoprecipitation with an anti-T7 mAb revealed that T7-Drb1-WT co-precipitated with either EGFP-TDP-43-WT or EGFP-TDP-mt (data not shown). To clarify whether Drb1 binds TDP-43 directly, a GST pulldown assay was performed using radiolabeled TDP-43 protein and bacterially expressed and purified GST-Drb1 fusion protein, both of which were prepared by nuclease treatment. Fig. 1D shows that GST-Drb1, but not GST, pulled down radiolabeled TDP-43, demonstrating that Drb1 binds to TDP-43 directly. These results suggest that Drb1 can be recruited to TDP-43 mutant-induced cytoplasmic aggregates through direct protein-protein interaction.
FIGURE 1.

Drb1 partially co-localizes with the cytoplasmic TDP-43 aggregates. A, schematics indicating constructs of EGFP-fused human TDP-43 (WT and a double deletion mutant) and T7 tag-fused human WT Drb1. GRD, glycine-rich domain. B, subcellular localization of EGFP-TDP-43-WT (a–c), EGFP-TDP-mt (d–f), or T7-Drb1-WT (g–i) fusion protein expressed in HeLa cells. T7-tagged Drb1 was co-expressed with EGFP-TDP-mt (j–l). HeLa cells were transfected with the plasmids and incubated on glass bottom dishes for 24 h to allow plasmid expression. Nuclei were stained with Hoechst 33258 (b and e, c and f in blue). Scale bars = 10 μm. C, co-IP of TDP-43 and Drb1. T7-tagged Drb1 was co-expressed with EGFP, EGFP-tagged TDP-43-WT, or TDP-mt in HeLa cells, and immunoprecipitation was performed with an anti-GFP antibody. T7-Drb1 was co-immunoprecipitated with EGFP-tagged TDP-43-WT and TDP-mt but not with EGFP. IB, immunoblot. HC, immunoglobulin heavy chain. D, GST pulldown experiment of 35S-labeled TDP-43 using GST fused to full-length Drb1 (GST-Drb1) or GST. Input, 2% of in vitro translated 35S-labeled proteins, and proteins released from glutathione beads are shown at the left and the autoradiography of the same gel at the right. TDP-43 was pulled down with GST-Drb1 but not with GST alone.
Drb1 Contains a Classical NLS in Its Carboxyl Terminus
To investigate the elements involved in the subcellular localization of Drb1, we transiently transfected HeLa cells with a plasmid encoding the wild-type or the deletion mutant of Drb1 fused to modified GFP, termed Clover (Ref. 26 and Fig. 2A). Wild-type Drb1 clearly showed nucleoplasmic localization (Fig. 2B, a–c). This localization was consistent with that of endogenous Drb1 protein reported previously in human neuronal cells (22, 24) or T7-tagged Drb1 (Fig. 1B). The C-terminal deletion mutant (2–400) localized exclusively in the cytoplasm (Fig. 2B, d–f), whereas the N-terminal deletion mutant (401–474) predominantly localized in the nucleoplasm (Fig. 2B, g–i). This indicates that the C-terminal region (401–474 amino acids) of Drb1 is necessary and sufficient for its nucleoplasmic localization. To study the mechanism involved in Drb1 nuclear localization, we identified the NLS in Drb1 using cNLS Mapper, an online program predicting classical NLS sequences (31). In agreement with our result, the program predicted a bipartite-type NLS in the C terminus of Drb1, which is well conserved from Drosophila to mammals (supplemental Fig. S1). Site-directed mutagenesis was performed to verify the predicted bipartite-type NLS (Fig. 2C). Each mutant was fused to Clover and tested for protein localization in HeLa cells. When both upstream and downstream clusters of basic residues in the predicted bipartite-type NLS were substituted with alanine residues, termed mtNLSc1+c2, this Drb1 mutant localized exclusively to the cytoplasm as expected (Fig. 2D, d–f). The substitution of the upstream cluster (mtNLSc1) presented the nucleoplasmic localization pattern as that of the wild type (Fig. 2D, g–i, compared with the wild type, Fig. 2D, a–c). The substitution of the downstream cluster (mtNLSc2) changed its localization exclusively to the cytoplasm (Fig. 2D, j–l). Interestingly, a single basic amino acid substitution in NLSc2 to a glycine residue (R470G) resulted in exclusive cytoplasmic localization (Fig. 2D, m–o). These results indicate that Drb1 NLS is present in the C terminus, probably as a monopartite but not as a bipartite NLS.
FIGURE 2.

