

Abstract
Progesterone or its analog, one of components of hormone replacement therapy, may attenuate the cardioprotective effects of estrogen. However, the underlying mechanisms have not been fully elucidated. Expression of CD36, a receptor for oxidized LDL (oxLDL) that enhances macrophage/foam cell formation, is activated by the transcription factor peroxisome proliferator-activated receptor γ (PPARγ). CD36 also functions as a fatty acid transporter to influence fatty acid metabolism and the pathophysiological status of several diseases. In this study, we determined that progesterone induced macrophage CD36 expression, which is related to progesterone receptor (PR) activity. Progesterone enhanced cellular oxLDL uptake in a CD36-dependent manner. Mechanistically, progesterone increased PPARγ expression and PPARγ promoter activity in a PR-dependent manner and the binding of PR with the progesterone response element in the PPARγ promoter. Specific deletion of macrophage PPARγ (MφPPARγ KO) expression in mice abolished progesterone-induced macrophage CD36 expression and cellular oxLDL accumulation. We also determined that, associated with gestation and increased serum progesterone levels, CD36 and PPARγ expression in mouse adipose tissue, skeletal muscle, and peritoneal macrophages were substantially activated. Taken together, our study demonstrates that progesterone can play dual pathophysiological roles by activating PPARγ expression, in which progesterone increases macrophage CD36 expression and oxLDL accumulation, a negative effect on atherosclerosis, and enhances the PPARγ-CD36 pathway in adipose tissue and skeletal muscle, a protective effect on pregnancy.
Keywords: atherosclerosis, fatty acid, macrophage, peroxisome proliferator-activated receptor (PPAR), progesterone, CD36, HRT
Introduction
Hormone replacement therapy (HRT,4 estrogen plus progestin) is applied to postmenopausal women to alleviate or/and prevent common complications of female menopause. The initial use of estrogen alone as estrogen replacement therapy for postmenopausal women was reported to cause irregular bleeding and increase the risk of endometrial hyperplasia and carcinoma (1, 2). Later studies demonstrated that addition of a synthetic progesterone (for example, progestogen or progestin, which can demonstrate progestational effects similar to progesterone), can reduce the risk of estrogen-induced bleeding, endometrial hyperplasia, and carcinoma (3). Progesterone is a steroid hormone predominantly produced by the corpus luteum after ovulation and exerts its primary action by activating progesterone receptor (PR) (4).
Compared with men at matched ages before menopause, women have much lower rates of cardiovascular disease, which can be attributed to endogenous estrogen production. Indeed, studies with animal models have confirmed the cardioprotective effects of estrogen. For example, treatment of ovariectomized rabbits or monkeys with estrogen reduces high-fat diet-induced atherosclerosis (5–7). However, both clinical observations and animal studies have demonstrated that addition of progestin in HRT attenuated the inhibitory effects of estrogen on atherosclerosis. Compared with placebo, HRT even increased cardiovascular events, which resulted in earlier termination of HRT clinical trials (8–11). In postmenopausal women with established coronary atherosclerosis, estrogen replacement therapy had little effect on progression of the disease, which suggests the importance of the timing of estrogen intervention in the reduction of atherosclerosis (12, 13).
CD36, an 88-kDa membrane glycoprotein, was first identified as the receptor for uptake of oxidized LDL (oxLDL) by monocytes/macrophages (14, 15). The binding and internalization of oxLDL by macrophage CD36 facilitates the formation of macrophage/foam cells, the prominent part of atherosclerotic lesions. In addition, the interaction between macrophages and oxLDL can trigger both proinflammatory and proatherogenic signaling responses, which further demonstrates that macrophage CD36 can play an important role in the development of atherosclerosis (16–18). Expression of CD36 can also be found in other cell type/tissues, such as platelets, cardiac myocytes, adipose tissue, and skeletal muscle, where CD36 mainly functions as a fatty acid transporter, thereby demonstrating different pathophysiological roles in energy storage/mobilization, inflammation, and insulin resistance. For instance, lack of CD36 expression impairs fatty acid uptake and adaptive fuel flexibility in mouse heart and skeletal muscle (19). In humans, either overexpression or deficiency of CD36 are associated with metabolic abnormalities in patients with metabolic syndrome, diabetes, and nonalcoholic fatty liver disease (20–23).
CD36 expression is transcriptionally regulated by peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated nuclear receptor, because there is a PPARγ-responsive element (PPRE) in the proximal region of the CD36 promoter (24). Thus, both natural and synthetic PPARγ ligands, such as prostaglandin J2 (PGJ2) and thiazolidinediones (a class of medicine-enhancing insulin sensitivity for type 2 diabetes treatment), can increase CD36 expression. Regulation of CD36 expression by some cytokines is also related to PPARγ activity (25–27).
Although the addition of progesterone in HRT may attenuate the cardioprotective effects of estrogen, the underlying molecular mechanisms have not been fully understood. In this study, we initially determined whether progesterone would activate macrophage CD36 expression/oxLDL accumulation and the involved mechanisms. Because of high progesterone production during pregnancy, we then determined changes of CD36 and PPARγ expression in skeletal muscle, adipose tissue, and peritoneal macrophages in pregnant mice, which may suggest the physiological relevance of PPARγ-CD36-mediated fatty acid metabolism for gestation.
Results
Progesterone Induces Macrophage CD36 Expression and Enhances Cellular oxLDL Accumulation
Compared with men, women have much lower rates of coronary heart disease before menopause, which is partially attributed to the cardioprotection of endogenous estrogen production. Estrogen has been reported to inhibit HIV proteases inhibitor-induced macrophage CD36 expression in an estrogen receptor α-dependent manner (28). Macrophage CD36 functions as an oxLDL receptor to facilitate cellular oxLDL accumulation and macrophage/foam cell formation. To determine whether progesterone is able to attenuate estrogen-inhibited CD36 expression, peritoneal macrophages isolated from female C57BL/6 wild-type mice were treated with estradiol in the absence or presence of progesterone, followed by determination of CD36 expression. Similarly, we determined that estrogen reduced CD36 expression (Fig. 1A; in general, one and two bands of CD36 protein were determined in peritoneal macrophages/animal tissues and in macrophage cell lines, respectively, by Western blotting). However, the inhibitory effect of estrogen on CD36 expression was attenuated by progesterone, which indicates that progesterone may play a role in the regulation of macrophage CD36 expression and cellular oxLDL accumulation.
FIGURE 1.

Progesterone induces macrophage CD36 protein expression. A, peritoneal macrophages were isolated from female C57BL/6 mice and treated with estradiol at the indicated concentrations or plus 100 nm progesterone overnight. a, p < 0.05; b, p < 0.01 versus control in the corresponding groups; ns, not significantly different (n = 3); ctrl, control. B–E, J774 cells at ∼95% confluence or peritoneal macrophages isolated from female C57BL/6 mice in serum-free medium were treated with progesterone at the indicated concentrations overnight (B and D) or with 50 nm progesterone for the indicated times (C and E). After treatment, cellular proteins were extracted and used to determine CD36 expression by Western blotting. All bands in the Western blots were scanned, and the density of the target band normalized by GAPDH was calculated with statistical analysis. a, p < 0.05; b, p < 0.01 versus control in the corresponding groups (n = 3). F, peritoneal macrophages isolated from female C57BL/6 mice were treated with 50 nm progesterone for the indicated times, followed by determination of CD36 protein expression in intact cells by immunofluorescent staining. Neg Control, negative control. Cells were added with normal rabbit IgG.
