

Abstract
A peptide segment that binds the active site of a serine protease in a substrate-like manner may behave like an inhibitor or a substrate. However, there is sparse information on which factors determine the behavior a particular peptide segment will exhibit. Here, we describe the first x-ray crystal structure of a nanobody in complex with a serine protease. The nanobody displays a new type of interaction between an antibody and a serine protease as it inserts its complementary determining region-H3 loop into the active site of the protease in a substrate-like manner. The unique binding mechanism causes the nanobody to behave as a strong inhibitor as well as a poor substrate. Intriguingly, its substrate behavior is incomplete, as 30–40% of the nanobody remained intact and inhibitory after prolonged incubation with the protease. Biochemical analysis reveals that an intra-loop interaction network within the complementary determining region-H3 of the nanobody balances its inhibitor versus substrate behavior. Collectively, our results unveil molecular factors, which may be a general mechanism to determine the substrate versus inhibitor behavior of other protease inhibitors.
Keywords: biophysics; biotechnology; serine protease; single-domain antibody (sdAb, nanobody); x-ray crystallography
Introduction
Serine proteases catalyze the hydrolysis of peptide bonds and are involved in numerous physiological processes, including digestion, blood clotting, fibrinolysis, complement activation, and turnover of the extracellular matrix (1). Neutralizing serine protease activity using orthosteric inhibitors, i.e. active site binding inhibitors, has been shown to be a successful therapeutic strategy for a number of pathological conditions, although the similar active site topology in all serine proteases increases the risk of off-target effects. Today, serine protease inhibitors are clinically used for therapy of several diseases, including thrombosis and bleeding disorders (2–4).
All serine proteases catalyze the same type of hydrolytic reaction utilizing the same biochemical mechanism. Serine protease-catalyzed hydrolysis of a scissile bond proceeds through a highly conserved mechanism involving two tetrahedral intermediates and an acyl-enzyme complex. The polypeptide substrate is aligned in the active site of the protease interacting with the substrate specificity pockets denoted S1-Sn and S1′-Sn′ on the acyl and leaving group side of the scissile bond, respectively (5). The P1 residue of the substrate binds into the S1 pocket, and its carbonyl oxygen atom is inserted into the so-called oxyanion hole (backbone amides of Ser-195 and Ser-193, chymotrypsinogen numbering). The catalytic triad (His-57, Asp-102, and Ser-195) in the protease generates the required nucleophile for the attack of the hydroxyl group of Ser-195 on the carbonyl group of the P1-P1′ scissile bond to form the first tetrahedral intermediate and later the acyl-enzyme. Following release of the P1′-leaving group, a water molecule performs a second nucleophilic attack, thereby completing the cycle (6).
Peptide segments that bind the active site of serine proteases in a substrate-like manner may behave like an inhibitor or substrate. However, there is little information on the molecular factors that determine the inhibitor or substrate behavior of such a peptide segment. Understanding such factors is of particular importance as a growing number of new protease inhibitors with a substrate-like binding mode are emerging. Such inhibitors can be derived from combinatorial phage-display libraries (7), extracted from plants (8, 9) or designed by protein engineering based on naturally occurring standard mechanism inhibitors or other scaffolds (10–17).
Intriguingly, inhibitory antibody fragments that insert one or several complementary determining regions (CDR)2 into the active site of serine proteases have recently been isolated. Structural studies demonstrated that the antibody fragments function as inhibitors instead of substrates as their CDR loops adopt non-substrate-like conformations at the protease active site (18–21). In this report, we describe a new type of serine protease inhibitor by developing a single-domain Camelid-derived antibody fragment, a so-called nanobody, which specifically targets the active site of the trypsin-like serine protease urokinase-type plasminogen activator (uPA). Nanobodies are ideally shaped for interacting with concave clefts such as an active site of an enzyme. Accordingly, they were found to primarily target the substrate-binding cleft of lysozyme by insertion of a long protruding loop (22–24).