Drb1 contains a classical nuclear localization signal in its carboxyl terminus. A, Clover-T7-fused WT Drb1 and the deletion mutants. Numbers indicate the relevant amino acids. B, subcellular localization of Clover-T7 fused WT (a–c), 2–400 (d–e), and 401–474 (g–h) Drb1 in HeLa cells. The cells were incubated for 24 h, followed by fixation and permeabilization as described previously. The nuclei were stained with Hoechst 33258 (b, e, and h and c, f, and i in blue). The representative subcellular localization of the expressed protein is shown on the right as nucleoplasm (N) and cytoplasm (C), respectively. Scale bars = 10 μm. C, a putative bipartite NLS and the amino acid substitution mutants in the C terminus of Drb1. c1 and c2 indicate cluster 1 and cluster 2 of positively charged amino acids residues of the putative bipartite NLS, respectively. The NLS was predicted by the NLS prediction program cNLS Mapper (31). D, subcellular localization of Drb1 WT and putative NLS mutants expressed as Clover-T7-fused proteins in HeLa cells. The nuclei were stained with Hoechst 33258. The representative subcellular localization of the expressed protein is shown on the right.
Drb1 Is a Nuclear-Cytoplasmic Shuttling Protein
TDP-43 and FUS/TLS, RNA-binding proteins related to ALS and FTLD, have been shown to shuttle between the nucleus and cytoplasm to function in RNA metabolism in both compartments under normal conditions (11, 32). Drb1 movement in and out of the nucleus has not been determined. To evaluate Drb1 nuclear-cytoplasmic shuttling, we established interspecies heterokaryon assays, commonly used to the study of RNA-binding protein transport. We established HeLa cells stably expressing Clover-fused non-shuttling protein (Clover-NPc-NLS) (29) or Clover-fused wild-type Drb1 (Clover-Drb1-WT, Fig. 3A). When HeLa cells stably expressing Clover-NPc-NLS were fused with NIH3T3 cells under translational inhibitor (CHX) treatment, the fluorescent signal of Clover-NPc-NLS was not observed in the NIH3T3 nucleus in heterokaryon cells (Fig. 3B, left panel), indicating that Clover-NPc-NLS has no shuttling ability. On the other hand, when HeLa cells stably expressing Clover-Drb1-WT were used, the fluorescent signal was clearly observed in the NIH3T3 nucleus in heterokaryon cells (Fig. 3, B, right panel, and C). Furthermore, when a human neuroblastoma cell line (SH-SY5Y cells) stably expressing Clover-Drb1-WT was used, Drb1 shuttling ability could be detected. No statistical difference was observed (p = 0.0573, Fisher's exact test) between non-neuronal HeLa cells and neuronal SH-SY5Y cells in term of shuttling efficiency (Fig. 3D). These results indicate that Drb1 is a nuclear-cytoplasmic shuttling protein whose shuttling activity functions in neuronal as well as non-neuronal cells.
FIGURE 3.

Drb1 shuttles between the nucleus and cytoplasm in both neuronal and non-neuronal cells. A, schematic of the Clover-Myc-fused nucleoplasmin core domain (NPc) with a classical NLS from SV40 large T antigen and Clover-T7-fused Drb1. B, nuclear-cytoplasmic shuttling of Drb1. Heterokaryon assay was performed using HeLa cells stably expressing nuclear Clover-NPc or Clover-Drb1 and (untransfected) NIH3T3 cells. Prior to cell fusion, de novo protein synthesis was inhibited by addition of CHX. After fusion, cell culture was continued in the presence of CHX. Representative examples of the subcellular distribution of Clover-NPc (left panel) or Clover-Drb1 (right panel) are shown as well as actin filaments (red) and Hoechst-stained nuclei (gray) photographs. Scale bar = 10 μm. Heterokaryon cells were recognized as cells including both the human nucleus and the murine nucleus. The murine nucleus indicates several heterochromatins observed as the dots. C, box plot of heterokaryon cells showing a Clover signal in the murine nucleus. p < 0.001 (Mann-Whitney U test). D, the heterokaryon assay was performed using human neuroblastoma SH-SY5Y cells stably expressing Clover-Drb1 as in A. A representative example of the subcellular distribution of Clover-Drb1 is shown (left panel). Using the results obtained in C, the threshold value was configured as the maximum value of the negative control to classify the cells as cells with shuttling of Drb1 or as cells without shuttling. According to this threshold, the nuclear-cytoplasmic shuttling efficiency of Drb1 showed no difference between neuronal SH-SY5Y cells and non-neuronal HeLa cells. p = 0.0573 (Fisher's exact test). The percentage of heterokaryon cells (NIH3T3 nucleus with HeLa or SH-SY5Y nucleus) indicates Drb1 shuttling. N.S., not significant. Scale bar, 10 μm.