To define it, we treated J774 cells, a murine macrophage cell line originally derived from a female mouse, and peritoneal macrophages, which were isolated from female C57BL/6 wild-type mice, with progesterone at different concentrations overnight. The results in Fig. 1, B and D, indicate that progesterone induced CD36 expression in a concentration-dependent manner. The time course study (Fig. 1, C and E) demonstrates that the induction occurred quickly, with maximum induction at 4 and 8 h after progesterone treatment in J774 cells and peritoneal macrophages, respectively. The time-dependent induction of CD36 protein expression by progesterone was also confirmed by immunofluorescent staining analysis in peritoneal macrophages (Fig. 1F).
CD36 is a cellular membrane protein and functions as the receptor for cellular oxLDL uptake (18). Associated with increased cellular CD36 protein expression, we determined by FACS assay that progesterone increased cell surface CD36 protein levels (Fig. 2A), which implies that progesterone treatment may enhance cellular oxLDL accumulation and foam cell formation. To determine this, after progesterone treatment, macrophages were assessed for cellular oxLDL uptake by Oil Red O staining. The results in Fig. 2B, top panels, indicate that progesterone substantially increased oxLDL accumulation. In the presence of anti-CD36 antibody, cellular oxLDL accumulation was abolished (Fig. 2B, bottom panels). Therefore, the induction of cellular oxLDL accumulation by progesterone mainly depends on CD36 expression.
FIGURE 2.
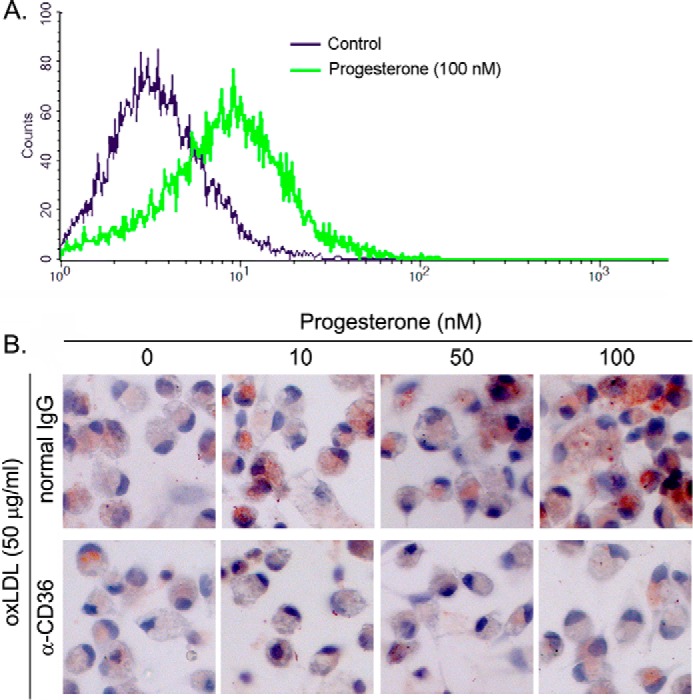
Progesterone increases macrophage surface CD36 protein levels and enhances cellular oxLDL accumulation in a CD36-dependent manner. A, peritoneal macrophages isolated from female C57BL/6 mice were treated with 100 nm progesterone overnight. After washing with PBS, surface CD36 protein was determined by FACS assay. B, after treatment and incubation with normal IgG or anti-CD36 antibody, peritoneal macrophages were used to conduct Oil Red O staining for determination of cellular oxLDL uptake. The cells were also stained with hematoxylin solution.
Progesterone Activates CD36 Transcription by Inducing PPARγ Expression
To determine whether macrophage CD36 expression can be correlated to PR activity, we initially treated J774 and RAW cells, two murine macrophage cell lines derived from a female mouse and a male mouse, respectively, with progesterone. In contrast to J774 cells, progesterone slightly reduced CD36 expression in RAW cells (Fig. 3A, top panel). Furthermore, we treated J774 macrophages with progesterone at different concentrations in the absence or presence of mifepristone, a potent PR antagonist. Although mifepristone alone had little effect on CD36 expression, it totally blocked progesterone-induced CD36 expression in J774 cells (Fig. 3A, bottom panel). Taken together, the above results suggest that induction of macrophage CD36 expression by progesterone is dependent on activation of PR or PR-targeted gene expression, which is able to control CD36 expression.
FIGURE 3.
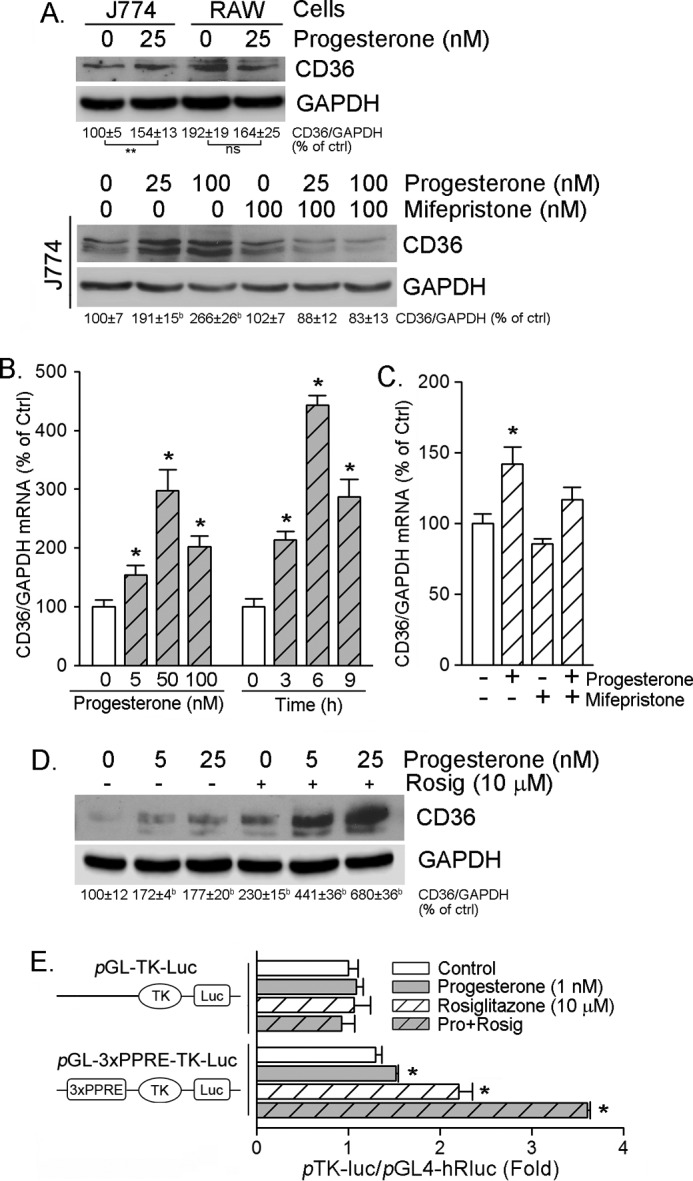
Progesterone activates CD36 transcription. A, J774 and RAW cells, two murine macrophage cell lines originally derived from a female and a male mouse, respectively, were treated with 25 nm progesterone overnight (top panel). J774 cells were treated with progesterone at the indicated concentrations in the absence or presence of 100 nm mifepristone, respectively, overnight (bottom panel). Expression of CD36 was determined by Western blotting. b and **, p < 0.01 versus control (ctrl) in the corresponding group; ns, not significantly different (n = 3). B and C, peritoneal macrophages isolated from female C57BL/6 wild-type mice received the following treatment overnight: progesterone at the indicated concentrations overnight (B, left panel) or with 50 nm progesterone for the indicated times (B, right panel); progesterone (50 nm), mifepristone (100 nm), or both (C). After treatment, total cellular RNA was extracted and used to determine CD36 mRNA expression by real-time RT-PCR. *, p < 0.05 versus control in the corresponding group (n = 3). D, peritoneal macrophages isolated from female C57BL/6 wild-type mice were treated with progesterone at the indicated concentrations in the absence or presence of rosiglitazone (Rosig, 10 μm) overnight. CD36 protein expression was determined by Western blotting. b, p < 0.01 versus control (n = 3). E, ∼90% confluent J774 cells were transfected with the indicated TK promoters plus Renilla (as an internal control) for 4 h, followed by the indicated treatment overnight. Activity of firefly and Renilla luciferases (Luc) in the cellular lysate was determined using the Dual-Luciferase reporter assay system. *, p < 0.05 versus control (n = 3). Pro, progesterone.
