

Abstract
Cyclooxygenase-2 (COX-2) catalyzes the oxygenation of arachidonic acid (AA) and endocannabinoid substrates, placing the enzyme at a unique junction between the eicosanoid and endocannabinoid signaling pathways. COX-2 is a sequence homodimer, but the enzyme displays half-of-site reactivity, such that only one monomer of the dimer is active at a given time. Certain rapid reversible, competitive nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to inhibit COX-2 in a substrate-selective manner, with the binding of inhibitor to a single monomer sufficient to inhibit the oxygenation of endocannabinoids but not arachidonic acid. The underlying mechanism responsible for substrate-selective inhibition has remained elusive. We utilized structural and biophysical methods to evaluate flufenamic acid, meclofenamic acid, mefenamic acid, and tolfenamic acid for their ability to act as substrate-selective inhibitors. Crystal structures of each drug in complex with human COX-2 revealed that the inhibitor binds within the cyclooxygenase channel in an inverted orientation, with the carboxylate group interacting with Tyr-385 and Ser-530 at the top of the channel. Tryptophan fluorescence quenching, continuous-wave electron spin resonance, and UV-visible spectroscopy demonstrate that flufenamic acid, mefenamic acid, and tolfenamic acid are substrate-selective inhibitors that bind rapidly to COX-2, quench tyrosyl radicals, and reduce higher oxidation states of the heme moiety. Substrate-selective inhibition was attenuated by the addition of the lipid peroxide 15-hydroperoxyeicosatertaenoic acid. Collectively, these studies implicate peroxide tone as an important mechanistic component of substrate-selective inhibition by flufenamic acid, mefenamic acid, and tolfenamic acid.
Keywords: crystal structure, cyclooxygenase (COX), electron paramagnetic resonance (EPR), endocannabinoid, structural biology, nonsteroidal anti-inflammatory drugs
Introduction
The cyclooxygenases (COX-1 and COX-2) convert arachidonic acid (AA)2 to prostaglandin H2 (1). Prostaglandin H2 is subsequently metabolized by downstream tissue-specific synthases into potent signaling molecules that play fundamental roles in both the regulation of physiological homeostasis as well as in disease states such as inflammation and cancer (2). COX-1 preferentially oxygenates AA, whereas COX-2 efficiently oxygenates a broad spectrum of fatty acid, ester, and amide substrates, including the endocannabinoids 1-arachidonoyl glycerol (1-AG), 2-arachidonoyl glycerol, and anandamide (3–8). 2-Arachidonoyl glycerol and anandamide are widely distributed in mammalian tissues and were the first characterized endogenous ligands for the cannabinoid receptors CB1 and CB2 (9). COX-2 oxygenates endocannabinoids using the same catalytic mechanism employed for AA, generating PG-glycerol esters and PG-ethanolamides (10–12). Endocannabinoid signaling plays a significant role in various physiological processes and has been implicated in pathologies ranging from anxiety and depression, to multiple sclerosis, Parkinson disease, and cancer (13). The unique ability to oxygenate endocannabinoids places COX-2 at a critical junction between the eicosanoid and endocannabinoid signaling systems.
COX-2 contains both a peroxidase active site and cyclooxygenase active site that are spatially distinct but functionally linked. The peroxidase active site contains a Fe3+-protoporphyrin IX heme moiety that upon reaction with peroxide is converted to an oxy-ferryl protoporphyrin IX cation radical. Electron transfer from Tyr-385 in the cyclooxygenase active site to the oxy-ferryl protoporphyrin IX cation radical reduces the heme back to the fully covalent oxy-ferryl protoporphyrin IX and generates a Tyr-385 radical in the cyclooxygenase active site. Reducing co-substrates such as phenol further reduces the oxy-ferryl heme by one electron to the resting ferric state. The Tyr-385 radical abstracts the pro-S hydrogen from carbon-13 of AA to initiate cyclooxygenase catalysis and two molecules of oxygen are then added to the substrate. A hydrogen atom is then transferred back from Tyr-385 to the peroxyl radical on carbon-15 to form prostaglandin G2 and regenerate the Tyr-385 radical. As the Tyr-385 radical is regenerated at the end of catalysis, a single turnover of peroxide in the peroxidase active site can sustain many turnovers of AA in the cyclooxygenase active site. This is referred to as the branched chain mechanism of catalysis (14). Although endocannabinoid substrates bind in a similar fashion to AA in the cyclooxygenase active site (16), endocannabinoid oxygenation is sensitive to peroxide tone and requires more turnover of hydroperoxide to sustain cyclooxygenase catalysis, suggesting that the branched chain mechanism is less efficient with endocannabinoid substrates (17).
COX-2 is inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs; for review, see Ref. 18). Prusakiewicz et al. (19) demonstrated that some NSAIDs, such as ibuprofen and mefenamic acid, act in a “substrate-selective” fashion in that they are weak competitive inhibitors of AA oxygenation but potent non-competitive inhibitors of endocannabinoid oxygenation by COX-2. These results were extended when it was demonstrated that the R-isomers of the propionic acid class of NSAIDs, such as R-flurbiprofen, are potent inhibitors of endocannabinoid oxygenation but have no effect on AA turnover (20). Substrate-selective inhibition of COX-2 has been shown to augment endocannabinoid levels in vivo, resulting in favorable psychological effects, including reduced anxiety (21). Thus, substrate-selective inhibitors of COX-2 hold promising potential to provide the beneficial effects of endocannabinoid augmentation such as reduced anxiety, depression, and pain while avoiding the negative gastric and cardiovascular consequences associated with the inhibition of AA oxygenation.
Although the phenomenon of substrate-selective inhibition of COX-2 is well established, the mechanism by which these NSAIDs inhibit endocannabinoid but not AA oxygenation remains unclear. COX enzymes are sequence homodimers composed of tightly associated monomers. Studies have shown that COX functions as a conformational heterodimer, with only one monomer of the homodimer active at a given time (22). Moreover, the monomers act in concert via an allosteric/catalytic couple, with the oxygenation of substrate in the “catalytic” monomer (Ecat) modulated by the binding of dietary nonsubstrate fatty acids, such as palmitic acid, and NSAIDs to the partner “allosteric” monomer (Eallo) (8, 19, 23, 24). A model of substrate-selective inhibition has been proposed where binding of an inhibitor to Eallo results in the allosteric inhibition of endocannabinoid but not AA oxygenation in the partner monomer, and AA inhibition only results when inhibitor binds to both monomers of the COX-2 homodimer (19). Although x-ray crystal structures of substrate-selective NSAIDs bound to COX-2 have been elucidated, extensive analyses and comparisons to substrate-bound crystal structures do not reveal protein conformational differences at the dimer interface and thus do not provide insight into the molecular details responsible for cross-talk between monomers (20, 25).
Although mefenamic acid is known to be a substrate-selective inhibitor of COX-2, the other fenamic acid inhibitors have yet to be investigated with respect to substrate selectivity. Moreover, there has been no structural investigation into the mode of binding of the fenamic acid inhibitors within the cyclooxygenase active site of COX-2. The fenamic acid inhibitors have similar chemical structures to the phenyl acetic acid class of inhibitors that include diclofenac and lumiracoxib. Structural studies have shown that both diclofenac and lumiracoxib bind with their carboxylate moieties interacting with the side chains of Tyr-385 and Ser-530 at the apex of the cyclooxygenase active site (26, 27). Here we report structural, biophysical, and functional studies designed to evaluate the ability of the remaining fenamic acids to inhibit human (hu) COX-2 in a substrate-selective manner.
Experimental Procedures
Materials
Meclofenamic acid sodium salt, diclofenac sodium salt, AA, 1-AG, and R-flurbiprofen were purchased from Cayman Chemical Co. (Ann Arbor, MI). FLAG peptide was purchased from GenScript (Piscataway, NJ). Tolfenamic acid, flufenamic acid, mefenamic acid, anti-FLAG M2 resin, and ethylene glycol were purchased from Sigma. Co3+-protoporphyrin IX and Fe3+-protoporphyrin IX were purchased from Frontier Scientific (Logan, UT). Decyl maltoside was purchased from Affymetrix (Santa Clara, CA). N-Octyl-β-d-glucopyranoside (βOG) was purchased from Inalco Pharmaceuticals (San Luis Obispo, CA). The QuikChangeTM Mutagenesis kit II was purchased from Agilent Technologies (Santa Clara, CA). HiTrapTM HP Chelating and HiPrepTM 16/60 Sephacryl S-300 HR chromatography columns were purchased from GE Healthcare. Oligos used for site-directed mutagenesis were purchased from Integrated DNA Technologies (Coralville, IA). Polyacrylic acid sodium salt 5100 and polyethylene glycol (PEG) 400 were purchased from Hampton Research (Aliso Viejo, CA). 15-Hydroperoxyeicosatetraenoic acid (15-HPETE) was generated and purified as described previously (28, 29).