A Double Leucine within the Drb1 Linker Region Is Necessary for Nuclear Export
Because Drb1 is a nuclear-cytoplasmic shuttling protein, it should contain an NES in addition to the NLS present at the C terminus. To investigate this issue, additional Drb1 deletion mutants (Fig. 4A) were tested. Clover was expressed in HeLa cells localized in both the nucleus and cytoplasm with almost equal signal intensity (Fig. 4B, a–c). Clover-fused wild-type Drb1 predominantly localized to the nucleus (Fig. 4B, d–f), and the Drb1 carboxyl-terminal deletion mutant (2–400) exclusively localized to the cytoplasm because of the loss of Drb1 NLS (Fig. 4B, g–i, see also Fig. 2B). An additional C-terminal deletion mutant (2–300) equally localized to both the cytoplasm and nucleus (Fig. 4B, j–l), similar to Clover, suggesting that Drb1 NES is present within the 301–400 amino acid region. The amino-terminal deletion mutant (301–474) showed nuclear localization because of the C-terminal NLS, like wild-type Drb1 (Fig. 4B, m–o). The linker region (301–400), a deletion mutant lacking both N-terminal and C-terminal regions, exhibited high fluorescence intensity in the cytoplasm, although a weak nuclear fluorescence signal was observed, probably because of the free diffusion of the Clover-fused proteins (Fig. 4B, p–r). These results indicate that the 301–400 linker region between RRM2 and RRM3 functions as an NES. The canonical NES sequence is known as a leucine-rich sequence (33). Drb1 does not contain a typical leucine-rich sequence in the 301–400 amino acid region, but we noted that Drb1 presents a double leucine in this region. To evaluate the significance of the double leucine as an NES, we substituted the leucine with an alanine in the Drb1 301–400 deletion mutant (301–400-mtLL). Substitution of the double leucine resulted in equal distribution of the Drb1 mutant protein in the nucleus and cytoplasm (Fig. 4B, s–u). This result suggests that the double leucine within the Drb1 linker region is necessary for NES activity.
FIGURE 4.

Dileucine in the linker region of Drb1 functions as a nuclear export signal. A, schematic of Drb1 deletion and missense mutants. B, subcellular localization of Clover (a–c) and Clover-T7 fused WT (d–f), 2–400 (g–i), 2–300 (j–l), 301–474 (m–o), 301–400 (p–r), and 301–400-mtLL (s–u) Drb1 in HeLa cells. The representative subcellular localization of the expressed protein is shown on the right as nucleoplasm (N) and cytoplasm (C), respectively. The nuclei were stained with Hoechst 33258. Scale bars = 10 μm.
Disruption of the Nuclear-Cytoplasmic Trafficking of Drb1 Induces the Formation of Cytoplasmic Aggregates
The disruption of nuclear-cytoplasmic transport in many proteins, i.e. mislocalization of proteins, is associated with the production of affected neurons in neurodegenerative diseases (34–37). In these neurons, protein aggregates formed by mislocalized proteins are observed in the cytoplasm. We tested whether disruption of the nuclear-cytoplasmic shuttling of Drb1 generated cytoplasmic aggregates of Drb1 in HeLa cells transiently expressing Clover-fused wild-type or NLS and/or NES mutants of Drb1. When each of the wild-type, NLS (R470G), or NES (mtLL) mutant Drb1 was expressed in HeLa cells, cytoplasmic aggregates were not observed even though the NLS mutant showed mislocalization from the nucleus to the cytoplasm (see Fig. 2D for the wild-type and NLS mutant and supplemental Fig. S2 for mtLL). Interestingly, the introduction of a double mutation in both Drb1 NLS and NES (mtLL/R470G, Fig. 5A) resulted in the induction of cytoplasmic aggregates in about 20–30% of transfected cells at 24 h post-transfection (Fig. 5, B and C). Notably, endogenous or mRuby-fused TDP-43 was co-localized with the cytoplasmic Drb1 aggregates (Fig. 5D). In agreement with these observation, both overexpressed and endogenous TDP-43 were co-immunoprecipitated with wild-type Drb1 and the double mutant, mtLL/R470G, even under nuclease treatment conditions (Fig. 5E and supplemental Fig. S3B). These results suggest that Drb1-based cytoplasmic aggregation can recruit TDP-43 to the aggregates via protein-protein interaction.
FIGURE 5.