To define the mechanisms by which progesterone induces CD36 expression, we initially determined macrophage CD36 mRNA expression in response to progesterone treatment. The results in both concentration and time course studies demonstrate that progesterone increased CD36 mRNA levels (Fig. 3B). In addition, similar to the effect on CD36 protein expression, mifepristone also inhibited progesterone-induced CD36 mRNA expression (Fig. 3C). Thus, these results imply that progesterone may activate CD36 transcription.
CD36 expression is transcriptionally activated by PPARγ because of the existence of a PPRE in the proximal region of the CD36 promoter (24). To determine whether PPARγ is involved in progesterone-induced CD36 expression, we initially treated macrophages with progesterone, rosiglitazone (a synthetic PPARγ ligand), or both. The results in Fig. 3D demonstrate that either progesterone or rosiglitazone alone induced CD36 expression, whereas co-treatment induced CD36 expression in a synergistic manner, which indicates an interaction between PPARγ and progesterone to regulate CD36 expression. We then constructed a promoter containing a tandem of three copies of the PPRE in the CD36 promoter into the pGL-TK-Luc vector and determined the effect of progesterone, rosiglitazone, or their co-treatment on the activity of this promoter (pGL-3xPPRE-TK-Luc). Similar to protein level, we determined that, although progesterone alone moderately activated PPRE-luciferase, it synergized rosiglitazone-activated PPRE motif activity. These findings suggest that activation of CD36 transcription by progesterone requires activation of PPARγ.
Interestingly, a previous study has demonstrated that PPARγ is a PR-targeted gene in granulosa cells of the preovulatory follicles during the ovulatory process. To define the role of PPARγ in progesterone-induced CD36 transcription, we determined whether progesterone can influence PPARγ expression. The results in Fig. 4A show that progesterone increased PPARγ protein expression in dose- and time-dependent manners. Furthermore, we determined that progesterone induced PPARγ mRNA expression (Fig. 4B), suggesting that progesterone induces PPARγ expression at the transcriptional level.
FIGURE 4.

Induction of CD36 expression by progesterone is completed by activating PPARγ expression. A and B, peritoneal macrophages isolated from female C57BL/6 wild-type mice were treated with progesterone at the indicated concentrations overnight or with 100 nm progesterone for the indicated times. Expression of PPARγ protein and mRNA was determined by Western blotting (A) and real time RT-PCR (B), respectively. a and *, p < 0.05; b and **, p < 0.01 versus control (ctrl) in the corresponding group (n = 3). C, J774 cells were transfected with DNA for pPPARγ or pPPARγ-PREmut plus Renilla, followed by treatment with progesterone, rosiglitazone, or both overnight. Total cellular lysate was extracted and used to determine firefly and Renilla luciferase activity. *, p < 0.05 versus pPPARγ alone (lane 1, n = 3). D, peritoneal macrophages isolated from female C57BL/6 wild-type mice were treated with 50 nm progesterone (Pro) overnight. Chromatin was then isolated from cells, followed by immunoprecipitation (IP) with normal IgG or anti-PR antibody as indicated. The PCR was conducted with the primers for the corresponding PRE in the PPARγ promoter. E and F, peritoneal macrophages were isolated from female PPARγfl/fl mice and MacPPARγ KO mice, respectively. The cells were treated with progesterone at the indicated concentrations overnight (E) or with 50 nm progesterone for the indicated times (F). After treatment, cellular proteins were extracted and used to determine CD36 and PPARγ protein expression by Western blotting. a, p < 0.05; b, p < 0.01 versus control; ns, not significantly different (n = 3). G, peritoneal macrophages isolated from female PPARγfl/fl and MφPPARγ KO mice were treated with progesterone at the indicated concentrations overnight. Cellular oxLDL accumulation was determined by Oil Red O staining.
By completing a sequence alignment analysis, we found a putative progesterone response element (PRE) in the proximal region of the PPARγ promoter (from +36 to +50, AGAGCAtggTGCCTT). The sequence of the conserved PRE motif is RGNACAnrnTGTNCY (R/r = G/A, Y = T/G), which is composed of hexameric half-sites separated by precisely three nucleotides exhibiting dyad symmetry (29). To determine the effect of progesterone on PPARγ transcription, we constructed a natural PPARγ promoter (pPPARγ) and a PPARγ promoter with PRE mutation (pPPARγ-PREmut) and then determined the activity of these two promoters in response to treatment. As shown in Fig. 4C, progesterone increased the natural PPARγ promoter activity (lane 2 versus lane 1), whereas it had little effect on the PPARγ promoter with PRE mutation (lane 6 versus lane 5). Because PPARγ transcription cannot be activated by itself, rosiglitazone had no effect on the activity of the natural or mutated PPARγ promoter (Fig. 4C, lanes 3 and 7) or on progesterone-induced natural PPARγ promoter activity (Fig. 4C, lane 4). These results demonstrate that the PRE plays an important role in the induction of PPARγ expression by progesterone. Furthermore, we determined increased interaction between the PRE and PR protein in response to progesterone treatment by a ChIP assay (Fig. 4D).