Production of Wild Type and Mutant Constructs of Human COX-2
His6 N580A huCOX-2 in pFastBac-1 was used to engineer Y385F and S530A mutant constructs using the QuikChange Mutagenesis kit II. To create the Y385F/S530A double mutant, we utilized Y385F huCOX-2 in pFastBac-1 as the template for mutagenesis. All constructs were sequence-verified. Expression of all constructs was carried out in insect cells using the baculovirus system as described in Vecchio et al. (15). For solubilization, the cell pellet from a 2-liter culture of Sf21 insect cells was resuspended in 50 mm Tris, pH 8.0, 300 mm NaCl, 1 mm 2-mercaptoethanol. The resuspended cells were lysed using a Microfluidizer and solubilized by adding decyl maltoside to a final concentration of 0.87% (w/v). The solubilization mixture was stirred for 60 min at 4 °C followed by centrifugation at 140,000 × g for 75 min. A two-step purification protocol consisting of affinity and size-exclusion chromatographic steps (15) was then utilized to produce purified wild type and mutant huCOX-2 in 25 mm Tris, pH 8.0, 150 mm NaCl, and 0.53% (w/v) βOG for kinetic and biophysical characterization.
For crystallization, we utilized a wild type huCOX-2 construct that had been engineered to contain a FLAG affinity tag at the N terminus and a deletion of residues 586–612 (Δ586) at the C terminus (30). The resulting FLAG Δ586 huCOX-2 construct was used to generate purified protein in βOG. Specifically, the supernatant resulting from the solubilization of the enzyme in decyl maltoside was loaded onto an anti-FLAG M2 affinity column (2.5 cm × 10 cm) equilibrated in 50 mm Tris, pH 7.4, 150 mm NaCl, 0.87% (w/v) decyl maltoside. After a washing step, utilizing 50 mm Tris, pH 7.4, 150 mm NaCl, and 0.53% (w/v) βOG, the protein was released from the resin by running 40 ml of the wash buffer containing 100 μg/ml FLAG peptide over the column. The eluted protein was dialyzed overnight against 50 mm Tris, pH 7.4, 150 mm NaCl, 0.53% (w/v) βOG and subsequently run over a HiPrep 16/60 Sephacryl S-300 HR column equilibrated in the same buffer utilized in dialysis. Peak fractions from the size-exclusion column were pooled and concentrated to 4 mg/ml for crystallization trials.
Crystallization of Human COX-2 Fenamate Complexes
To generate crystals of huCOX-2 bound with meclofenamic acid, FLAG Δ586 huCOX-2 was reconstituted with a 2-fold molar excess of Co3+-protoporphyrin IX and crystallized using the sitting drop vapor diffusion method in conjunction with streak seeding. Specifically, 3 μl of the Co3+-reconstituted enzyme was mixed with 3 μl of 29–34% polyacrylic acid 5100, 100 mm HEPES, pH 7.5, 20 mm MgCl2. The resulting drops were equilibrated against wells containing 500 μl of the same buffer for 48 h at 25 °C. Crystal nucleation was induced by streak seeding the equilibrated drops with pulverized apoenzyme crystals grown in similar conditions reported previously for murine muCOX-2 (6, 15, 16, 25, 31). The seeded drops were resealed and incubated at 25 °C. Large, single crystals appeared 2–3 weeks after streak seeding. Crystals were then harvested from the drops and soaked in a cryoprotectant solution consisting of 35% (v/v) polyacrylic acid 5100, 100 mm HEPES, pH 7.5, 20 mm MgCl2, 0.6% (w/v) βOG, 10% (v/v) ethylene glycol, and 1 mm meclofenamic acid for 20 min followed by flash-freezing directly in a gaseous nitrogen stream at 100 K.
We were unsuccessful in obtaining crystals of huCOX-2 bound with mefenamic acid, flufenamic acid, and tolfenamic acid using standard co-crystallization methods in conjunction with the polyacrylic acid 5100 mixture or the streak seeding method described above. As such, we utilized the 1536 condition tailored membrane protein screen (32) in the High-Throughput Crystallization Laboratory at the Hauptman-Woodward Institute (33) to search for new leads that would facilitate crystallization. Although numerous crystallization leads were identified, one lead was successfully optimized and converted from microbatch-under-oil format to the sitting drop vapor-diffusion format. To generate the remaining complexes, Co3+-protoporphyrin IX reconstituted huCOX-2 at 4 mg/ml was incubated with a 5-fold molar excess of inhibitor followed by crystallization using the sitting drop vapor diffusion method. Specifically, 2 μl of the huCOX-2 inhibitor complex was mixed with 2 μl of 27–32% (v/v) PEG 400, 100 mm HEPES, pH 7.0, 300 mm ammonium phosphate. The resulting drops were equilibrated against wells containing 500 μl of the same buffer at 25 °C, and crystals appeared within 1–2 weeks of setup. Crystals were subsequently harvested from drops and soaked in a cryoprotectant solution consisting of 32% (v/v) PEG 400, 100 mm HEPES, pH 7.0, 300 mm ammonium phosphate, 0.6% (w/v) βOG, and 10% (v/v) glycerol followed by flash-freezing directly in a gaseous nitrogen stream at 100 K.
Structure Solution and Refinement
Diffraction data were collected at the Advanced Photon Source (Argonne, IL). Specifically, data for the FLAG Δ586 huCOX-2 meclofenamic acid complex were collected on beamline 23ID-B utilizing a MAR mosaic 300 CCD detector, whereas data for FLAG Δ586 huCOX-2 complexed with mefenamic acid, flufenamic acid, and tolfenamic acid were collected on beamline 17ID-B utilizing a Dectris Pilatus 6M pixel array detector. All diffraction data were integrated, scaled, and merged using HKL2000 (34). Data collection statistics are detailed in Table 1.
TABLE 1.
Crystallographic statistics
| Crystallographic parameters | huCOX-2 meclofenamate | huCOX-2 mefenamate | huCOX-2 tolfenamate | huCOX-2 flufenamate |
|---|---|---|---|---|
| Space group | I222 | I222 | I222 | I222 |
| No. in asymmetric unit | 2 | 2 | 2 | 2 |
| Unit cell length (Å) a | ||||
| a | 120.51 | 127.67 | 126.67 | 126.92 |
| b | 134.05 | 149.69 | 149.76 | 149.33 |
| c | 179.38 | 188.39 | 185.73 | 184.77 |
| α = β = γ (°) | 90.00 | 90.00 | 90.00 | 90.00 |
| Wavelength (Å) | 1.033 | 1.000 | 1.000 | 1.000 |
| Resolution (Å) | 40.00-2.41 | 30.00-2.35 | 30.00-2.45 | 30.00-2.50 |
| Highest res. shell (Å) | 2.45-2.41 | 2.39-2.35 | 2.49-2.45 | 2.54-2.50 |
| Rmergea | 8.2 (64.4)b | 5.1 (57.5) | 7.4 (72.7) | 8.2 (67.4) |
| Rmeas | 9.2 (73.2) | 5.6 (64.2) | 8.3 (82.2) | 9.0 (74.2) |
| Rpim | 4.1 (32.9) | 2.3 (27.8) | 3.7 (37.9) | 3.6 (30.8) |
| Total observations | 270,696 | 420,148 | 314,885 | 370,585 |
| Total unique | 56,332 | 75,533 | 64,987 | 60,238 |
| I/σ(I) | 16.0 (2.0) | 30.1 (2.5) | 19.2 (2.2) | 22.4 (2.5) |
| Completeness (%) | 98.6 (100) | 99.3 (94.9) | 99.8 (99.7) | 100 (100) |
| Multiplicity | 4.9 (4.8) | 5.6 (5.1) | 4.9 (4.6) | 6.2 (5.7) |
| Wilson B-factor (Å2) | 36.27 | 29.38 | 36.75 | 37.12 |
| No. atoms in refinement | 9,671 | 9,744 | 9,646 | 9,620 |
| R | 17.07 (21.68) | 18.47 (23.48) | 17.95 (23.98) | 18.54 (24.48) |
| Rfree | 21.17 (27.74) | 21.11 (25.52) | 21.96 (28.51) | 22.38 (28.45) |
| Ave. B factor, protein (Å2) | 42.40 | 42.10 | 45.10 | 44.00 |
| Ave. B factor, ligand (Å2) | 38.60 | 28.55 | 33.97 | 38.25 |
| Ave. B factor, solvent (Å2) | 37.40 | 35.90 | 39.20 | 37.40 |
| Mean positional error (Å) | 0.28 | 0.32 | 0.30 | 0.33 |
| r.m.s.d.c bond length (Å) | 0.010 | 0.005 | 0.004 | 0.006 |
| r.m.s.d. bond angle (°) | 0.85 | 0.83 | 0.82 | 0.88 |
| Ramachandran plot | ||||
| Allowed (%) | 98.00 | 97.00 | 97.00 | 97.00 |
| Generous (%) | 2.00 | 2.91 | 3.00 | 3.00 |
| Disallowed (%) | 0 | 0.09 | 0 | 0 |
a Rmerge, Rmeas, and Rpim were calculated as described in Evans 56.
b Values in parentheses are for the outermost resolution shell.
c r.m.s.d. is root mean square deviation.