Drb1 with a double mutation in both the NLS and NES is prone to aggregate in the cytoplasm and to recruit TDP-43 into the aggregates. A, schematic of Clover-T7-fused Drb1 involving NLS/NES mutations. B, representative image of Clover-Drb1-mtLL/R470G in HeLa cells. Cells were transfected for 24 h, followed by fixation and permeabilization as described previously. Nuclei were stained by Hoechst 33258. Arrowheads indicate cytoplasmic aggregation of Clover-Drb1-mtLL/R470G. Scale bar = 10 μm. C, the percentages of cells with Clover-fused Drb1 aggregates in the cytoplasm were counted in more than 100 transfected cells. Three independent experiments were performed. Error bars show standard deviation. D, representative images of the Clover-Drb1-mtLL/R470G expression construct transiently transfected without (top panel) or with (bottom panel) the TDP-WT-mRuby2 expression construct in HeLa cells. Localization of endogenous TDP-43 was visualized by indirect immunofluorescence (b and c in red). Scale bars = 10 μm. E, co-IP of the Drb1 NLS/NES mutant and TDP-43. The Clover-tagged Drb1 construct (WT or mtLL/R470G mutant) was transfected with the TDP-43 expression construct in HeLa cells, and IP was performed with an anti-GFP antibody (for the Clover tag). The expression level of TDP-43 protein increased ∼1.8-fold by overexpression (supplemental Fig. S3A). TDP-43 was co-immunoprecipitated with Clover-Drb1 WT and mtLL/R470G but not with Clover. IB, immunoblot. HC, immunoglobulin heavy chain.
Cytoplasmic Drb1 Aggregation Induces a Reduction of Mitochondrial Membrane Potential
To evaluate the effect of cytoplasmic Drb1 aggregation on cell function, Δψm was assessed by measurement of TMRE fluorescence. In several cellular models of neurodegenerative diseases, including ALS, a decrease in mitochondrial membrane potential is observed in the preapoptotic state (38). TMRE fluorescence was detected 24 h after HeLa cells were transfected with Clover-fused wild-type Drb1 or the mutant-expressing plasmids, and the fluorescence intensity was measured. Neither wild-type nor R470G Drb1 expression influenced the mitochondrial membrane potential as the Clover expression control (Fig. 6, A, a–d, and B). In cells expressing the double Drb1 mutant (mtLL/R470G), a significant reduction of mitochondrial membrane potential was observed only in cells with cytoplasmic Drb1 aggregates (Fig. 6, A, e–f, and B). These results indicate that the reduction of mitochondrial membrane potential is not due to the effect of overexpression or just cytoplasmic mislocalization of Drb1 but to the formation of cytoplasmic Drb1 aggregates. Furthermore, we tested the effect of the TDP-43 mutant on mitochondrial membrane potential. Cells expressing EGFP-TDP-mt with cytoplasmic TDP-43 aggregates slightly reduced the mitochondrial membrane potential, although a significant difference was not observed (supplemental Fig. S4). When EGFP-TDP-43-M337V, an ALS-related mutant (6), was overexpressed in HeLa cells, we observed a significant reduction of the mitochondrial membrane potential. However, we could not find any cytoplasmic aggregates in the cells. EGFP-TDP-43-M337V was localized to the nucleoplasm in these cells, similar to EGFP-TDP-43-WT (data not shown). This suggests that the reduction of mitochondrial membrane potential by this TDP-43 mutant occurs via mechanisms distinct from those observed with the Drb1 mutant.
FIGURE 6.
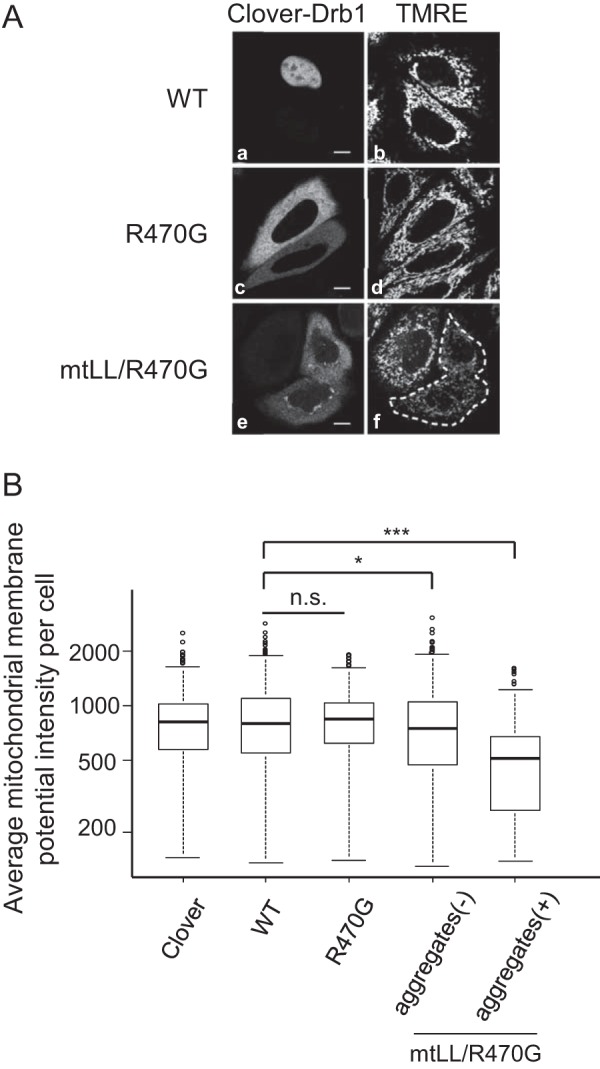
Induction of cytoplasmic Drb1 aggregation decreases the mitochondrial membrane potential. A, Δψm in HeLa cells transiently expressing Clover-Drb1-WT, Clover-Drb1-R470G, and Clover-Drb1-mtLL/R470G. The mitochondrial membrane potential was visualized by the fluorescent indicator TMRE. B, box plot of average Δψm fluorescence intensity in HeLa cells expressing Clover or Clover-Drb1 fusion protein (WT, R470G, or mtLL/R470G). Clover-Drb1-mtLL/R470G-expressing cells were divided into two classes by the presence of cytoplasmic aggregates (aggregates (−) and aggregates (+)). *, p < 0.05; ***, p < 0.001; n.s., not significant.