Here, we report the first x-ray crystal structure of a nanobody in complex with a serine protease. The crystal structure demonstrates that the nanobody binds to the active site of uPA in a substrate-like manner by inserting its protruding CDR-H3 loop. Specificity of the nanobody toward uPA is achieved by its interaction with the surface-exposed 37s loop of uPA. Combining alanine scanning mutagenesis, activity assays, proteolysis experiments, and surface plasmon resonance, we demonstrate that the nanobody acts as a strong inhibitor and as a poor substrate as it becomes slowly cleaved at the P1-P1′ peptide bond in the CDR-H3. Introducing mutations that weaken an intra-loop interaction network in the CDR-H3 converted the nanobody from a poor to a good substrate, thus suggesting that the rigid conformation of the CDR-H3 ensures a slow rate of hydrolysis. Collectively, our results reveal determinants decisive for inhibitor versus substrate behavior of the nanobody and suggest a general molecular mechanism by which a peptide segment that binds in a substrate-like manner to the active site of a serine protease behaves as an inhibitor or a substrate.
Results
Inhibition of Human uPA by an Anti-uPA Nanobody
A nanobody library was generated by immunizing an Alpaca (Vicugna pacos) with zymogen single-chain uPA. Screening the library with the active two-chain uPA as bait, we identified a nanobody (Nb4) that potently inhibited the proteolytic activity of uPA toward the macromolecular substrate plasminogen as well as uPA's amidolytic activity toward the small peptidic chromogenic substrate pyro-Glu-Gly-Arg-p-nitroanilide (CS-61(44)) (Fig. 1, a and b). We next tested the specificity of Nb4 toward a panel of human trypsin-like serine proteases and uPA from other species. Our results revealed that Nb4 is highly specific toward uPA under the given conditions, as it inhibited >90% of the catalytic activity of uPA from human, dog, and rabbit, although it inhibited only 50% of the catalytic activity of mouse uPA. It did not inhibit other human trypsin-like serine proteases or rat uPA measurably (Fig. 1c).
FIGURE 1.

Characterization of an anti-uPA inhibitory nanobody. a, Nb4 or a control nanobody at the indicated concentration was incubated with uPA (0.25 nm) before adding plasminogen (100 nm) and the chromogenic plasmin substrate S-2251 (0.5 mm). The IC50 value was calculated by non-linear regression. b, Nb4 or a control nanobody at the indicated concentration was incubated with uPA (2 nm) before addition of the chromogenic substrate CS-61(44) (47 μm). The inhibitory constant (Ki) was determined by non-linear regression. c, Nb4 (4 μm) were incubated with uPA-related trypsin-like serine proteases or uPA from other species. Chromogenic substrates were added to measure the effect of Nb4 on the amidolytic activity of the proteases. Error bars show the standard deviation from three experiments.
Crystal Structure of Nb4 Reveals a Protruding CDR-H3 Loop
The x-ray crystal structure of Nb4 was determined to a resolution of 2.16 Å (Rfree = 0.218 and Rwork = 0.177) by molecular replacement with four molecules in the asymmetric unit (Table 1). Only molecule B is described, as all residues of the CDR-H3 are visible in the electron density of this molecule (see “Experimental Procedures”). The crystal structure of Nb4 revealed a typical immunoglobulin domain with a scaffold consisting of two β-sheets presenting three antigen-binding CDR loops (Fig. 2a). In addition to the highly conserved Cys-22–Cys-96 disulfide bond, the structure revealed that Nb4 contains an additional disulfide bond between Cys-50 in the beginning of the CDR-H2 and Cys-104 in the middle of the CDR-H3. The Cys-50–Cys-104 disulfide bond stabilizes the 18-amino acid-long CDR-H3 loop by dividing it into two parts, an N-terminal part consisting of Asp-99 to Leu-103 and a C-terminal part consisting of Thr-105 to Val-116. The C-terminal part of the CDR-H3 forms a protruding loop in which the conformation of the loop is stabilized by multiple intra-loop interactions (Fig. 2b).