Although all four huCOX-2 inhibitor complexes crystallized with two molecules in the asymmetric unit in orthorhombic space group I222, the unit cell parameters for crystals grown in polyacrylic acid 5100 and PEG 400 differ significantly (Table 1). Given these observations, difference Fourier maps were not utilized to generate initial phases, and extensive care was taken to remove model bias during structure solution. Each huCOX2 inhibitor structure was solved by molecular replacement methods utilizing PHASER (35) and a truncated muCOX-2 search model derived from PDB entry 3HS5 (15). After molecular replacement, the structures were rebuilt with Arp/wArp (36) using the primary sequence for huCOX-2 and the “automated model building starting from experimental phases” option (37). Iterative cycles of manual rebuilding and refinement were then performed using COOT (38) and PHENIX (39), respectively. Riding hydrogens were included in the model during refinement in PHENIX. Translation-libration-screw (TLS) refinement (40) using parameters obtained from the TLSMD web server (41) was applied in the final stages of refinement. The final refinement statistics for each structure are listed in Table 1. Despite the differences in unit cell parameters, the overall tertiary structure of the huCOX-2 sequence homodimer in each structure is identical to that observed previously (42). Moreover, there are no significant differences observed between monomers when they are compared with root mean square deviations within and between structures of ∼0.2 Å. Simulated annealing omit maps were generated using PHENIX, and model validation was carried out using MOLPROBITY (43). The figurs were produced using PyMOL (Version 1.7.0.0; Schrodinger, LLC). Coordinates and structure factors for the huCOX-2 flufenamic acid, huCOX-2 meclofenamic acid, huCOX-2 mefenamic acid, and huCOX-2 tolfenamic acid complexes have been deposited in the Protein Data Bank with IDs 5IKV, 5IKQ, 5IKR, and 5IKT, respectively.
Cyclooxygenase Activity Assays
Cyclooxygenase activity was measured using a Clark type oxygen electrode as described in Vecchio et al. (15). The assays were performed at 37 °C utilizing cuvettes containing 100 mm Tris, pH 8.0, 1 mm phenol, 5 μm Fe3+-protoporphyrin IX, and 100 μm AA or 1-AG as substrate. Reactions were initiated via the addition of 5 μg of huCOX-2, and activity was recorded as the maximal rate of oxygen consumption. For inhibition studies, inhibitors were dissolved in DMSO and added to the reaction cuvette such that the final concentration of DMSO was <0.1% of the reaction volume. When appropriate, kinetic profiles of cyclooxygenase activity versus substrate concentration were fit with a model of substrate-dependent inhibition using GraphPad Prism 5.0 (La Jolla, CA).
Tryptophan Fluorescence Quenching
Tryptophan fluorescence measurements were performed at 25 °C using a Fluoromax-4 spectrofluorometer. Wild type huCOX-2 was diluted to 0.26 μm in 25 mm Tris, pH 8.0, 150 mm NaCl, 0.53% (w/v) βOG. Tryptophan fluorescence was monitored with λex 283 nm and λem 335 nm. Excitation and emission slits widths were set to 2.5 mm and 10 mm, respectively. Fluorescence intensity was sampled every 100 ms. Inhibitors dissolved in DMSO were injected into the stirred protein solution to a final concentration of 1 μm. The rate of mefenamic and meclofenamic acid binding (k) was determined by fitting fluorescence intensity versus time to a plateau followed by one-phase dissociation equation in GraphPad Prism 5.
Thermal Shift Assay
Inhibitor-induced melting temperature shifts were determined using a Stratagene Mx3005P real-time PCR instrument (Stratagene, La Jolla, CA) and the thiol reactive fluorescent dye 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) as described previously (25). Wild type and mutant huCOX-2 constructs were diluted to a concentration of 1 μm in 25 mm Tris, pH 8.0, 150 mm NaCl, 0.53% (w/v) βOG. Inhibitors were added to the diluted protein to a final concentration of 50 μm and incubated on ice for 30 min. CPM was added to a final concentration of 25 μm, and thermal denaturation was performed by increasing the temperature from 25 °C to 99 °C in 0.5 °C increments over 45 min. CPM fluorescence was monitored with an ALEXA filter using a λex of 350 nm and a λem of 440 nm. Melting temperatures (Tm) were determined as the maximum of the first derivative plot of fluorescence intensity versus temperature. The change in melting temperature (ΔTm) induced by inhibitor binding was calculated as the difference between the Tm of inhibitor-bound protein and protein alone.
UV-Visible Spectroscopy
To determine the effect that tolfenamic acid had on the formation of heme oxidation states, wild type huCOX-2 (3 μm monomer) in 100 mm Tris, pH 8.0, was complexed with 3 μm Fe3+-protoporphyrin IX in a stirred quartz cuvette at 25 °C. Tolfenamic acid was added to a final concentration of 10 μm before initiation of peroxidase activity. The formation of oxy-ferryl protoporphyrin IX was stimulated by the addition of hydrogen peroxide to a final concentration of 15 μm. Absorbance spectra were recorded every 10 s using an Agilent 8453 UV-visible spectrophotometer equipped with an Agilent 89090a Peltier temperature controller. The ability of the inhibitors to act as reducing co-substrates was evaluated by reconstituting wild type huCOX-2 (3 μm monomer) in 100 mm Tris, pH 8.0, with 3 μm Fe3+-protoporphyrin IX in a stirred quartz cuvette at 25 °C. Hydrogen peroxide was added to a final concentration of 15 μm, and the peroxidase reaction was allowed to proceed for ∼45 s before the addition of 9 μm mefenamic, tolfenamic, or flufenamic acid. Reduction of compound II back to the resting ferric state was monitored at 409 nm, with spectra being recorded every 0.5 s.
EPR Spectroscopy
EPR studies were conducted at the Advanced Center for Electron Resonance Technology (ACERT) at Cornell University. Wild type huCOX-2 (54 μm) was complexed with a 1.5-fold molar excess of Fe3+-protoporphyrin IX followed by the addition of glycerol to a final concentration of 25% (v/v). Tyrosyl radicals were generated by reacting huCOX-2 with a 5-fold excess of hydrogen peroxide and mixing for 30 s on ice before flash-freezing in a dry ice/ethanol bath followed by liquid nitrogen. When appropriate, inhibitors dissolved in DMSO were injected immediately after the 30-s incubation and briefly mixed for 2–3 s before freezing. X-band EPR spectra were recorded on a Bruker Elexsys E500 spectrometer at 120 K and 0.064 milliwatt power, 100 kHz modulation frequency, 2G modulation amplitude, and a time constant of 10.24 ms.
Results
Fenamic Acid Binding to Human COX-2
For structural characterization of the huCOX-2 fenamate complexes, we utilized a construct that had residues 586–612 deleted from the C terminus of the enzyme (30). This region of the C terminus corresponds to the 27-amino acid instability motif involved in the degradation of COX-2 (44, 45). After reconstituting huCOX-2 with Co3+-protoporphyrin IX, we generated crystals of the enzyme in complex with mefenamic acid, flufenamic acid, and tolfenamic acid via co-crystallization. To generate huCOX-2 in complex with meclofenamic acid, we soaked the inhibitor directly into preformed crystals. The crystal structures of huCOX-2 in complex with flufenamic acid, meclofenamic acid, mefenamic acid, and tolfenamic acid were determined to resolutions of 2.5, 2.4, 2.35, and 2.45 Å, respectively, utilizing synchrotron radiation (Table 1). Despite the differences in cell parameters for the meclofenamic acid complex compared with the other three complexes, all four structures crystallized in space group I222. Moreover, the overall domain architecture of the huCOX-2 dimer is conserved in each structure, and there are no significant structural differences observed between monomers of the complexes when they are superimposed.
In each crystal structure we observed interpretable electron density corresponding to bound inhibitor within the cyclooxygenase channel of both monomers of the huCOX-2 dimer (Fig. 1). All four of the fenamic acid inhibitors bound in an inverted orientation, with their carboxylate groups located near the side chains of Tyr-385 and Ser-530 at the apex of the channel, similar to that seen previously in crystal structures of diclofenac and lumiracoxib bound to murine COX-2 (26, 27, 46). In this orientation hydrogen bonds are formed between the carboxylate group of the inhibitor and the phenolic oxygen of Tyr-385 and the hydroxyl oxygen of Ser-530. Outside of these two hydrogen bonds, the remaining contacts between inhibitor and the residues lining the cyclooxygenase channel are via van der Waals interactions.
FIGURE 1.