Discussion
Several RNA-binding proteins are known to play crucial roles in forming cytoplasmic aggregates in ALS/FTLD pathology. Drb1 is an RNA-binding protein that was recently reported as a novel component of TDP-43-positive cytoplasmic inclusion bodies in patients with ALS and FTLD (21, 22). In this study, we disclose a role of Drb1 in forming cytoplasmic aggregates relating to the process of neuronal cytotoxicity. First, we verified the co-localization and mutual interaction between Drb1 and TDP-43 in an in vitro cellular model that forms cytoplasmic TDP-43 aggregates. We found that mutations in Drb1 nuclear-cytoplasmic trafficking elements tended to induce TDP-43-positive cytoplasmic aggregates. Furthermore, only cells with cytoplasmic Drb1 aggregates, but not cells just presenting cytoplasmic mislocalized Drb1, presented with a decreased mitochondrial membrane potential, implying mitochondrial dysfunction. From these results, it is likely that the novel cytotoxic mechanism triggered by the perturbation of Drb1 subcellular localization system contributes to ALS/FTLD pathology.
We recapitulated the co-localization of Drb1, which predominantly localizes to the nucleus, into TDP-43-induced cytoplasmic aggregates in an in vitro culture cell model. Co-immunoprecipitation experiments using nuclease-treated lysates (Fig. 1C) suggest the physical interaction between wild-type Drb1 and mutant TDP-43. Additionally, GST pulldown analysis (Fig. 1D) confirms a direct interaction between Drb1 and TDP-43. Very recently, Li et al. (39) reported that Drb1 interacted with TDP-43 indirectly via RNA. Although there are discrepancies between our results and theirs, the direct or indirect binding of Drb1 to TDP-43 aggregates in the cytoplasm of ALS-affected neural cells seems to surpass the nuclear import of cytoplasmic Drb1. Conversely, we also demonstrated that wild-type TDP-43 can be recruited to mutant Drb1 (mtLL/R470G)-induced cytoplasmic aggregates in HeLa cells. Although whether Drb1-induced aggregates have similar features as aggregates observed in affected neurons from patients with ALS/FTLD remains to be determined, this result allows us to highlight the importance of the interaction between Drb1 and TDP-43 in cytoplasmic aggregates for cell toxicity despite the different origin of the aggregates. Simultaneously, we speculate that there is a potential dominant role of Drb1 in ALS/FTLD disease pathology. To date, no mutation related to ALS/FTLD in the DRB1 gene has been reported. Further genetic studies focusing on the DRB1 gene in patients with ALS/FTLD are warranted to understand the molecular mechanism underlying ALS/FTLD pathogenesis.
We demonstrated that Drb1, which predominantly localized to the nucleus, is a nuclear-cytoplasmic shuttling protein in both neuronal and non-neuronal cells. This suggests that Drb1 is involved in RNA metabolism, which occurs both in the nucleus and cytoplasm in normal conditions. Other ALS-linked RNA-binding proteins such as TDP-43 (11), FUS/TLS (32), and hnRNP A1 (29) also shuttle in both cellular compartments under normal conditions. Thus, nuclear-cytoplasmic trafficking may be a common feature among ALS-linked RNA-binding proteins.
Conserved basic residues at the C terminus (amino acids 469–472, KRQR) of Drb1 function as a monopartite-type NLS, whereas a double leucine in the linker region functions as an NES. Li et al. (39) also reported that Drb1 has a bipartite-type NLS that includes the upstream basic amino acids (454–472) in addition to the NLS region identified in this report (see Fig. 2C for clusters 1 and 2). However, we concluded that the cluster 1 of Drb1 is not required for nuclear localization because the substitution of basic residues in cluster 2 (Lys-469, Arg-470, and Arg-472) to alanine resulted in exclusive cytoplasmic localization of the Drb1 mutant in our experiment. This result suggests that the cluster 1 does not contribute to nuclear transport. ALS-linked RNA-binding proteins such as TDP-43, FUS/TLS, and hnRNP A1 contain distinct NLSs. TDP-43 contains a classical bipartite-type NLS that is composed of two separate clusters of basic amino acids residues (14). FUS/TLS contains a non-classical NLS, known as PY-NLS, that consists of overall basic characteristics and a central hydrophobic or basic motif followed by a C-terminal R/H/KX(2–5)PY (where X is any residues) consensus sequence in the C terminus of FUS/TLS (40). HnRNP A1 contains a 38-residues signal near its C terminus, termed M9 (28).