To further confirm that the induction of CD36 expression by progesterone is dependent on activation of PPARγ expression, we isolated peritoneal macrophages from specific macrophage PPARγ knockout (MφPPARγ KO) mice and the corresponding controls (PPARγfl/fl), respectively, and treated cells with progesterone at different concentrations. As shown in Fig. 4E, lack of PPARγ expression reduced the basal level of CD36 protein, which demonstrates the importance of PPARγ in the activation of CD36 expression. More importantly, progesterone induced CD36 expression in macrophages isolated from PPARγfl/fl mice, which was associated with induction of PPARγ expression (Fig. 4E, left half). In contrast, progesterone had little effect on CD36 expression in macrophages isolated from MφPPARγ KO mice (Fig. 4E, right half). Similarly, the results of the time course study demonstrate that induction of CD36 expression by progesterone occurred in macrophages isolated from PPARγfl/fl mice but not cells isolated from MφPPARγ KO mice (Fig. 4F).
Next, we determined whether the induction of macrophage oxLDL accumulation by progesterone also relies on induction of PPARγ expression. We treated peritoneal macrophages isolated from PPARγfl/fl mice and MφPPARγ KO mice, respectively, with progesterone at different concentrations, followed by determination of cellular oxLDL accumulation by Oil Red O staining. Similar to cells isolated from wild-type mice (Fig. 2B), progesterone increased cellular oxLDL accumulation in a CD36-dependent manner in cells isolated from PPARγfl/fl mice (Fig. 4G, top half). However, progesterone had little effect on cellular oxLDL uptake in cells isolated from MφPPARγ KO mice (Fig. 4G, bottom half). Taken together, the results in Fig. 4 suggest that induction of macrophage CD36 expression and oxLDL accumulation by progesterone is mainly determined by induction of PPARγ expression.
Fatty acid binding protein 4 (FABP4) is another target molecule transcriptionally activated by PPARγ in macrophages (30). We also determined by Western blotting and immunofluorescent staining whether progesterone treatment can induce FABP4 expression. Similar to CD36, progesterone increased macrophage FABP4 expression (Fig. 5). Therefore, there might be a common inductive effect of progesterone on the expression of PPARγ targeted genes.
FIGURE 5.

Progesterone induces macrophage FABP4 expression. A and B, peritoneal macrophages isolated from female C57BL/6 wild-type mice were treated with progesterone (100 nm) for the indicated times (A) or at the indicated concentrations overnight (B). After treatment, the cells were used to extract total cellular proteins for determination of FABP4 expression by Western blotting. a, p < 0.05; b, p < 0.01 versus control (ctrl) (n = 3). C, after the indicated treatment, FABP4 protein expression in intact cells was determined by immunofluorescent staining.
Progesterone Induces CD36 and PPARγ Expression in Vivo
To investigate whether progesterone can induce macrophage CD36 expression in vivo, female PPARγfl/fl mice or MφPPARγ KO mice were injected intraperitoneally with vehicle (corn oil) or progesterone solution for 4 consecutive days, followed by collection of peritoneal macrophages and adipose tissue. Similar to the results of the in vitro study (Fig. 4E), progesterone increased CD36 expression in peritoneal macrophages collected from PPARγfl/fl mice, whereas it had little effect on CD36 expression in cells collected from MφPPARγ KO mice (Fig. 6A). Therefore, progesterone induces CD36 expression in vivo in a PPARγ-dependent manner as well. Because PPARγ expression is normal in adipose tissue of MφPPARγ KO mice, we determined that treatment of animals with progesterone induced CD36 and FABP4 expression in adipose tissue of both PPARγfl/fl mice and MφPPARγ KO mice normally at similar degrees (Fig. 6B). Correspondingly, we determined that progesterone activated PPARγ expression in both peritoneal macrophages and adipose tissue of PPARγfl/fl mice (Fig. 6C).
FIGURE 6.

Progesterone induces CD36 and PPARγ expression in vivo. A–C, female PPARγfl/fl mice and MφPPARγ KO mice were injected subcutaneously with vehicle (corn oil) or progesterone solution (1 mg/mouse) for 4 consecutive days. One day after the last injection, peritoneal macrophages and adipose tissue were individually collected from each mouse and used to extract total cellular proteins for determination of CD36 (A and B), FABP4 (B), and PPARγ (C) expression by Western blotting. *, p < 0.05 versus control in the corresponding group (n = 3). D–I, female C57BL/6 wild-type mice in estrus were mated with adult male C57BL/6 wild-type mice overnight and examined for the appearance of a vaginal plug the following morning, which was defined as P0. On P11 or P17, mice were sacrificed, followed by collection of blood, adipose tissue, skeletal muscle, and peritoneal macrophages. D, serum progesterone levels were determined using a progesterone ELISA kit. *, p < 0.05 versus control (n = 5). E–I, adipose tissue, skeletal muscle, and peritoneal macrophages were used to extract total cellular proteins and total RNA for determination of CD36 protein expression (E–G) by Western blotting. *, p < 0.05; **, p < 0.01 versus control (n = 3). CD36 mRNA (H) and PPARγ mRNA (I) expression was determined by real-time RT-PCR. *, p < 0.05; **, p < 0.01 versus control (n = 5).
To investigate the nature of progesterone on induction of CD36 and PPARγ expression, we collected serum, adipose tissue, skeletal muscle, and peritoneal macrophages from female wild-type mice at different time points of pregnancy. Associated with increased serum progesterone levels during gestation (Fig. 6D), expression of CD36 protein in adipose tissues (Fig. 6E), skeletal muscle (Fig. 6F), and peritoneal macrophages (Fig. 6G) was substantially elevated. Similarly, expression of CD36 mRNA in adipose tissue, skeletal muscle, and peritoneal macrophages was increased (Fig. 6H). In addition, we determined that PPARγ mRNA expression in the above tissue samples was increased during gestation (Fig. 6I). Taken together, the results in Figs. 6, D–I, demonstrate that expression of CD36 and PPARγ in different tissues of pregnant mice is activated, which may enhance fatty acid metabolism, a high requirement for gestation.
Discussion
Estrogen can reduce the risk of coronary heart disease, which might be attenuated by progestin, a synthetic progesterone that is included in HRT. Several negative effects of progesterone on cardioprotection of estrogen have been reported. In vitro, progesterone reduces the expression of extracellular superoxide dismutase and manganese superoxide dismutase induced by estrogen, whereas progesterone activates NADPH oxidase. Therefore, the presence of progesterone increases the production of reactive oxygen species in vascular smooth muscle cells (31). Activation of angiotensin II type 1 receptor (AT1) has been implicated in the pathogenesis of cardiovascular disease. Estrogen inhibits AT1 expression and angiotensin II-induced reactive oxygen species production in vascular smooth muscle cells. However, these estrogen actions are substantially attenuated by progesterone (32). The inhibitory effects of estrogen on serum LDL levels or LDL oxidation, expression of cell adhesion molecules, chemokines/cytokines, and C-reactive protein are also abolished by progesterone (33). In this study, we determined that treatment of macrophages with progesterone induced CD36 expression (Fig. 1), a molecule facilitating foam cell formation. At the cellular level, we observed that progesterone increased macrophage oxLDL accumulation in a CD36-depenednt manner (Fig. 2B). Furthermore, we determined that induction of macrophage CD36 by progesterone is completed by activating PPARγ expression (Fig. 4). Therefore, our study demonstrates another negative effect of progesterone on the cardiovascular system.