TABLE 1.
X-ray data collection and model refinement statistics
| Apo-Nb4 | uPA-Nb4 | |
|---|---|---|
| Data collection | ||
| Space group | P43212 | C121 |
| Cell dimensions | ||
| a, b, c (Å) | 99.83, 99.83, 127.33 | 186.985, 77.749, 61.454 |
| α, β, γ (°) | 90, 90, 90 | 90, 102.4, 90 |
| Resolution (Å) | 47.28–2.16 (2.24–2.16)a | 25.66–1.97 (2.02–1.97) |
| Rsym or Rmerge | 0.085 (0.691) | 0.058 (0.364) |
| I/σI | 22.42 (4.18) | 19.38 (2.33) |
| Completeness (%) | 99.9 (99.9) | 97.4 (92.6) |
| Redundancy | 14.5 (14.2) | 4.2 (3.7) |
| Refinement | ||
| Resolution (Å) | 47.28–2.16 | 25.66–1.97 |
| No. of reflections | 35,146 (3431) | 59,384 (2804) |
| Rwork/Rfree | 0.177/0.218 | 0.177/0.207 |
| No. of atoms | 4219 | 6216 |
| Protein | 3735 | 5802 |
| Ligand/ion | 24 | 127 |
| Water | 460 | 287 |
| B-factors | ||
| Protein | 37.80 | 29.14 |
| Ligand/ion | 69.60 | 62.01 |
| Water | 46.30 | 31.68 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.007 | 0.0187 |
| Bond angles (°) | 1.09 | 2.05 |
| Ramachandran favored (%) | 98 | 98.2 |
| Ramachandran allowed (%) | 2 | 1.8 |
| Ramachandran outliers (%) | 0 | 0 |
a Numbers in parentheses refer to the highest resolution shells.
FIGURE 2.
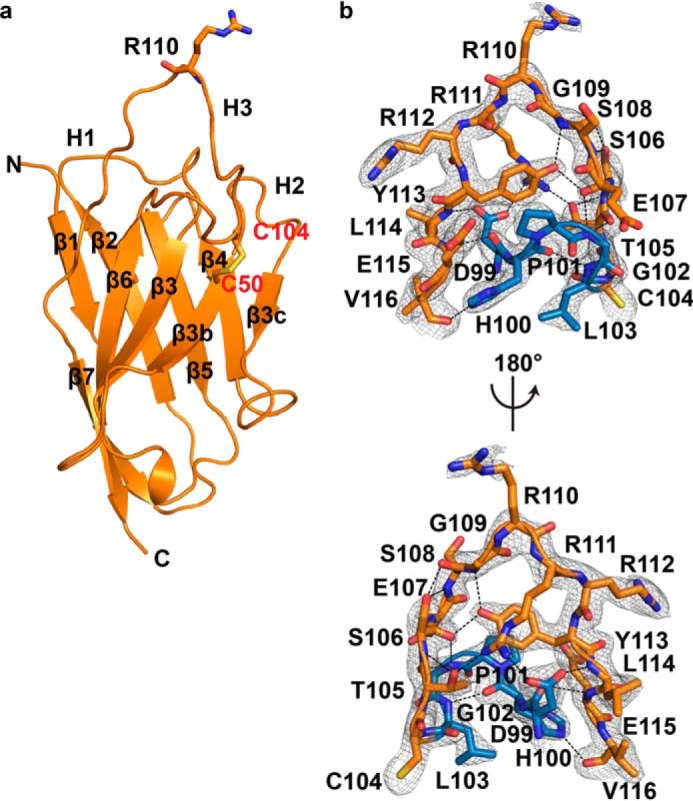
Crystal structure of Nb4. a, typical β-strand arrangement of the immunoglobulin domain of Nb4 is indicated along with the three CDR loops (H1, H2, and H3). The exposed Arg-110 at the tip of the CDR-H3 loop is show as sticks. The extra Cys-50–Cys-104 disulfide bond is indicated in red. b, CDR-H3 (DHPGLCTSESGRRRYLEV) is shown as sticks. The N-terminal part (99DHPGL103) is colored blue, and the C-terminal part (105TSESGRRRYLEV116) is colored orange. Black dashed lines show the intra-loop hydrogen bond network. The 2mFo − DFc electron density map (grey mesh) is contoured at 1σ over mean.