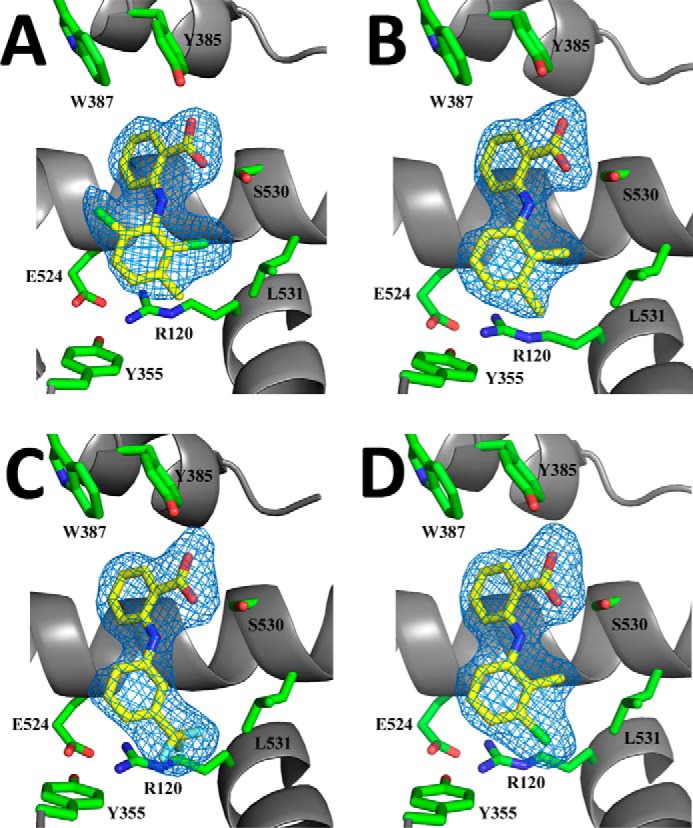
Fenamic acid derivatives bound in the cyclooxygenase channel of human COX-2. Simulated annealing FO - FC omit maps (blue), contoured at 3σ for meclofenamic acid (A), mefenamic acid (B), flufenamic acid (C), and tolfenamic acid (D) bound within the cyclooxygenase channel of monomer B in each complex. For each drug, carbon, nitrogen, and oxygen atoms are colored yellow, blue, and red, respectively. The chlorine atoms in meclofenamic acid and tolfenamic acid are colored green, whereas the fluorine atoms in flufenamic acid are colored cyan. Residues lining the cyclooxygenase channel are labeled accordingly.
We utilized a thermal shift assay in conjunction with a thiol-based fluorescent dye to quantify the contribution that Tyr-385 and Ser-530 had on the binding of the fenamic acid inhibitors in the cyclooxygenase channel. 50 μm concentrations of inhibitor was added to wild type and mutant constructs of huCOX-2, including Y385F, S530A, and the Y385F/S530A double mutant followed by the addition of CPM. Thermal denaturation was then carried out as described under “Experimental Procedures” (Fig. 2). The binding of each of the fenamic acid inhibitors to wild type huCOX-2 resulted in a 17–20 °C shift in enzyme melting temperature, indicating that inhibitor binding significantly stabilizes the enzyme compared with the unliganded state. When the Y385F huCOX-2 construct was utilized, melting temperatures were decreased ∼7 °C on average compared with wild type enzyme, thus providing a measure of the contribution that the hydrogen bond between the carboxylate group and Tyr-385 plays in inhibitor binding. Conversely, fenamic acid binding to S530A huCOX-2 resulted in the same shifts in melting temperature observed for the wild type enzyme despite the loss of a hydrogen bond between Ser-530 and the carboxylate group. These results are in line with previous studies that ruled out Ser-530 as an important determinant for time-dependent inhibition of muCOX-2 by meclofenamic acid and competitive inhibition of muCOX-2 by mefenamic acid (26). The Y385F/S530A double mutant had an additive affect on the thermal stability of the mutant construct, with melting temperatures decreased ∼14 °C on average compared with wild type enzyme. Clearly, removal of the hydrogen bonding partners for the fenamic acid carboxylate group decreases inhibitor affinity for the cyclooxygenase channel. Similar trends in melting temperatures were observed for diclofenac, a close structural homolog of meclofenamic acid, with the exception of the Y385F/S530A double mutant, which exhibited no shift in melting temperature upon inhibitor binding.
FIGURE 2.
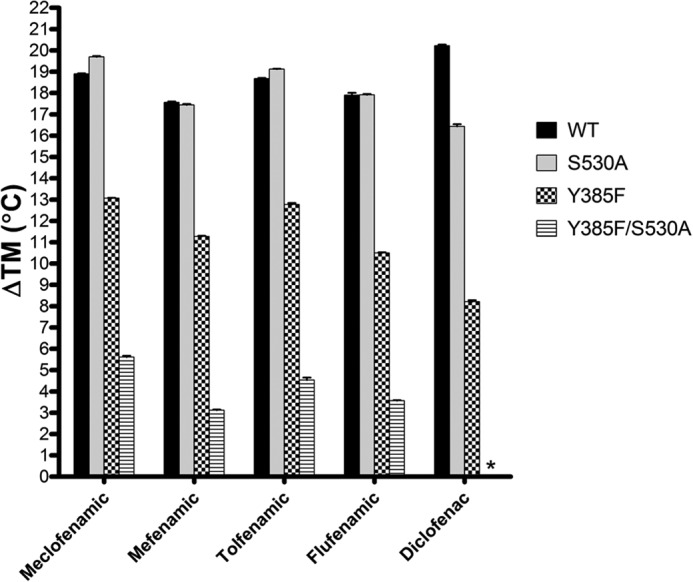
Thermal melting temperature shifts induced by fenamic acid inhibitor binding to COX-2. huCOX-2 was incubated with 50 μm concentrations of the indicated inhibitor before measuring the melting temperature as described under “Experimental Procedures.” Results are reported as the difference in melting temperature (ΔTm) between ligand bound and apo states of COX-2. Melting temperature measurements were made in triplicate ± S.E.
Substrate-selective Inhibition of Human COX-2 by Fenamic Acids
Previous studies have shown that mefenamic acid is a weak competitive inhibitor of AA oxygenation but acts as a potent non-competitive inhibitor of 2-arachidonoyl glycerol oxygenation (19). To examine the substrate-selective inhibition properties of flufenamic acid, meclofenamic acid, and tolfenamic acid, we measured instantaneous cyclooxygenase inhibition using an oxygen electrode. The reaction cuvette contained 500 nm inhibitor and either 100 μm AA or 1-AG as the substrate, with the reaction initiated via the addition of enzyme. At 500 nm, none of the fenamic acids inhibited the oxygenation of AA (Fig. 3). Meclofenamic acid did not inhibit 1-AG oxygenation, whereas flufenamic acid, mefenamic acid, and tolfenamic acid all resulted in decreased oxygenation of 1-AG (Fig. 3). The degree to which these three inhibitors reduced the oxygenation of 1-AG differed, with mefenamic acid resulting in an ∼50% decrease in the oxygenation, whereas flufenamic acid and tolfenamic acid resulted in virtually complete inhibition of substrate oxygenation.
FIGURE 3.
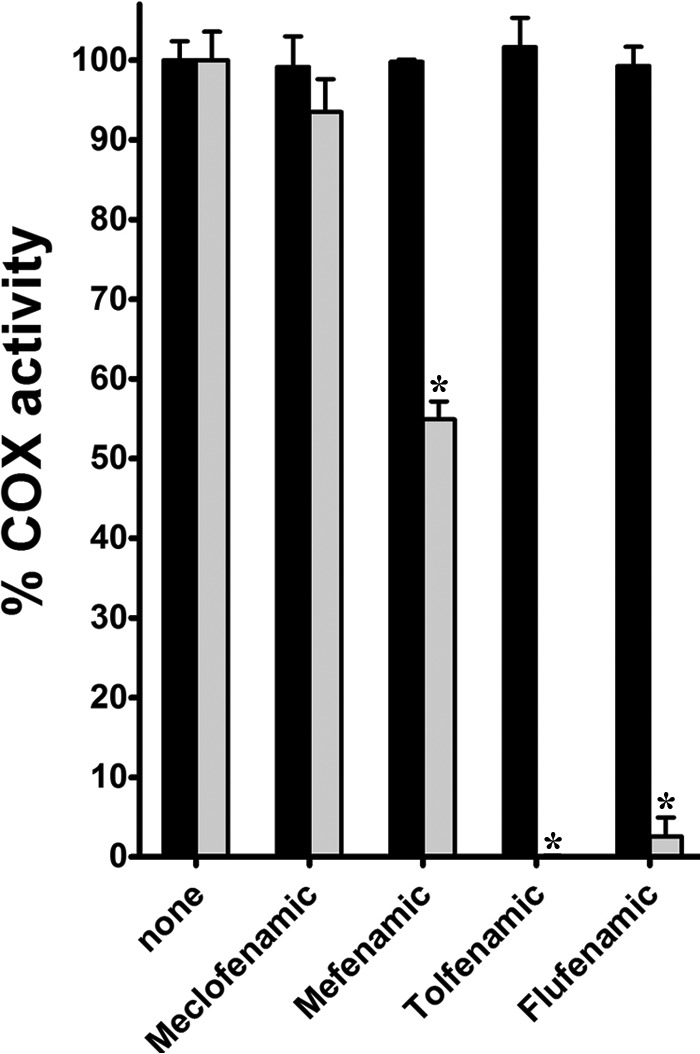
Instantaneous inhibition of AA and 1-AG oxygenation by fenamic acid derivatives. Shown is a bar graph depicting the relative cyclooxygenase activity remaining after injection of 5 μg purified huCOX-2 into reaction cuvettes containing 500 nm inhibitor and AA or 1-AG as described under “Experimental Procedures.” Measurements were determined in triplicate, and activity was recorded as the maximal rate of oxygen consumption ± S.D. Oxygenation of AA or 1-AG in the absence of inhibitor was normalized to 100% activity. *, p < 0.05, as determined by t test.