Drb1 does not contain a classical NES, a leucine-rich stretch of hydrophobic amino acids (41). Instead, the double leucine motif in the linker region participates in nuclear export. This two-tandem hydrophobic residue is highly conserved among the known vertebrate Drb1 homologue as LL or LI. FUS/TLS contains a classical leucine-rich NES (42). TDP-43 also contains a relatively variable leucine-rich NES sequence (14). They are both sensitive to leptomycin B, an inhibitor of CRM1, which is a receptor of classical NES. The M9 motif of hnRNP A1 functions as an NES in addition to the NLS but is independent of CRM1 (28). The Drb1NES presents little similarity with classical leucine-rich NESs or the M9 sequence, suggesting that it is a novel NES sequence. Taken together, these ALS-linked proteins, including Drb1, are nuclear-cytoplasmic shuttling proteins, but their trafficking machineries, such as NLSs and NESs or its receptors, seem to be variable.
The fact that only Drb1 mutated in both NLS and NES forms cytoplasmic aggregates under normal conditions is unexpected. The Drb1 NLS mutant showed cytoplasmic localization but did not form aggregates. This suggests that Drb1 cytoplasmic mislocalization is necessary but insufficient for the formation of cytoplasmic inclusion bodies. On the other hand, the Drb1 NES mutant localized to the nucleus similar to the wild-type because of the NLS but did not induce remarkable aggregates in the nucleus. However, a double NLS and NES Drb1 mutant formed Drb1 cytoplasmic aggregates. Thus, the double leucine motif of Drb1 seems to be a region in the cytoplasm sensitive to protein folding, and the disruption of the motif elicits protein misfolding, a possible key mechanism for the occurrence of cytoplasmic aggregates observed in ALS/FTLD-affected neurons (43). Unlike other ALS-linked RNA-binding proteins, Drb1 does not contain prion-like domains, which can accelerate aggregation with amyloid-like formation (44). Alternatively, it has been proposed that Drb1 contains a homo-oligomer assembly domain, which is crucial for Drb1 self-association (39). The homo-oligomer assembly domain locates 12 amino acids upstream of the double leucine motif. Therefore, Drb1 misfolding elicited by NES disruption may facilitate self-aggregation via the homo-oligomer assembly domain. Interestingly, Drb1 mutant (mtLL/R470G)-inducing cytoplasmic aggregates decreased the mitochondrial membrane potential, whereas cytoplasmic mislocalization of the Drb1 mutant (R470G) did not affect the mitochondrial membrane potential. Mitochondria play pivotal roles in a number of cellular processes, including energy production, Ca2+ homeostasis, and lipid metabolism. Mitochondria are also the main source of reactive oxygen species and function as the initiation site of the intrinsic apoptotic cascade. Therefore, damages to mitochondrial function can lead to bioenergetic failure, oxidative stress, or apoptosis. There is abundant evidence of mitochondrial dysfunction in ALS (45). Because the decrease of mitochondrial membrane potential precedes mitochondrial dysfunction, our cellular model suggests that the emergence of cytoplasmic Drb1 aggregates causes mitochondrial dysfunction. Thus, further studies are needed to examine how the Drb1 mutant leads to mitochondrial dysfunction.
In conclusion, we demonstrated that Drb1 is a nuclear-cytoplasmic shuttling protein. We also verified the interaction between Drb1 and TDP-43 in both cellular models of TDP-43-derived and Drb1-derived cytoplasmic aggregates. The emergence of Drb1-induced cytoplasmic aggregates can decrease the mitochondrial membrane potential in cells. These results provide insights into the common concept of RNA-binding proteins involved in neurodegenerative diseases.
Author Contributions
T. Mashiko, E. S., K. Kasashima, and H. E. designed the experiments. T. Mashiko and E. S. performed the experiments, analyzed the data, and wrote the manuscript with assistance from all other authors. K. Kasashima, K. T., and K. Kuroiwa provided technical expertise and interpreted the data. Y. N. provided important materials and expertise. T. Matsuura and T. H. coordinated the study. H. E. supervised the project. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Chiyoko Kato and Eriko Ohta for experimental assistance, members of the H. E. laboratory for valuable discussions, and Dr. Makiko Mieno for statistical advice. The TDP-43 plasmid and its mutants were provided by Dr. Takashi Nonaka (Tokyo Metropolitan Organization for Medical Research), and the NPc-NLS-REV-NES construct was provided by Dr. Toshinobu Fujiwara and Dr. Hiroshi Sakamoto. We thank Editage for English language editing.