Besides macrophages, CD36 is also expressed by other cell type/tissues where CD36 has different functions. Soluble CD36 is considered a novel marker of insulin resistance, and a high level of soluble CD36 level is associated with an increased risk of type 2 diabetes (34, 35). Interestingly, a spontaneous CD36 deletion is also associated with insulin resistance, defective fatty acid metabolism, and hypertension in a spontaneously hypertensive rat model (36), whereas the transgenic rescue of CD36 expression ameliorates insulin resistance and lowers serum fatty acid levels (37). Similarly, lack of CD36 expression in humans can be linked to insulin resistance and abnormal fatty acid metabolism (38). As a fatty acid transporter, particularly in adipose tissue and muscle, deficiency of CD36 expression leads to a defect in fatty acid uptake, which may contribute to insulin resistance (39), and suggests that CD36 is metabolically protective in these tissues. We determined, for the first time, that CD36 expression in multiple tissues was substantially increased during pregnancy (Fig. 6, E–I). Although it is important to maintain appropriate fatty acid metabolism in normal energy homeostasis, insulin sensitivity, and metabolic health, there is substantially increased fatty acid mobilization from adipocytes and fatty acid delivery to other tissues, particularly the skeletal muscle, for uptake and oxidation during pregnancy. In addition, it has been reported that expression of CD36 and scavenger receptor type A is required for fetal protection against microbial attack (40). Thus, the high CD36 expression in multiple tissues during pregnancy suggests its physiological role.
In our study, we determined that progesterone induced CD36 expression by activating the PPARγ pathway (Fig. 4). High PPARγ expression in multiple tissues was also determined during pregnancy (Fig. 6I). PPARγ plays an important role in different biological processes. Interestingly, the interaction between PPARγ and progesterone or PR has been determined in the reproductive system. In the ovary, activation of PPARγ can regulate steroidogenesis, including progesterone production or release, differentiation, and tissue remodeling (41, 42). Meanwhile, it has been determined that PPARγ is a target of PR in the ovary and that ovarian PPARγ expression plays a critical role in ovulation (43). Lack of PR expression results in impaired induction of PPARγ expression in the tissue in the context of superovulation. Meanwhile, conditional knockout PPARγ expression in the ovary results in failure of preovulatory follicles to rupture and decline of released eggs from ovaries under conditions of superovulation (43). In our study, we determined that expression of PPARγ and CD36 in different tissues was simultaneously activated during pregnancy, which may imply a physiological importance of PPARγ/CD36-mediated lipid metabolism for reproduction.
Taken together, in this study, we determined the dual pathophysiological roles of progesterone. Progesterone activates macrophage CD36 expression and cellular oxLDL accumulation by activating the PPARγ pathway, which demonstrates one of the negative effects of progesterone on the cardiovascular system by inducing foam cell formation. However, activated PPARγ and CD36 expression in multiple tissues during pregnancy can enhance PPARγ/CD36-mediated fatty acid metabolism and possibly protect the animals against microbial attack, which suggests an important physiological role of progesterone.
Experimental Procedures
Reagents
Progesterone was purchased from Sigma-Aldrich (St. Louis, MO). LipofectamineTM 2000 was purchased from Invitrogen. Rabbit anti-CD36 polyclonal antibody (catalog no. NB400–145) was purchased from Novus Biologicals (Littleton, CO). Rabbit anti-PPARγ polyclonal antibody (catalog no. 16643-1-AP) was purchased from Proteintech Group Inc. (Chicago, IL). Rabbit anti-GAPDH and PR polyclonal antibodies (catalog nos. sc-25778 and sc-539, respectively), goat anti-FABP4 polyclonal antibody (catalog no. sc-18661), FITC-conjugated goat anti-rabbit IgG, and FITC-conjugated rabbit anti-goat IgG were purchased from Santa Cruz Biotechnology (Dallas, Texas). The mouse progesterone ELISA kit was purchased from Elabscience Biotechnology (Wuhan, China). Isolation of LDL and preparation of oxLDL were completed as described previously (44).
In Vivo Study
The protocol for animal study was approved by the Ethics Committee of Nankai University and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. C57BL/6 wild-type mice were purchased from the Animal Center of Nanjing University (Nanjing, China). The specific macrophage PPARγ-deficient (MφPPARγ KO) mice and the corresponding control (PPARγfl/fl) mice were generated as described previously (45).
To determine the effect of progesterone on CD36 expression in vivo, female PPARγfl/fl mice and MφPPARγ KO mice were intraperitoneally injected with vehicle (corn oil) or progesterone solution in corn oil (1 mg/mouse) for 4 consecutive days. Mice were then sacrificed in a CO2 chamber 1 day after the last progesterone injection, followed by collection of individual tissue samples and determination of protein expression by Western blotting.
To investigate whether progesterone can play a physiological role for gestation by activating CD36 and PPARγ expression, female C57BL/6 wild-type mice in estrus were mated with adult male mice overnight and examined for the appearance of a vaginal plug in the following morning, which was defined as day 0 of pregnancy (P0). The mice were sacrificed on P11 or P17. Blood, adipose tissue, skeletal muscle, and peritoneal macrophages were collected, followed by isolation of serum and extraction of total protein and RNA, respectively. Serum progesterone levels were determined using the progesterone ELISA assay kit. Expression of protein and RNA in tissue samples was determined by Western blotting and real-time RT-PCR, respectively.
Cell Culture
J774 and RAW cells were purchased from the ATCC (Manassas, VA) and cultured in complete RPMI 1640 medium (without phenol red) containing 10% hormone-free (dextran-coated, charcoal-treated) FBS and 50 μg/ml penicillin/streptomycin. The cells were switched to serum-free medium at ∼90% confluence for 2 h, followed by treatment.
Peritoneal macrophages were collected from female mouse abdomen by lavage with PBS. The cells were cultured in complete RPMI 1640 medium (hormone- and phenol red-free) for 2 h, and then all floating cells were removed. The adhesive cells, which were determined as macrophages by immunofluorescent staining with anti-MOMA2 (a marker for macrophages) antibody, were cultured in complete RPMI 1640 medium for another 2 days and then received treatment in serum-free medium.
Western blotting, FACS, and Immunofluorescent Staining
After treatment, cells were washed with PBS and lysed in ice-cold lysis buffer (50 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 1 mm PMSF, 50 mm sodium fluoride, 1 mm sodium orthovanadate, and 50 μg/ml aprotinin/leupeptin). After extraction, total cellular proteins were used to determine CD36, PPARγ or FABP4 protein expression by Western blotting as described previously (46).