To further investigate substrate-selective inhibition by flufenamic acid, mefenamic acid, and tolfenamic acid, we evaluated the inhibition of 1-AG oxygenation at various substrate and inhibitor concentrations. All three inhibitors exhibited dose-dependent inhibition of 1-AG oxygenation (Fig. 4). Analysis of the calculated kinetic parameters for flufenamic acid and tolfenamic acid show that both the Vmax and Km decrease as the inhibitor concentration increases (Table 2). This type of behavior is most consistent with uncompetitive inhibition, suggesting that the fenamic acids inhibit 1-AG oxygenation either by binding to the enzyme-substrate complex or by interacting with the enzyme intermediates generated upon reaction with substrate.
FIGURE 4.
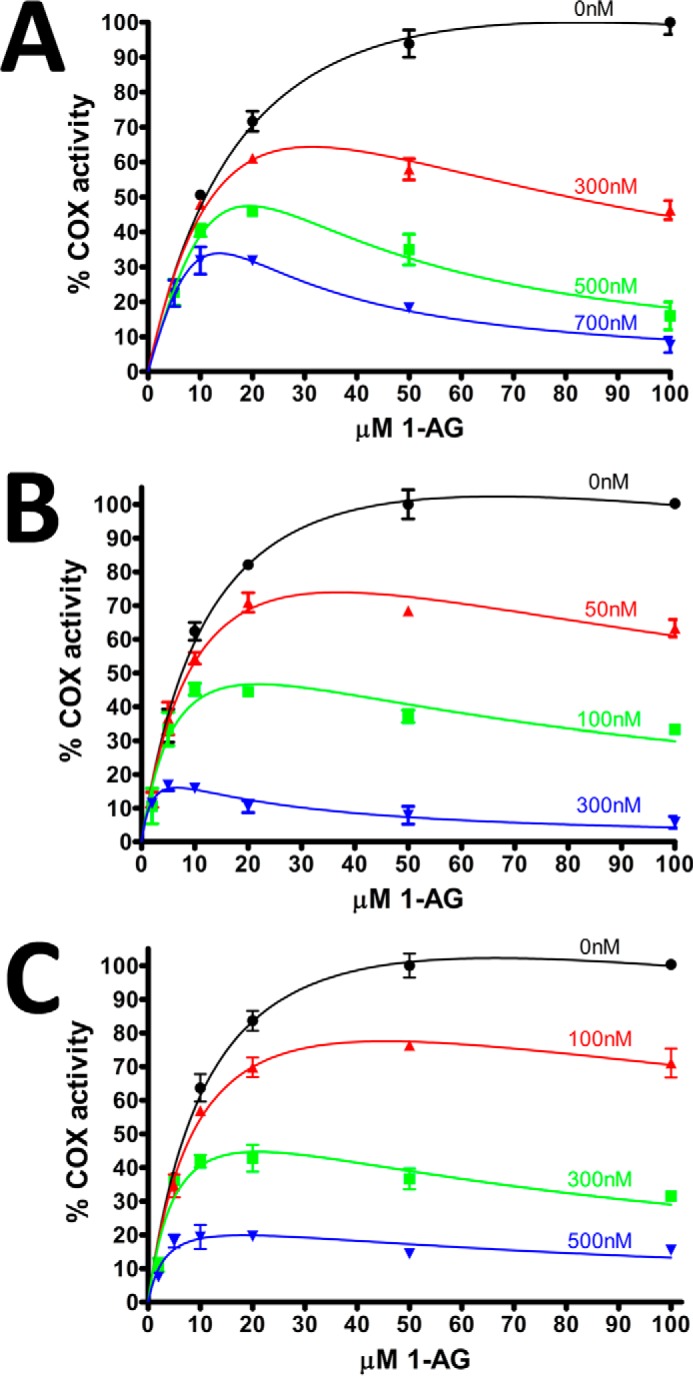
Instantaneous inhibition of 1-AG oxygenation by varying concentrations of fenamic acids. Plots of instantaneous inhibition of huCOX-2 by mefenamic acid (A), tolfenamic acid (B), and flufenamic acid (C). Cyclooxygenase activity was measured using an oxygen electrode as described under “Experimental Procedures.” Kinetic profiles were subsequently fit to a model of substrate-inhibition in GraphPad Prism 5. All measurements were made in duplicate ± S.D.
TABLE 2.
Calculated kinetic parameters for flufenamic acid and tolfenamic acid inhibition of huCOX-2 as a function of inhibitor concentration
Measurements were carried out in duplicate ± S.D.
| Tolfenamic acid |
Flufenamic acid |
||||
|---|---|---|---|---|---|
| [Inhibitor] | Km | VMAX | [Inhibitor] | Km | VMAX |
| nm | μm | >nmol O2/min | nm | μm | nmol O2/min |
| 0 | 14.4 ± 2.6 | 146.7 ± 14.1 | 0 | 12.8 ± 1.9 | 141.6 ± 10.7 |
| 50 | 12.0 ± 2.9 | 121.8 ± 16.6 | 100 | 10.1 ± 1.9 | 111.8 ± 10.3 |
| 100 | 7.3 ± 3.2 | 78.0 ± 17.5 | 300 | 6.0 ± 2.4 | 70.1 ± 13.5 |
| 300 | 2.4 ± 1.4 | 27.5 ± 7.1 | 500 | 3.5 ± 1.7 | 27.4 ± 5.2 |
We next monitored the quenching of wild type huCOX-2 tryptophan fluorescence in the presence of the fenamic acids to evaluate the time-dependent nature of inhibitor binding (Fig. 5). Earlier investigations revealed that only rapidly reversible, competitive compounds were substrate-selective inhibitors of COX-2 (19, 20, 27, 47). Both flufenamic acid and tolfenamic acid quenched the native tryptophan fluorescence of huCOX-2 very rapidly. The binding of these two inhibitors occurred almost completely within the mixing time of the instrument, thus precluding an accurate assessment of binding rate. Mefenamic acid also bound rapidly to huCOX-2, with a rate constant of 0.18 ± 0.01 s−1. Conversely, the binding of meclofenamic acid was an order of magnitude slower (0.02 ± 0.01 s−1), with maximal quenching reached ∼4 min post mixing. Taken together, our results indicate that flufenamic acid and tolfenamic acid are time-independent substrate-selective inhibitors of COX-2, consistent with previous studies carried out utilizing mefenamic acid (19). Our results are also consistent with meclofenamic acid acting as a time-dependent inhibitor lacking substrate selectivity.
FIGURE 5.
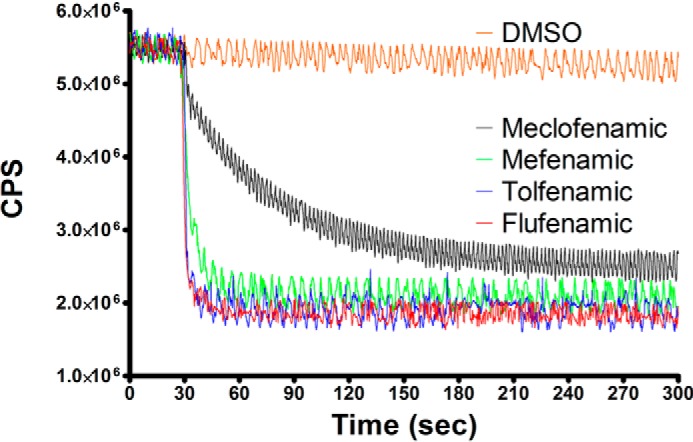
Tryptophan fluorescence quenching of huCOX-2 by fenamic acids. Fenamic acid derivatives were added after 30 s to 0.26 μm huCOX-2 to a final concentration of 1 μm, and native tryptophan fluorescence was monitored with λex 283 nm and λem 335 nm as described under “Experimental Procedures.” Measurements were carried out in triplicate.