uPA-Nb4 Complex Crystal Structure Reveals That Nb4 Binds to uPA in a Substrate-like Manner
The x-ray crystal structure of the catalytic domain of human uPA in complex with Nb4 was determined to a resolution of 1.97 Å (Rfree = 0.207 and Rwork = 0.177) with two complexes in the asymmetric unit (Table 1). The following describes the complex between molecule S (Nb4) and molecule B (uPA). The structure of the complex revealed that Nb4 inserts the CDR-H3 into the active site region of uPA utilizing the S4-S3′ substrate-binding pockets (Fig. 3a). The side chain of the P1 Arg-110 residue of Nb4 inserts into the S1 specificity pocket to interact with Asp-189, Ser-190, and Gly-219 at the bottom of the pocket (Fig. 3b). The carbonyl group of Arg-110 occupies the oxyanion hole by contacting the backbone amides of Gly-193 and Ser-195, whereas the backbone amide of Arg-110 interacts with the side chain of Ser-195 and backbone carbonyl of Ser-214. This extensive network of interactions stably secures Arg-110 at the S1 pocket and oxyanion hole of uPA.
FIGURE 3.
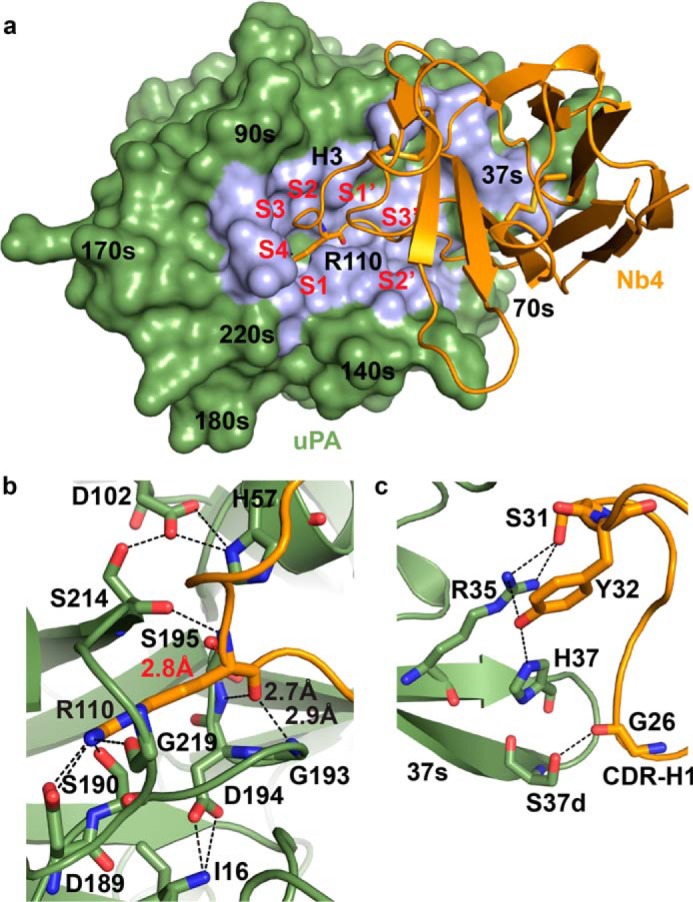
Crystal structure of the uPA-Nb4 complex. a, interaction of Nb4 (orange) with uPA (green) is primarily mediated by the CDR-H3, which interacts in a substrate-like manner with the substrate-binding pockets in the active site region of uPA (S4-S3′). The Nb4-uPA interaction surface is colored light blue. b, enlarged view of the P1-S1 interactions. The P1 Arg-110 of Nb4 (orange) inserts into the S1 pocket of uPA (green) to form interactions with Asp-189 at the bottom of the pocket. The carbonyl oxygen atom of Arg-110 is aligned in a substrate-like manner in the oxyanion hole, and the distance from the oxygen atom of the catalytic Ser-195 in uPA to the P1 carbonyl group in Nb4 is 2.8 Å (indicated in red). c, exosite interactions between the CDR-H1 of Nb4 (orange) and the 37s loop of uPA (green). Polar interactions are indicated as black dashed lines.