To analyze cell surface CD36 protein levels, ∼1 × 106 cells from each sample were blocked for 30 min at room temperature with PBS containing 5% goat serum. After washing, cells were incubated with rabbit anti-CD36 antibody for 1 h at room temperature, followed by incubation with goat anti-rabbit FITC-conjugated IgG for 45 min. After washing with PBS, cells were subjected to flow cytometric evaluation.
To determine CD36 or FABP4 expression by immunofluorescent staining, mouse peritoneal macrophages were cultured on coverslips in a 24-well plate. After treatment, cells were washed twice with PBS and then fixed with 4% paraformaldehyde for 30 min at room temperature. The cells were washed twice with PBS and then blocked with 2% BSA for 2 h at room temperature, followed by incubation with anti-CD36 rabbit or anti-FABP4 goat polyclonal antibody overnight at 4 °C. After the primary antibody was removed by washing with PBS for 30 min, cells were incubated with FITC-conjugated goat anti-rabbit IgG or rabbit anti-goat IgG for 2 h at room temperature. After washing with PBS, the coverslips were stained with DAPI solution for determination of nuclei. The coverslips were observed, and the images were photographed with a fluorescence microscope.
Real-time RT-PCR
After treatment, cells were lysed in RNAzolTM B (Tel-Test, Inc.). The lysate was mixed well with chloroform and centrifuged for 10 min at 12,000 rpm at 4 °C. The top aqueous phase was collected and mixed with an equal volume of isopropanol to precipitate total cellular RNA. The cDNA was synthesized with 1 μg of total RNA using a reverse transcriptase kit purchased from New England Biolabs (Ipswich, MA). Real-time PCR was conducted using SYBR Green PCR Master Mix (Bio-Rad) and the following primers: CD36, 5′-TTTCCTCTGACATTTGCAGGTCTA-3′ (forward) and 5′-AAAGGCATTGGCTGGAAGAA-3′ (reverse); PPARγ, 5′-TGACTTGAACGACCAAGTAACTC-3′ (forward) and 5′-CTAGTACAAGTCCTTGTAGATCTC-3′ (reverse); and GAPDH, 5′-ACCCAGAAGACTGTGGATGG-3′ (forward) and 5′-ACACATTGGGGGTAGGAACA-3′ (reverse). CD36 or PPARγ mRNA expression was normalized by GAPDH mRNA in the corresponding sample.
Oil Red O Staining
Mouse peritoneal macrophages were plated on coverslips in a 24-well plate. After treatment, cells were incubated with rabbit anti-CD36 polyclonal antibody or normal rabbit IgG (0.3 μg/sample) for 1 h. Cells were then incubated with 50 μg/ml oxLDL for 3 h, fixed with 4% paraformaldehyde for 30 min, washed twice with PBS, and stained with Oil Red O solution (0.3% Oil Red O in 60% isopropanol) for 50 min at room temperature, followed by washing twice with water. Cells were then restained with hematoxylin solution for 30 s, kept in water for 5 min, and photographed with a microscope (Leica, DM5000B).
Determination of Promoter Activity
The natural mouse PPARγ promoter (pPPARγ, from −670 to +247), including the PRE (AGAGCAtggTGCCTT, locates from +36 to +50; the conserved sequence of PRE is RGNACAnrnTGTNCY (R/r = G/A, Y = T/G)) was generated by PCR with mouse genomic DNA and the following primers: forward, 5′-CACGCTCGAGTTTGGATAGCAGTAAC-3′; reverse, 5′-ACGTAAGCTTTAGGGTTCTATGCTGA-3′. After the sequence was confirmed, the PCR product was digested with XhoI and HindIII followed by ligation into the pGL4 luciferase reporter vector, transformed, and amplified. The promoter with the PRE mutation (pPPARγ-PREmut) was constructed with pPPARγ DNA and the following primers with PRE mutation: pPPARγ-PREmut forward, 5′-cctgttgacccTAAGTAtcgCACCTGcgctgatgc-3; pPPARγ-PREmut reverse, 5′-gcatcagcgCAGGTGcgaTACTTAgggtcaacagg-3′. The letters in italic are mutated nucleotides in the PRE of PPARγ.
A tandem of three copies of the consensus PPRE in the mouse CD36 promoter (pGL-3xPPRE-TK-Luc) was constructed with the pGL-TK-Luc plasmid (Clontech) with the following oligonucleotides: forward, 5′-CAAATGTAGGTGATGGGTCTTCACCAGGTGATGGGTCTTCACCAGGTGATGGGTCTTCACC-3′; reverse, 5′-GGTGTAGACCCATCACCTGGTGAAGACCCATCACCTGGTGAAGACCCATCACCTACATTTG-3′. The underlined sequence is the PPRE or DR1 (the direct repeated hexanucleotides were separated by one of any nucleotide) in the CD36 promoter.
All constructs were verified by sequencing. To analyze PPARγ promoter or CD36 3xPPRE promoter activity, ∼95% confluent J774 cells in 48-well plates were transfected with DNA for the promoter and Renilla (for internal normalization). After 4-h transfection, the transfected cells received treatment overnight. Cells were then lysed, and the cellular lysate was used to determine the activity of firefly and Renilla luciferases using the Dual-Luciferase reporter assay system (Promega, Madison, WI).
ChIP Assay
After treatment, cells were cross-linked by addition of formaldehyde and sonication in lysis buffer (50 mm Hepes-KOH (pH 7.5), 140 mm NaCl, 1% Triton X-100, 1 mm EDTA, 0.1% sodium deoxycholate, 0.1% SDS and protease inhibitors of aprotinin/leupeptin) to fragment DNA into an average size of 500–1000 bp. The input PCR was conducted with DNA extracted from the sonicated chromatin after reversal of the cross-linking. Based on the input, immunoprecipitation was conducted with the same amount of chromatin from each sample and rabbit anti-PR polyclonal antibody, followed by PCR. The primers for PRE (from +36 to +50) ChIP assay were as follows: forward, 5′-AAGCTCGATGACCATAAGCCTTT-3′; reverse, 5′-AAGCATCCCTTGCAGCAACA-3′.
Data Analysis
All experiments were repeated at least three times, and representative results are presented. The data are presented as mean ± S.E., and the statistical results were obtained by Student's t test using Prism (GraphPad Software). The differences were considered significant at p < 0.05.
Author Contributions
J. H. and Y. D. designed the study. X. Y., W. Z., Y. C., Y. Li, L. S., Y. Liu, M. L., M. Y., and X. L. performed the experiments. J. H. and Y. D. prepared the manuscript.
This work was supported by National Science Foundation of China Grants 81473204 (to J. H.), 81573427 (to Y. D.), and 31400694 (to Y. C.); Program for Changjiang Scholars and Innovative Research Team in University Grant IRT13023 and 111 Project Grant B08011 (to J. H.); and International Science and Technology Cooperation Program of China Grant 2015DFA30430 (to J. H., Y. D., and Y. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- HRT
- hormone replacement therapy
- PR
- progesterone receptor
- oxLDL
- oxidized LDL
- PPARγ
- peroxisome proliferator-activated receptor γ
- PPRE
- peroxisome proliferator-activated receptor γ-responsive element
- PRE
- progesterone response element
- P
- day of pregnancy.