Fenamic Acids as Reducing Co-substrates of the Heme Moiety
Various compounds, including NSAIDs, have been shown to act as reducing co-substrates for the peroxidase reaction carried out by COX enzymes (48). After the reaction with peroxide, the heme moiety is oxidized from its resting ferric state to an oxy-ferryl porphyrin cation radical denoted as Compound I. After electron transfer from Tyr-385, Compound I is reduced to the oxy-ferryl state with a fully covalent porphyrin ring, denoted as Compound II. A subsequent one-electron reduction of Compound II by reducing co-substrate generates the resting ferric state of the heme moiety. To probe the ability of the fenamic acids to reduce Compound II, we utilized UV-visible spectroscopy to monitor the soret peak and α/β bands of the heme moiety following reaction with peroxide substrate. When Fe3+-protoporphyrin IX-reconstituted huCOX-2 was reacted with 10-fold excess peroxide, we observed the characteristic shift in soret peak absorbance from 409 nm to 415 nm and the formation of α/β bands at 525 nm and 555 nm (Fig. 6A). When the same reaction was performed in the presence of tolfenamic acid, no shift in the soret peak absorbance or formation of α/β bands was observed (Fig. 6B). This result suggests that tolfenamic acid acts as a reducing co-substrate by donating an electron to reduce Compound II back to the resting ferric state.
FIGURE 6.

Effect of fenamic acids on the heme oxidation states in huCOX-2. Visible spectra for Fe3+-protoporphyrin IX reconstituted huCOX-2 in the absence (A) and presence (B) of 10 μm tolfenamic acid. Spectra were recorded every 10 s. The red line denotes the resting state of the heme moiety before the addition of hydrogen peroxide. C, reconstituted huCOX-2 at a concentration of 3 μm monomer was reacted with 15 μm hydrogen peroxide for 45 s to generate compound II. After compound II was generated, fenamic acids were injected to a final concentration of 9 μm. The return of the heme moiety to the resting state was monitored by observing the increase in absorbance at 409 nm.
From the experiment described above, we could not differentiate whether or not tolfenamic acid was acting as a reducing co-substrate or simply preventing the formation of higher oxidation states of the heme entirely. Therefore, a second experiment was conducted wherein Compound II was generated by reacting Fe3+-protoporphyrin IX-reconstituted huCOX-2 with peroxide for 40 s followed by the addition of flufenamic acid, mefenamic acid, or tolfenamic acid. Soret peak absorbance at 409 nm was monitored to follow the return of the heme moiety to the resting ferric state. Compared with a control where no inhibitor was added to the reaction, all three inhibitors induced a rapid return of soret peak absorbance at 409 nm (Fig. 6C). This result demonstrates that all three fenamic acid inhibitors tested act as reducing co-substrates that donate an electron to reduce Compound II back to the resting ferric state.
Fenamic Acids Quench Tyrosyl Radicals
Previous electron spin resonance (ESR) studies have demonstrated that peroxidase catalysis generates tyrosyl radicals on Tyr-385 and Tyr-504 in huCOX-2 (29, 49). Furthermore, the binding of certain NSAIDs to huCOX-2 can either destabilize (flurbiprofen and diclofenac) or stabilize (naproxen) radicals formed on Tyr-385 and Tyr-504 (50). Based on the inverted binding orientation observed for the fenamic acids within the cyclooxygenase channel in the crystal structures described above, we hypothesized that binding of this class of inhibitors to huCOX-2 would destabilize preformed tyrosyl radicals on Tyr-385 and Tyr-504. To test this hypothesis, tyrosyl radicals were generated on Tyr-385 and Tyr-504 in Fe3+-protoporphyrin IX-reconstituted huCOX-2 by reacting the enzyme with hydrogen peroxide on ice for 30 s. Mefenamic acid or tolfenamic acid was then injected into the reaction mixture containing preformed tyrosyl radicals and briefly mixed before freezing. Both mefenamic acid and tolfenamic acid induced a decrease in the observed amplitude of the tyrosyl radical ESR signal, indicating that both inhibitors quenched the preformed tyrosyl radicals (Fig. 7, A and B). Double integration of the resultant spectra revealed that mefenamic acid and tolfenamic acid quenched preformed tyrosyl radicals in a dose-dependent fashion (Fig. 7C). Moreover, there was no significant change in line shape associated with the decrease in ESR amplitude, consistent with previous observations that utilized other classes of NSAIDs (50). The wide singlet continuous wave ESR spectrum of tyrosyl radicals in wild type huCOX-2 is composed of contributions from both Tyr-385 radical (wide doublet) and Tyr-504 radical (narrow singlet) (51). The lack of a change in ESR line shape with increasing concentrations of mefenamic acid and tolfenamic acid indicates that the proportions of the Tyr-385 radical and Tyr-504 radical remains constant as the radicals are quenched.
FIGURE 7.

Fenamic acid inhibitors quench preformed huCOX-2 tyrosyl radicals. Tyrosyl radicals were generated by reacting reconstituted huCOX-2 with hydrogen peroxide in ice for 30 s. Varying concentrations of mefenamic acid (A) and tolfenamic acid (B) were then added and mixed for 2–3 s before freezing the sample and collecting ESR spectra. The arrows indicate the decrease in ESR spectra amplitude as the inhibitor concentration is increased. C, calculation of the total radical yield, based on double integration of the spectra in A and B. The double integral of the spectrum with no added inhibitor was normalized to 100% radical yield.
Exogenous Lipid Hydroperoxide Alleviates Substrate-selective Inhibition
Compared with AA, the oxygenation of endocannabinoids by COX-2 is more sensitive to peroxide tone (17). Higher levels of peroxide are required to sustain maximal rates of endocannabinoid oxygenation compared with AA oxygenation. The ability of fenamic acids to reduce both tyrosyl radicals and higher oxidation states of heme in COX-2 pushes the enzyme into the resting state, thus requiring another turnover of peroxide in order to generate a tyrosyl radical to sustain endocannabinoid oxygenation. We reasoned that adding excess peroxide would serve to alleviate the inhibition of endocannabinoid oxygenation that is achieved with relatively low concentrations of fenamic acids. To test this hypothesis, we analyzed the extent of inhibition of 1-AG oxygenation by tolfenamic acid in the presence of increasing concentrations of the lipid hydroperoxide 15-HPETE. The addition of 15-HPETE had no significant effect on the rate of 1-AG oxygenation in the absence of tolfenamic acid. However, the ability of tolfenamic acid to inhibit 1-AG oxygenation was significantly diminished in the presence of 15-HPETE (Fig. 8A). The addition of 15-HPETE resulted in a concentration-dependent recovery in the rate of 1-AG oxygenation to ∼70% that observed in the absence of tolfenamic acid. Similar results were observed with mefenamic acid, flufenamic acid, and the non-fenamic acid substrate-selective inhibitor R-flurbiprofen (Fig. 8, B–D). Collectively, these results demonstrate that the substrate-selective inhibition of endocannabinoid oxygenation by fenamic acids is dependent upon the peroxide tone and that substrate-selective inhibition can be attenuated via the addition of excess lipid hydroperoxide.
FIGURE 8.

15-HPETE alleviates inhibition of 1-AG by substrate-selective inhibitors. Shown is a bar graph depicting the relative cyclooxygenase activity of huCOX-2 in the absence and presence of 100 nm tolfenamic acid (Tolf, A), 500 nm mefenamic acid (B), 300 nm flufenamic acid (C), and 20 μm R-flurbiprofen (D) and varying concentrations of 15-HPETE. Measurements were carried out in triplicate ± S.E.
Discussion
The x-ray crystal structures of huCOX-2 in complex with flufenamic acid, meclofenamic acid, mefenamic acid, and tolfenamic acid reveal that all four inhibitors bind in an inverted orientation within the cyclooxygenase channel of each monomer of the homodimer. In this orientation the inhibitor's carboxylate group forms hydrogen bonds with the side chains of Tyr-385 and Ser-530 at the apex of the channel similar to that observed in the crystal structures of muCOX-2 in complex with diclofenac and lumiracoxib (26, 27). All four of the fenamic acid inhibitors bound in nearly identical orientations within the cyclooxygenase channel regardless of their ability to inhibit COX-2 in a time-dependent or substrate-selective fashion. The crystal structures presented here failed to provide insight into the residues responsible for signaling across the dimer interface, as we did not observe protein conformational differences between monomers when they were compared. However, by virtue of their interactions with Tyr-385 at the apex of the channel, the structures did provide insight into why the fenamic acids have a propensity to reduce the tyrosine radical required for substrate oxygenation.
Crystal structures of muCOX-2 in complex with the substrate-selective inhibitors ibuprofen, R-naproxen, and R-flurbiprofen reveal that the carboxylate group of the inhibitor forms a hydrogen bond with the side chain of Arg-120 at the opening of the cyclooxygenase channel (20, 25). Subsequent mutation of Arg-120 abolishes the ability of these compounds to inhibit in a substrate-selective manner, indicating that stabilization of the carboxylate moiety is an important determinant for these inhibitors. Additionally, no shift in melting temperature was observed when ibuprofen binding to R120A muCOX-2 was evaluated using the thermal shift assay (25). Conversely, the inverted binding orientation observed in the crystal structures and the ability of the fenamic acids to bind to the Y385F/S530A huCOX-2 double mutant indicates that stabilization of the carboxylate group is less important for substrate-selective inhibition by this class of drugs.