The CDR-H1 loop of Nb4 is also involved in binding to uPA by forming exosite interactions with the surface-exposed 37s loop of uPA (Fig. 3c). A list of polar contacts is given in Table 2.
TABLE 2.
Contact list between Nb4 and uPA from PISA server
| Polar contacts between uPA (chain B) and Nb4 (chain S) | |||
|---|---|---|---|
| CDR/framework | Nb4 | uPA | Distance |
| Å | |||
| Framework1 | Gly-26(O) | Ser-37d(OG) | 2.68 |
| CDR-H1 | Ser-31(OG) | Arg-35(NH1) | 3.27 |
| Ser-31(OG) | Arg-35(NH2) | 3.26 | |
| Tyr-32(OH) | Arg-35(NH1) | 3.15 | |
| Tyr-32(OH) | His-37(NE2) | 3.23 | |
| CDR-H3 | Ser-106(OG) | His-99(NE2) | 2.89 |
| Glu-107(OE1) | Arg-217(NH1) | 3.87 | |
| Glu-107(OE2) | Arg-217(NH1) | 3.00 | |
| Ser-108(OG) | Leu-97b(O) | 2.58 | |
| Ser-108(O) | Gly-216(N) | 3.00 | |
| Gly-109(O) | Gln-192(NE2) | 2.88 | |
| Arg-110(O) | Ser-195(N) | 2.87 | |
| Arg-110(O) | Gly-193(N) | 2.74 | |
| Arg-110(N) | Ser-214(O) | 3.07 | |
| Arg-110(NH1) | Asp-189(OD1) | 2.82 | |
| Arg-110(NH1) | Asp-189(OD2) | 3.42 | |
| Arg-110(NH1) | Ser-190(OG) | 2.81 | |
| Arg-110(NH2) | Asp-189(OD2) | 2.85 | |
| Arg-110(NH2) | Asp-189(OD1) | 3.79 | |
| Arg-110(NH2) | Gly-219(O) | 2.93 | |
| Arg-111(NH2) | Asp-60a(OD2) | 3.83 | |
| Arg-112(N) | Val-41(O) | 3.08 | |
| Tyr-113(O) | Tyr-151(OH) | 3.71 | |
| Glu-114[OE1) | Lys-143(NZ) | 2.81 | |
| Glu-114(OE2) | Lys-143(NZ) | 3.61 | |
Inhibitory Activity of Nb4 Depends on a Stable Conformation of the CDR-H3 Loop
To validate the structure model, we generated a panel of uPA and Nb4 mutants by alanine substitution of residues at the CDR-H3 of Nb4 and at the 37s, 60s, 90s, 140s, and 180s loops of uPA. Next, we determined and compared the Ki values of wild-type and mutant enzymes for Nb4 inhibition of uPA activity toward the chromogenic substrate CS-61(44). Overall, the analysis validated the importance of interactions observed in the complex crystal structure, as alanine substitutions of interface residues in both uPA and Nb4 with alanine resulted in increased Ki values (Table 3). Alanine substitution of the P1 Arg-110 in Nb4 completely abolished the inhibitory activity. Moreover, we observed a >4000-fold increase in Ki when the P1′ Arg-111 is mutated to alanine. Although Arg-111 does not interact with uPA extensively, our crystal structure suggests that it may exert its effect on the inhibitory activity of Nb4 by stabilizing the conformation of the CDR-H3 loop through a salt bridge with Asp-99 and cation-π interaction with Tyr-113 (Fig. 4a). Similar to Arg-111, Asp-99 does not form any close interaction with uPA, yet substitution of Asp-99 to alanine resulted in a >4000-fold increase in Ki, further stressing the importance of the Arg-111–Asp-99 ionic pair for the inhibitory activity of Nb4.