References
- 1. Smith D. C., Prentice R., Thompson D. J., and Herrmann W. L. (1975) Association of exogenous estrogen and endometrial carcinoma. N. Engl. J. Med. 293, 1164–1167 [DOI] [PubMed] [Google Scholar]
- 2. Sturdee D. W., Wade-Evans T., Paterson M. E., Thom M., and Studd J. W. (1978) Relations between bleeding pattern, endometrial histology, and oestrogen treatment in menopausal women. Br. Med. J. 1, 1575–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Padwick M. L., Pryse-Davies J., and Whitehead M. I. (1986) A simple method for determining the optimal dosage of progestin in postmenopausal women receiving estrogens. N. Engl. J. Med. 315, 930–934 [DOI] [PubMed] [Google Scholar]
- 4. Diep C. H., Daniel A. R., Mauro L. J., Knutson T. P., and Lange C. A. (2015) Progesterone action in breast, uterine, and ovarian cancers. J. Mol. Endocrinol. 54, R31–R53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hanke H., Hanke S., Finking G., Muhic-Lohrer A., Mück A. O., Schmahl F. W., Haasis R., and Hombach V. (1996) Different effects of estrogen and progesterone on experimental atherosclerosis in female versus male rabbits: quantification of cellular proliferation by bromodeoxyuridine. Circulation 94, 175–181 [DOI] [PubMed] [Google Scholar]
- 6. Hanke H., Hanke S., Bruck B., Brehme U., Gugel N., Finking G., Mück A. O., Schmahl F. W., Hombach V., and Haasis R. (1996) Inhibition of the protective effect of estrogen by progesterone in experimental atherosclerosis. Atherosclerosis 121, 129–138 [DOI] [PubMed] [Google Scholar]
- 7. Adams M. R., Register T. C., Golden D. L., Wagner J. D., and Williams J. K. (1997) Medroxyprogesterone acetate antagonizes inhibitory effects of conjugated equine estrogens on coronary artery atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 17, 217–221 [DOI] [PubMed] [Google Scholar]
- 8. Hulley S., Grady D., Bush T., Furberg C., Herrington D., Riggs B., and Vittinghoff E. (1998) Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women: Heart and Estrogen/Progestin Replacement Study (HERS) Research Group. JAMA 280, 605–613 [DOI] [PubMed] [Google Scholar]
- 9. Rossouw J. E., Anderson G. L., Prentice R. L., LaCroix A. Z., Kooperberg C., Stefanick M. L., Jackson R. D., Beresford S. A., Howard B. V., Johnson K. C., Kotchen J. M., Ockene J., and Writing Group for the Women's Health Initiative Investigators (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA 288, 321–333 [DOI] [PubMed] [Google Scholar]
- 10. Hodis H. N., Mack W. J., Azen S. P., Lobo R. A., Shoupe D., Mahrer P. R., Faxon D. P., Cashin-Hemphill L., Sanmarco M. E., French W. J., Shook T. L., Gaarder T. D., Mehra A. O., Rabbani R., Sevanian A., et al. (2003) Hormone therapy and the progression of coronary-artery atherosclerosis in postmenopausal women. N. Engl. J. Med. 349, 535–545 [DOI] [PubMed] [Google Scholar]
- 11. Hsia J., Criqui M. H., Rodabough R. J., Langer R. D., Resnick H. E., Phillips L. S., Allison M., Bonds D. E., Masaki K., Caralis P., Kotchen J. M., and Women's Health Initiative Investigators (2004) Estrogen plus progestin and the risk of peripheral arterial disease: the Women's Health Initiative. Circulation 109, 620–626 [DOI] [PubMed] [Google Scholar]
- 12. Herrington D. M., Reboussin D. M., Brosnihan K. B., Sharp P. C., Shumaker S. A., Snyder T. E., Furberg C. D., Kowalchuk G. J., Stuckey T. D., Rogers W. J., Givens D. H., and Waters D. (2000) Effects of estrogen replacement on the progression of coronary-artery atherosclerosis. N. Engl. J. Med. 343, 522–529 [DOI] [PubMed] [Google Scholar]
- 13. Anderson G. L., Limacher M., Assaf A. R., Bassford T., Beresford S. A., Black H., Bonds D., Brunner R., Brzyski R., Caan B., Chlebowski R., Curb D., Gass M., Hays J., Heiss G., et al. (2004) Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291, 1701–1712 [DOI] [PubMed] [Google Scholar]
- 14. Endemann G., Stanton L. W., Madden K. S., Bryant C. M., White R. T., and Protter A. A. (1993) CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 268, 11811–11816 [PubMed] [Google Scholar]
- 15. Nicholson A. C., Frieda S., Pearce A., and Silverstein R. L. (1995) Oxidized LDL binds to CD36 on human monocyte-derived macrophages and transfected cell lines: evidence implicating the lipid moiety of the lipoprotein as the binding site. Arterioscler. Thromb. Vasc. Biol. 15, 269–275 [DOI] [PubMed] [Google Scholar]
- 16. Febbraio M., Podrez E. A., Smith J. D., Hajjar D. P., Hazen S. L., Hoff H. F., Sharma K., and Silverstein R. L. (2000) Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Invest. 105, 1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Febbraio M., Guy E., and Silverstein R. L. (2004) Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 2333–2338 [DOI] [PubMed] [Google Scholar]
- 18. Rahaman S. O., Lennon D. J., Febbraio M., Podrez E. A., Hazen S. L., and Silverstein R. L. (2006) A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 4, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glatz J. F., Luiken J. J., and Bonen A. (2010) Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 90, 367–417 [DOI] [PubMed] [Google Scholar]
- 20. Love-Gregory L., Sherva R., Sun L., Wasson J., Schappe T., Doria A., Rao D. C., Hunt S. C., Klein S., Neuman R. J., Permutt M. A., and Abumrad N. A. (2008) Variants in the CD36 gene associate with the metabolic syndrome and high-density lipoprotein cholesterol. Hum. Mol. Genet. 17, 1695–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corpeleijn E., van der Kallen C. J., Kruijshoop M., Magagnin M. G., de Bruin T. W., Feskens E. J., Saris W. H., and Blaak E. E. (2006) Direct association of a promoter polymorphism in the CD36/FAT fatty acid transporter gene with type 2 diabetes mellitus and insulin resistance. Diabet. Med. 23, 907–911 [DOI] [PubMed] [Google Scholar]
- 22. Sun Y., Scavini M., Orlando R. A., Murata G. H., Servilla K. S., Tzamaloukas A. H., Schrader R., Bedrick E. J., Burge M. R., Abumrad N. A., and Zager P. G. (2010) Increased CD36 expression signals monocyte activation among patients with type 2 diabetes. Diabetes Care 33, 2065–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greco D., Kotronen A., Westerbacka J., Puig O., Arkkila P., Kiviluoto T., Laitinen S., Kolak M., Fisher R. M., Hamsten A., Auvinen P., and Yki-Järvinen H. (2008) Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1281–1287 [DOI] [PubMed] [Google Scholar]