A single turnover at the peroxidase active site is required to generate a catalytic radical centered on Tyr-385 at the apex of the cyclooxygenase channel. The incipient tyrosyl radical initiates cyclooxygenase catalysis through the abstraction of the 13-pro-S hydrogen from AA (or 1-AG) and is subsequently regenerated upon formation of the prostaglandin G2 product. The tyrosyl radical can continue to be involved in multiple H abstractions from AA in the absence of another turnover at the peroxidase site until the enzyme undergoes suicide inactivation (1). Although little is known about the chemical changes that occur to the enzyme during suicide inactivation, the number of cyclooxygenase turnovers can vary from 10 to 1000s before its inactivation, depending upon the reaction conditions (51).
Recent studies have shown that a higher concentration of peroxide substrate is required to activate and maintain the cyclooxygenase activity for oxygenation of endocannabinoid substrates compared with the concentration needed for AA oxygenation (17). The peroxidase activation efficiencies of prostaglandin G2 and prostaglandin G2 glycerol ester are likely similar. However, Musee and Marnett (17) suggest that release of a prostaglandin G2-G peroxyl radical before oxidation of Tyr-385 would result in inactive enzyme that would need to be activated by turnover of another molecule of hydroperoxide at the peroxidase active site. As a consequence, feedback activation of peroxidase catalysis by the hydroperoxide products generated during cyclooxygenase catalysis is more critical during the oxygenation of endocannabinoids versus AA. The biophysical data presented above reveal that flufenamic acid, mefenamic acid, and tolfenamic acid bind rapidly to the enzyme, act as reducing co-substrates through the donation of an electron to reduce Compound II back to the resting ferric state, and quench preformed tyrosyl radicals. Thus, the substrate-selective fenamic acids effectively “reset” the enzyme such that another turnover at the peroxidase active site is required to reinitiate cyclooxygenase catalysis.
We show that the addition of 15-HPETE alleviates substrate-selective inhibition by flufenamic acid, mefenamic acid, and tolfenamic acid, presumably through its turnover in the peroxidase active site, resulting in the subsequent generation of tyrosyl radical in the cyclooxygenase active site. One alternative explanation for the rescue of endocannabinoid oxygenation upon the addition of 15-HPETE is that the peroxide modifies the fenamic acid scaffold such that they no longer serve as effective inhibitors. 15-HPETE did not alter the UV-visible spectra of tolfenamic acid as previously seen when fenamic acids are oxidized (data not shown) (53). Moreover, 15-HPETE effectively rescues endocannabinoid oxygenation in the presence of R-flurbiprofen, a substrate-selective inhibitor that is chemically unrelated to the fenamic acids. Taken together, our data strongly suggest that the inhibition of endocannabinoid oxygenation by the fenamic acids is dependent upon peroxide tone (Fig. 9).
FIGURE 9.
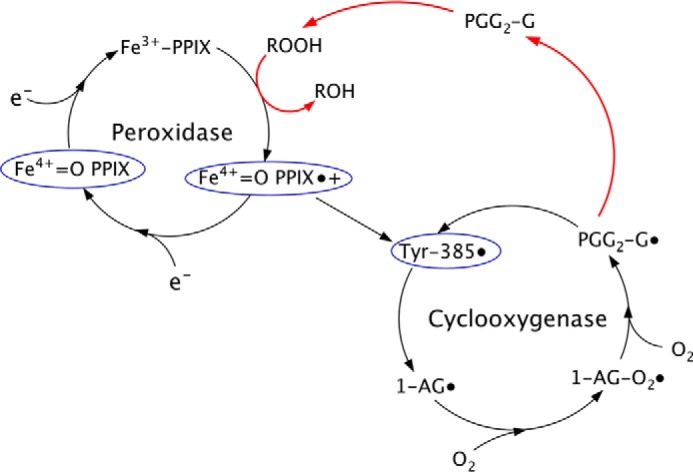
Effect of fenamic acids on endocannabinoid oxygenation. Fenamic acid inhibitors reduce higher oxidation states of the heme moiety and tyrosyl radicals (blue circles), resetting COX-2 back to the resting state. Feedback activation by peroxides (red arrow) is more important for sustaining endocannabinoid oxygenation than AA oxygenation. This feedback activation pathway can be stimulated by the addition of 15-HPETE in order to overcome the reductive effects of the fenamic acid inhibitors and alleviate substrate-selective inhibition. PGG2-G, prostaglandin G2 glycerol ester.
The role of reducing co-substrates in cyclooxygenase catalysis is enigmatic. Reducing co-substrates have a stimulatory effect on AA oxygenation at low concentrations, but above a certain threshold this stimulatory effect is lost. Computer simulations based on the branched chain mechanism suggest that this stimulatory effect is a result of extending the catalytic lifetime of COX-2 before the onset of suicide inactivation (54). However, endocannabinoid oxygenation requires higher levels of peroxide to maintain peak cyclooxygenase activity, suggesting that the branched chain mechanism is less efficient with endocannabinoids compared with AA. It may be the case that reducing co-substrates have differential effects on cyclooxygenase catalysis when endocannabinoids versus AA are utilized as the cyclooxygenase substrate. Previous investigations utilizing horseradish peroxidase in the presence of mefenamic acid have revealed that, upon reaction with hydrogen peroxide, transient mefenamic acid radicals are generated (53). By donating an electron to oxy-ferryl heme in COX-2, it is likely that transient fenamic acid cation radicals are generated. Additional studies are required to determine the effect that these radicals have on feedback activation of peroxidase activity to sustain endocannabinoid oxygenation.
The discovery that substrate-selective inhibition of endocannabinoid oxygenation by COX-2 is dependent on peroxide tone has important implications in vivo. The observed effect of peroxide tone on substrate-selective inhibition by fenamic acid inhibitors is reminiscent of similar effects seen with acetaminophen. Acetaminophen is a more potent inhibitor of prostaglandin formation in tissues with low peroxide tone, such as nervous tissue (55). The beneficial anxiolytic and analgesic effects observed with substrate-selective COX-2 inhibitors is directly correlated with increased levels of endocannabinoids in the central nervous system (21). As peroxide tone is low in the central nervous system, the fenamic acid inhibitors would be expected to be more effective substrate-selective inhibitors within this tissue.
Author Contributions
B. J. O. and M. G. M. coordinated the study and wrote the paper. B. J. O. carried out all experiments. Both authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Elka R. Georgieva and Peter P. Borbat for assistance with ESR spectroscopic measurements. X-ray diffraction experiments were conducted at the Advanced Photon Source. Use of IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by companies of the Industrial Macromolecular Crystallography Association through a contract with the Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM115386. ESR spectroscopic measurements were carried out at the National Biomedical Center for Advanced Electron Spin Resonance Technology, supported by National Institutes of Health grant P41 GM103521. GM/CA CAT is funded in whole or in part with federal funds from the National Institutes of Health Grants Y1-CO-1020 (NCI) and Y1-GM-1104 NIGMS). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5IKV, 5IKQ, 5IKR, and 5IKT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AA
- arachidonic acid
- 1-AG
- 1-aracidonoyl glycerol
- 15-HPETE
- 15-hydroperoxyeicosatetraenoic acid
- βOG
- n-octyl-β-d-glucopyranoside
- CPM
- 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin
- ESR
- electron spin resonance
- hu-
- human
- mu-
- murine
- PEG
- polyethylene glycol
- NSAID
- nonsteroidal anti-inflammatory drugs.