TABLE 3.
Km determinations for wild-type uPA and variants and Ki for inhibition of Nb4 mutants and uPA mutants
The data represent the mean ± S.D. of three independent experiments.
| Km | Ki | Ki (mut)/Ki (WT) | |
|---|---|---|---|
| mm | nm | ||
| uPA variant | |||
| Wild type | 0.12 ± 0.02 | 1.20 ± 0.08 | 1 |
| R35A | 0.11 ± 0.01 | 20.7 ± 0.4 | 17 |
| H37A | 0.12 ± 0.01 | 19.0 ± 3.0 | 16 |
| S37dA | 0.09 ± 0.01 | 0.34 ± 0.06 | 0.3 |
| D60aA | 0.09 ± 0.01 | 10.0 ± 1.0 | 8 |
| Y60bA | 0.09 ± 0.01 | 1.0 ± 0.2 | 0.75 |
| H99A | 0.46 ± 0.13 | 168 ± 6.0 | 140 |
| K143A | 0.12 ± 0.01 | 82 ± 6.0 | 68 |
| Y151A | 0.36 ± 0.02 | 5.8 ± 0.4 | 5 |
| Q192A | 0.21 ± 0.03 | 10.3 ± 0.3 | 8.5 |
| Nb4 variant | |||
| Wild type | 1.80 ± 0.15 | 1 | |
| S31A | 2.1 ± 0.1 | 1.1 | |
| D99A | >4000 | >4000 | |
| S106A | 61 ± 4.0 | 33 | |
| E107A | 3.6 ± 0.1 | 1.5 | |
| S108A | 11 ± 1.0 | 6 | |
| R110A | >4000 | >4000 | |
| R111A | >4000 | >4000 | |
| R112A | 2.6 ± 0.1 | 1.4 | |
| Y113A | 61 ± 4.0 | 33 | |
FIGURE 4.
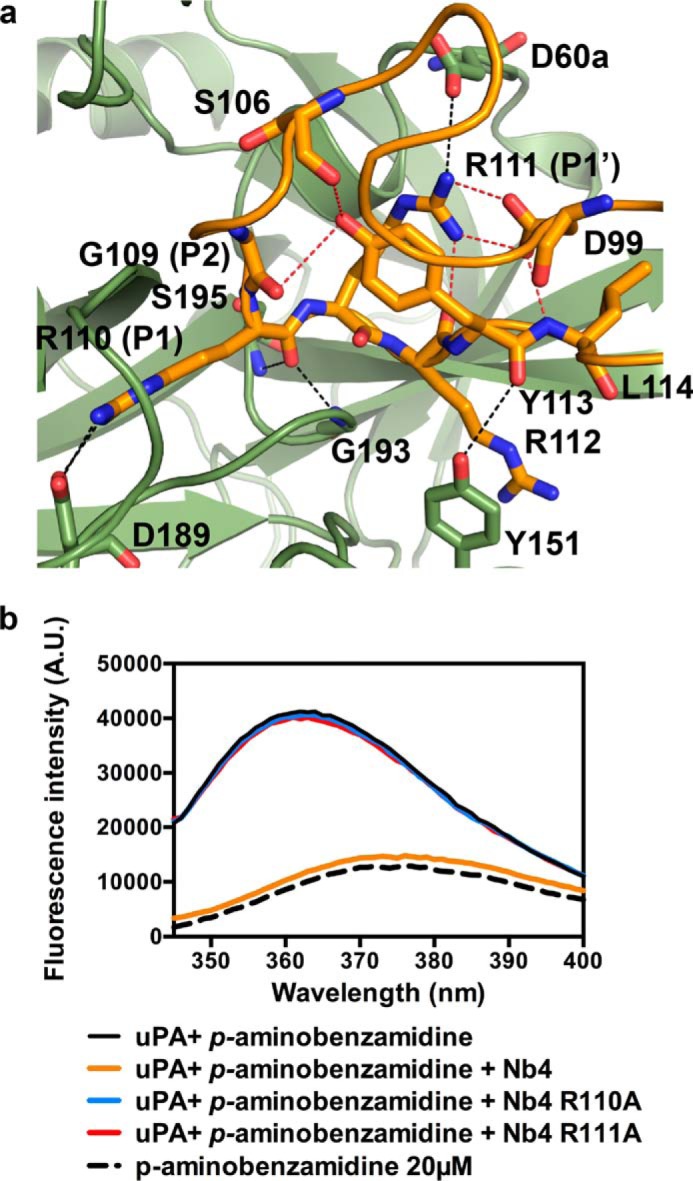
Intra-loop interaction network in the CDR-H3 of Nb4. a, shown is the CDR-H3 of Nb4 (orange) and the catalytic site of uPA (green). Red dashed lines indicate the intra-loop interaction network involving Asp-99, Arg-111, and Tyr-113. Black dashed lines indicate the interactions between the residues involved in the intra-loop hydrogen bond network and uPA. b, emission spectra of p-aminobenzamidine (20 μm, black dashed curve) in solution or when bound to uPA (0.4 μm, black solid curve) and in the presence of 500 nm wild-type Nb4 (orange), Nb4 R110A (blue), or Nb4 R111A (red). The excitation wavelength used was 335 nm. The curves are representative of three individual experiments.
Effect of P1 and P1′ Substitution on the Ability of Nb4 to Displace p-Aminobenzamidine from the S1 Pocket in uPA