This work was supported by Jichi Medical University graduate student start-up grants for young investigators, a Jichi Medical University graduate student research award, a Jichi Medical University young investigator award, and the MEXT-Supported Program for Strategic Research Foundation at Private Universities 2011–2015 and 2013–2017. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S4.
- ALS
- amyotrophic lateral sclerosis
- FTLD
- frontotemporal lobar degeneration
- FUS/TLS
- fused in sarcoma/translated in liposarcoma
- RRM
- RNA recognition motif
- NLS
- nuclear localization signal
- NES
- nuclear export signal
- EGFP
- enhanced GFP
- IP
- immunoprecipitation
- CHX
- cycloheximide
- TMRE
- tetramethylrhodamine ethyl ester
- IQR
- interquartile range
- Δψm
- mitochondrial membrane potential.
References
- 1. Kiernan M. C., Vucic S., Cheah B. C., Turner M. R., Eisen A., Hardiman O., Burrell J. R., and Zoing M. C. (2011) Amyotrophic lateral sclerosis. Lancet 377, 942–955 [DOI] [PubMed] [Google Scholar]
- 2. Neary D., Snowden J. S., Gustafson L., Passant U., Stuss D., Black S., Freedman M., Kertesz A., Robert P. H., Albert M., Boone K., Miller B. L., Cummings J., and Benson D. F. (1998) Frontotemporal lobar degeneration. Neurology 51, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 3. Strong M. J., Grace G. M., Freedman M., Lomen-Hoerth C., Woolley S., Goldstein L. H., Murphy J., Shoesmith C., Rosenfeld J., Leigh P. N., Bruijn L., Ince P., and Figlewicz D. (2009) Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 131–146 [DOI] [PubMed] [Google Scholar]
- 4. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., and Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 5. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 6. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rutherford N. J., Zhang Y.-J., Baker M., Gass J. M., Finch N. A., Xu Y.-F., Stewart H., Kelley B. J., Kuntz K., Crook R. J. P., Sreedharan J., Vance C., Sorenson E., Lippa C., Bigio E. H., et al. (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4, 10.1371/journal.pgen.1000193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kwiatkowski T. J. Jr., Bosco D. A., Leclerc A. L., Tamrazian E., Vanderburg C. R., Russ C., Davis A., Gilchrist J., Kasarskis E. J., Munsat T., Valdmanis P., Rouleau G. A., Hosler B. A., Cortelli P., de Jong P. J., et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 [DOI] [PubMed] [Google Scholar]
- 9. Vance C., Rogelj B., Hortobagyi T., De Vos K. J., Nishimura A. L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., Ganesalingam J., Williams K. L., Tripathi V., Al-Saraj S., Al-Chalabi A., et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ling S.-C., Polymenidou M., and Cleveland D. W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y.-F., Petrucelli L., Buratti E., and Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 12. Nonaka T., Arai T., Buratti E., Baralle F. E., Akiyama H., and Hasegawa M. (2009) Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett. 583, 394–400 [DOI] [PubMed] [Google Scholar]
- 13. Winton M. J., Van Deerlin V. M., Kwong L. K., Yuan W., Wood E. M., Yu C.-E., Schellenberg G. D., Rademakers R., Caselli R., Karydas A., Trojanowski J. Q., Miller B. L., and Lee V. M. (2008) A90V TDP-43 variant results in the aberrant localization of TDP-43 in vitro. FEBS Lett. 582, 2252–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., and Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blokhuis A. M., Groen E. J., Koppers M., van den Berg L. H., and Pasterkamp R. J. (2013) Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson B. S., Snead D., Lee J. J., McCaffery J. M., Shorter J., and Gitler A. D. (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pesiridis G. S., Lee V. M., and Trojanowski J. Q. (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, R156–R162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lattante S., Rouleau G. A., and Kabashi E. (2013) TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat. 34, 812–826 [DOI] [PubMed] [Google Scholar]
- 19. Tamada H., Sakashita E., Shimazaki K., Ueno E., Hamamoto T., Kagawa Y., and Endo H. (2002) cDNA cloning and characterization of Drb1, a new member of RRM-type neural RNA-binding protein. Biochem. Biophys. Res. Commun. 297, 96–104 [DOI] [PubMed] [Google Scholar]
- 20. Ray D., Kazan H., Cook K. B., Weirauch M. T., Najafabadi H. S., Li X., Gueroussov S., Albu M., Zheng H., Yang A., Na H., Irimia M., Matzat L. H., Dale R. K., Smith S. A., et al. (2013) A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Konno T., Tada M., Shiga A., Tsujino A., Eguchi H., Masuda-Suzukake M., Hasegawa M., Nishizawa M., Onodera O., Kakita A., and Takahashi H. (2014) C9ORF72 repeat-associated non-ATG-translated polypeptides are distributed independently of TDP-43 in a Japanese patient with c9ALS. Neuropathol. Appl. Neurobiol. 40, 783–788 [DOI] [PubMed] [Google Scholar]