- 24. Nagy L., Tontonoz P., Alvarez J. G., Chen H., and Evans R. M. (1998) Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell 93, 229–240 [DOI] [PubMed] [Google Scholar]
- 25. Han J., Hajjar D. P., Tauras J. M., Feng J., Gotto A. M. Jr., and Nicholson A. C. (2000) Transforming growth factor-β1 (TGF-β1) and TGF-β2 decrease expression of CD36, the type B scavenger receptor, through mitogen-activated protein kinase phosphorylation of peroxisome proliferator-activated receptor-γ. J. Biol. Chem. 275, 1241–1246 [DOI] [PubMed] [Google Scholar]
- 26. Feng J., Han J., Pearce S. F., Silverstein R. L., Gotto A. M. Jr., Hajjar D. P., and Nicholson A. C. (2000) Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-γ. J. Lipid Res. 41, 688–696 [PubMed] [Google Scholar]
- 27. Huang J. T., Welch J. S., Ricote M., Binder C. J., Willson T. M., Kelly C., Witztum J. L., Funk C. D., Conrad D., and Glass C. K. (1999) Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382 [DOI] [PubMed] [Google Scholar]
- 28. Allred K. F., Smart E. J., and Wilson M. E. (2006) Estrogen receptor-α mediates gender differences in atherosclerosis induced by HIV protease inhibitors. J. Biol. Chem. 281, 1419–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lieberman B. A., Bona B. J., Edwards D. P., and Nordeen S. K. (1993) The constitution of a progesterone response element. Mol. Endocrinol. 7, 515–527 [DOI] [PubMed] [Google Scholar]
- 30. Sun L., Nicholson A. C., Hajjar D. P., Gotto A. M. Jr., and Han J. (2003) Adipogenic differentiating agents regulate expression of fatty acid binding protein and CD36 in the J744 macrophage cell line. J. Lipid Res. 44, 1877–1886 [DOI] [PubMed] [Google Scholar]
- 31. Wassmann K., Wassmann S., and Nickenig G. (2005) Progesterone antagonizes the vasoprotective effect of estrogen on antioxidant enzyme expression and function. Circ. Res. 97, 1046–1054 [DOI] [PubMed] [Google Scholar]
- 32. Nickenig G., Strehlow K., Wassmann S., Bäumer A. T., Albory K., Sauer H., and Böhm M. (2000) Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation 102, 1828–1833 [DOI] [PubMed] [Google Scholar]
- 33. Koh K. K., and Sakuma I. (2004) Should progestins be blamed for the failure of hormone replacement therapy to reduce cardiovascular events in randomized controlled trials? Arterioscler. Thromb. Vasc. Biol. 24, 1171–1179 [DOI] [PubMed] [Google Scholar]
- 34. Handberg A., Levin K., Højlund K., and Beck-Nielsen H. (2006) Identification of the oxidized low-density lipoprotein scavenger receptor CD36 in plasma: a novel marker of insulin resistance. Circulation 114, 1169–1176 [DOI] [PubMed] [Google Scholar]
- 35. Handberg A., Norberg M., Stenlund H., Hallmans G., Attermann J., and Eriksson J. W. (2010) Soluble CD36 (sCD36) clusters with markers of insulin resistance, and high sCD36 is associated with increased type 2 diabetes risk. J. Clin. Endocrinol. Metab. 95, 1939–1946 [DOI] [PubMed] [Google Scholar]
- 36. Aitman T. J., Glazier A. M., Wallace C. A., Cooper L. D., Norsworthy P. J., Wahid F. N., Al-Majali K. M., Trembling P. M., Mann C. J., Shoulders C. C., Graf D., St Lezin E., Kurtz T. W., Kren V., Pravenec M., et al. (1999) Identification of CD36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat. Genet. 21, 76–83 [DOI] [PubMed] [Google Scholar]
- 37. Pravenec M., Landa V., Zidek V., Musilova A., Kren V., Kazdova L., Aitman T. J., Glazier A. M., Ibrahimi A., Abumrad N. A., Qi N., Wang J. M., St Lezin E. M., and Kurtz T. W. (2001) Transgenic rescue of defective CD36 ameliorates insulin resistance in spontaneously hypertensive rats. Nat. Genet. 27, 156–158 [DOI] [PubMed] [Google Scholar]
- 38. Kuwasako T., Hirano K., Sakai N., Ishigami M., Hiraoka H., Yakub M. J., Yamauchi-Takihara K., Yamashita S., and Matsuzawa Y. (2003) Lipoprotein abnormalities in human genetic CD36 deficiency associated with insulin resistance and abnormal fatty acid metabolism. Diabetes Care 26, 1647–1648 [DOI] [PubMed] [Google Scholar]
- 39. Hajri T., Han X. X., Bonen A., and Abumrad N. A. (2002) Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Invest. 109, 1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oz H. S., Ebersole J. L., and de Villiers W. J. (2011) The macrophage pattern recognition scavenger receptors SR-A and CD36 protect against microbial induced pregnancy loss. Inflamm. Res. 60, 93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Froment P., Gizard F., Defever D., Staels B., Dupont J., and Monget P. (2006) Peroxisome proliferator-activated receptors in reproductive tissues: from gametogenesis to parturition. J. Endocrinol. 189, 199–209 [DOI] [PubMed] [Google Scholar]
- 42. Komar C. M., Braissant O., Wahli W., and Curry T. E. Jr. (2001) Expression and localization of PPARs in the rat ovary during follicular development and the periovulatory period. Endocrinology 142, 4831–4838 [DOI] [PubMed] [Google Scholar]
- 43. Kim J., Sato M., Li Q., Lydon J. P., Demayo F. J., Bagchi I. C., and Bagchi M. K. (2008) Peroxisome proliferator-activated receptor γ is a target of progesterone regulation in the preovulatory follicles and controls ovulation in mice. Mol. Cell Biol. 28, 1770–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han J., Zhou X., Yokoyama T., Hajjar D. P., Gotto A. M. Jr., and Nicholson A. C. (2004) Pitavastatin downregulates expression of the macrophage type B scavenger receptor, CD36. Circulation 109, 790–796 [DOI] [PubMed] [Google Scholar]
- 45. Yang X., Yao H., Chen Y., Sun L., Li Y., Ma X., Duan S., Li X., Xiang R., Han J., and Duan Y. (2015) Inhibition of glutathione production induces macrophage CD36 expression and enhances cellular-oxidized low density lipoprotein (oxLDL) uptake. J. Biol. Chem. 290, 21788–21799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Y., Duan Y., Kang Y., Yang X., Jiang M., Zhang L., Li G., Yin Z., Hu W., Dong P., Li X., Hajjar D. P., and Han J. (2012) Activation of liver X receptor induces macrophage interleukin-5 expression. J. Biol. Chem. 287, 43340–43350 [DOI] [PMC free article] [PubMed] [Google Scholar]