References
- 1. Smith W. L., Urade Y., and Jakobsson P. J. (2011) Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 111, 5821–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hata A. N., and Breyer R. M. (2004) Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 103, 147–166 [DOI] [PubMed] [Google Scholar]
- 3. Laneuville O., Breuer D. K., Xu N., Huang Z. H., Gage D. A., Watson J. T., Lagarde M., DeWitt D. L., and Smith W. L. (1995) Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2: formation of 12-hydroxy-(9Z,13E/Z,15Z)-octadecatrienoic acids from α-linolenic acid. J. Biol. Chem. 270, 19330–19336 [DOI] [PubMed] [Google Scholar]
- 4. Rouzer C. A., and Marnett L. J. (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 111, 5899–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowlinson S. W., Crews B. C., Lanzo C. A., and Marnett L. J. (1999) The binding of arachidonic acid in the cyclooxygenase active site of mouse prostaglandin endoperoxide synthase-2 (COX-2): a putative l-shaped binding conformation utilizing the top channel region. J. Biol. Chem. 274, 23305–23310 [DOI] [PubMed] [Google Scholar]
- 6. Vecchio A. J., Orlando B. J., Nandagiri R., and Malkowski M. G. (2012) Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 287, 24619–24630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wada M., DeLong C. J., Hong Y. H., Rieke C. J., Song I., Sidhu R. S., Yuan C., Warnock M., Schmaier A. H., Yokoyama C., Smyth E. M., Wilson S. J., FitzGerald G. A., Garavito R. M., Sui de X., Regan J. W., and Smith W. L. (2007) Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J. Biol. Chem. 282, 22254–22266 [DOI] [PubMed] [Google Scholar]
- 8. Yuan C., Sidhu R. S., Kuklev D. V., Kado Y., Wada M., Song I., and Smith W. L. (2009) Cyclooxygenase allosterism, fatty acid-mediated cross-talk between monomers of cyclooxygenase homodimers. J. Biol. Chem. 284, 10046–10055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bisogno T. (2008) Endogenous cannabinoids: structure and metabolism. J. Neuroendocrinol 20, 1–9 [DOI] [PubMed] [Google Scholar]
- 10. Kozak K. R., Crews B. C., Ray J. L., Tai H. H., Morrow J. D., and Marnett L. J. (2001) Metabolism of prostaglandin glycerol esters and prostaglandin ethanolamides in vitro and in vivo. J. Biol. Chem. 276, 36993–36998 [DOI] [PubMed] [Google Scholar]
- 11. Kozak K. R., Prusakiewicz J. J., and Marnett L. J. (2004) Oxidative metabolism of endocannabinoids by COX-2. Curr. Pharm. Des. 10, 659–667 [DOI] [PubMed] [Google Scholar]
- 12. Kozak K. R., Rowlinson S. W., and Marnett L. J. (2000) Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J. Biol. Chem. 275, 33744–33749 [DOI] [PubMed] [Google Scholar]
- 13. Pacher P., Bátkai S., and Kunos G. (2006) The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 58, 389–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei C., Kulmacz R. J., and Tsai A. L. (1995) Comparison of branched-chain and tightly coupled reaction mechanisms for prostaglandin H synthase. Biochemistry 34, 8499–8512 [DOI] [PubMed] [Google Scholar]
- 15. Vecchio A. J., Simmons D. M., and Malkowski M. G. (2010) Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 285, 22152–22163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vecchio A. J., and Malkowski M. G. (2011) The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J. Biol. Chem. 286, 20736–20745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Musee J., and Marnett L. J. (2012) Prostaglandin H synthase-2-catalyzed oxygenation of 2-arachidonoylglycerol is more sensitive to peroxide tone than oxygenation of arachidonic acid. J. Biol. Chem. 287, 37383–37394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blobaum A. L., and Marnett L. J. (2007) Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 50, 1425–1441 [DOI] [PubMed] [Google Scholar]
- 19. Prusakiewicz J. J., Duggan K. C., Rouzer C. A., and Marnett L. J. (2009) Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 48, 7353–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duggan K. C., Hermanson D. J., Musee J., Prusakiewicz J. J., Scheib J. L., Carter B. D., Banerjee S., Oates J. A., and Marnett L. J. (2011) (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 7, 803–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hermanson D. J., Gamble-George J. C., Marnett L. J., and Patel S. (2014) Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol. Sci. 35, 358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan C., Rieke C. J., Rimon G., Wingerd B. A., and Smith W. L. (2006) Partnering between monomers of cyclooxygenase-2 homodimers. Proc. Natl. Acad. Sci. U.S.A. 103, 6142–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dong L., Vecchio A. J., Sharma N. P., Jurban B. J., Malkowski M. G., and Smith W. L. (2011) Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J. Biol. Chem. 286, 19035–19046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kudalkar S. N., Nikas S. P., Kingsley P. J., Xu S., Galligan J. J., Rouzer C. A., Banerjee S., Ji L., Eno M. R., Makriyannis A., and Marnett L. J. (2015) 13-Methylarachidonic acid is a positive allosteric modulator of endocannabinoid oxygenation by cyclooxygenase. J. Biol. Chem. 290, 7897–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orlando B. J., Lucido M. J., and Malkowski M. G. (2015) The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 189, 62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rowlinson S. W., Kiefer J. R., Prusakiewicz J. J., Pawlitz J. L., Kozak K. R., Kalgutkar A. S., Stallings W. C., Kurumbail R. G., and Marnett L. J. (2003) A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 278, 45763–45769 [DOI] [PubMed] [Google Scholar]
- 27. Windsor M. A., Valk P. L., Xu S., Banerjee S., and Marnett L. J. (2013) Exploring the molecular determinants of substrate-selective inhibition of cyclooxygenase-2 by lumiracoxib. Bioorg Med. Chem Lett 23, 5860–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Graff G., Anderson L. A., and Jaques L. W. (1990) Preparation and purification of soybean lipoxygenase-derived unsaturated hydroperoxy and hydroxy fatty acids and determination of molar absorptivities of hydroxy fatty acids. Anal. Biochem. 188, 38–47 [DOI] [PubMed] [Google Scholar]
- 29. Orlando B. J., Borbat P. P., Georgieva E. R., Freed J. H., and Malkowski M. G. (2015) Pulsed dipolar spectroscopy reveals that tyrosyl radicals are generated in both monomers of the cyclooxygenase-2 dimer. Biochemistry 54, 7309–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lucido M. J., Orlando B. J., Vecchio A. J., and Malkowski M. G. (2016) Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry 55, 1226–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vecchio A. J., and Malkowski M. G. (2011) The structure of NS-398 bound to cyclooxygenase-2. J. Struct. Biol. 176, 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koszelak-Rosenblum M., Krol A., Mozumdar N., Wunsch K., Ferin A., Cook E., Veatch C. K., Nagel R., Luft J. R., Detitta G. T., and Malkowski M. G. (2009) Determination and application of empirically derived detergent phase boundaries to effectively crystallize membrane proteins. Protein Sci. 18, 1828–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luft J. R., Snell E. H., and Detitta G. T. (2011) Lessons from high-throughput protein crystallization screening: 10 years of practical experience. Expert Opin Drug. Discov 6, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 35. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen S. X., Ben Jelloul M., Long F., Vagin A., Knipscheer P., Lebbink J., Sixma T. K., Lamzin V. S., Murshudov G. N., and Perrakis A. (2008) ARP/wARP and molecular replacement: the next generation. Acta Crystallogr. D Biol. Crystallogr. 64, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 41. Painter J., and Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 [Google Scholar]
- 42. Luong C., Miller A., Barnett J., Chow J., Ramesha C., and Browner M. F. (1996) Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 3, 927–933 [DOI] [PubMed] [Google Scholar]
- 43. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mbonye U. R., Wada M., Rieke C. J., Tang H. Y., Dewitt D. L., and Smith W. L. (2006) The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. J. Biol. Chem. 281, 35770–35778 [DOI] [PubMed] [Google Scholar]
- 45. Mbonye U. R., Yuan C., Harris C. E., Sidhu R. S., Song I., Arakawa T., and Smith W. L. (2008) Two distinct pathways for cyclooxygenase-2 protein degradation. J. Biol. Chem. 283, 8611–8623 [DOI] [PubMed] [Google Scholar]
- 46. Blobaum A. L., Xu S., Rowlinson S. W., Duggan K. C., Banerjee S., Kudalkar S. N., Birmingham W. R., Ghebreselasie K., and Marnett L. J. (2015) Action at a distance: mutations of peripheral residues transform rapid reversible inhibitors to slow, tight binders of cyclooxygenase-2. J. Biol. Chem. 290, 12793–12803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Windsor M. A., Hermanson D. J., Kingsley P. J., Xu S., Crews B. C., Ho W., Keenan C. M., Banerjee S., Sharkey K. A., and Marnett L. J. (2012) Substrate-selective inhibition of cyclooxygenase-2: development and evaluation of achiral profen probes. ACS Med. Chem. Lett. 3, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Markey C. M., Alward A., Weller P. E., and Marnett L. J. (1987) Quantitative studies of hydroperoxide reduction by prostaglandin H synthase: reducing substrate specificity and the relationship of peroxidase to cyclooxygenase activities. J. Biol. Chem. 262, 6266–6279 [PubMed] [Google Scholar]
- 49. Rogge C. E., Liu W., Wu G., Wang L. H., Kulmacz R. J., and Tsai A. L. (2004) Identification of Tyr504 as an alternative tyrosyl radical site in human prostaglandin H synthase-2. Biochemistry 43, 1560–1568 [DOI] [PubMed] [Google Scholar]
- 50. Wu G., Tsai A. L., and Kulmacz R. J. (2009) Cyclooxygenase competitive inhibitors alter tyrosyl radical dynamics in prostaglandin H synthase-2. Biochemistry 48, 11902–11911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsai A. L., and Kulmacz R. J. (2010) Prostaglandin H synthase: resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 493, 103–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deleted in proof
- 53. Muraoka S., and Miura T. (2003) Inactivation of creatine kinase during the interaction of mefenamic acid with horseradish peroxidase and hydrogen peroxide: participation by the mefenamic acid radical. Life Sci. 72, 1897–1907 [DOI] [PubMed] [Google Scholar]
- 54. Bambai B., and Kulmacz R. J. (2000) Prostaglandin H synthase: effects of peroxidase cosubstrates on cyclooxygenase velocity. J. Biol. Chem. 275, 27608–27614 [DOI] [PubMed] [Google Scholar]
- 55. Aronoff D. M., Oates J. A., and Boutaud O. (2006) New insights into the mechanism of action of acetaminophen: Its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clin. Pharmacol. Ther 79, 9–19 [DOI] [PubMed] [Google Scholar]
- 56. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]