- 22. Collins M., Riascos D., Kovalik T., An J., Krupa K., Krupa K., Hood B. L., Conrads T. P., Renton A. E., Traynor B. J., and Bowser R. (2012) The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 124, 717–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hans F., Fiesel F. C., Strong J. C., Jäckel S., Rasse T. M., Geisler S., Springer W., Schulz J. B., Voigt A., and Kahle P. J. (2014) UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase y regulate TDP-43 protein ubiquitination. J. Biol. Chem. 289, 19164–19179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bakkar N., Kousari A., Kovalik T., Li Y., and Bowser R. (2015) RBM45 modulates the antioxidant response in amyotrophic lateral sclerosis through interactions with KEAP1. Mol. Cell. Biol. 35, 2385–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasashima K., Terashima K., Yamamoto K., Sakashita E., and Sakamoto H. (1999) Cytoplasmic localization is required for the mammalian ELAV-like protein HuD to induce neuronal differentiation. Genes Cells 4, 667–683 [DOI] [PubMed] [Google Scholar]
- 26. Lam A. J., St-Pierre F., Gong Y., Marshall J. D., Cranfill P. J., Baird M. A., McKeown M. R., Wiedenmann J., Davidson M. W., Schnitzer M. J., Tsien R. Y., and Lin M. Z. (2012) Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 9, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 28. Michael W. M., Choi M., and Dreyfuss G. (1995) A nuclear export signal in hnRNP A1: a signal-mediated, temperature-dependent nuclear protein export pathway. Cell 83, 415–422 [DOI] [PubMed] [Google Scholar]
- 29. Piñol-Roma S., and Dreyfuss G. (1992) Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature 355, 730–732 [DOI] [PubMed] [Google Scholar]
- 30. Kanda Y. (2013) Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 48, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kosugi S., Hasebe M., Tomita M., and Yanagawa H. (2009) Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U.S.A. 106, 10171–10176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zinszner H., Sok J., Immanuel D., Yin Y., and Ron D. (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 110, 1741–1750 [DOI] [PubMed] [Google Scholar]
- 33. la Cour T., Kiemer L., Mølgaard A., Gupta R., Skriver K., and Brunak S. (2004) Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17, 527–536 [DOI] [PubMed] [Google Scholar]
- 34. Patel V. P., and Chu C. T. (2011) Nuclear transport, oxidative stress, and neurodegeneration. Int. J. Clin. Exp. Pathol. 4, 215–229 [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang K., Donnelly C. J., Haeusler A. R., Grima J. C., Machamer J. B., Steinwald P., Daley E. L., Miller S. J., Cunningham K. M., Vidensky S., Gupta S., Thomas M. A., Hong I., Chiu S.-L., Huganir R. L., et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kinoshita Y., Ito H., Hirano A., Fujita K., Wate R., Nakamura M., Kaneko S., Nakano S., and Kusaka H. (2009) Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 68, 1184–1192 [DOI] [PubMed] [Google Scholar]
- 37. Nagara Y., Tateishi T., Yamasaki R., Hayashi S., Kawamura M., Kikuchi H., Iinuma K. M., Tanaka M., Iwaki T., Matsushita T., Ohyagi Y., and Kira J. (2013) Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1α in amyotrophic lateral sclerosis. Brain Pathol. 23, 534–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carrì M. T., Ferri A., Battistoni A., Famhy L., Gabbianelli R., Poccia F., and Rotilio G. (1997) Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 414, 365–368 [DOI] [PubMed] [Google Scholar]
- 39. Li Y., Collins M., Geiser R., Bakkar N., Riascos D., and Bowser R. (2015) RBM45 homo-oligomerization mediates association with ALS-linked proteins and stress granules. Sci. Rep. 5, 14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee B. J., Cansizoglu A. E., Süel K. E., Louis T. H., Zhang Z., and Chook Y. M. (2006) Rules for nuclear localization sequence recognition by karyopherinβ2. Cell 126, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bogerd H. P., Fridell R. A., Benson R. E., Hua J., and Cullen B. R. (1996) Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol. Cell. Biol. 16, 4207–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kino Y., Washizu C., Aquilanti E., Okuno M., Kurosawa M., Yamada M., Doi H., and Nukina N. (2011) Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 39, 2781–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J.-P., Brady S. T., and Brown R. H. Jr. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. King O. D., Gitler A. D., and Shorter J. (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cozzolino M., and Carrì M. T. (2012) Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.