To further verify the structural and mutagenesis information suggesting that Arg-110 and Arg-111 play critical roles in the inhibitory activity of Nb4, we performed fluorescence spectroscopy to measure the binding of the reversible ligand p-aminobenzamidine to the S1 pocket of uPA in the presence or absence of wild-type Nb4, Nb4 R110A, and Nb4 R111A alanine mutants. Binding of p-aminobenzamidine to the S1 pocket of uPA resulted in a high intensity fluorescent signal with emission λmax at 362 nm when excited at 335 nm, whereas the low intensity signal of p-aminobenzamidine in solution has λmax at 375 nm (Fig. 4b). Addition of Nb4 displaced p-aminobenzamidine from the S1 pocket as the intensity of the signal decreased and a red shift occurred in a similar manner for free p-aminobenzamidine in solution. Mutation of Arg-110 and Arg-111 in Nb4 to alanine completely abolished the ability of the nanobody to displace p-aminobenzamidine. The inability of the P1 Nb4 R110A mutant to displace p-aminobenzamidine is in complete agreement with the crystal structure. In contrast, the P1′ Nb4 R111A mutant also failed to displace p-aminobenzamidine, verifying that Arg-111 is essential for the inhibitory activity of Nb4, thus supporting the result from the activity based assays.
Inhibitor Versus Substrate Behavior of Nb4 Is Balanced by the Intra-loop Interactions Network within the CDR-H3
Given the substrate-like binding mode of Nb4, we next investigated the possibility that Nb4 acts as a uPA substrate. uPA was incubated with 3.5-fold molar excess of Nb4 for up to 96 h at 37 °C, and the mixture was then resolved by SDS-PAGE. The results revealed that Nb4 was slowly hydrolyzed by uPA at the putative scissile bond between Arg-110 and Arg-111 (Fig. 5). Unexpectedly, a fraction of Nb4 remained intact and inhibitory toward uPA after 6 h of incubation, indicating that the reaction may have reached a cleavage-resynthesis equilibrium (Fig. 5, a and b). To better understand what molecular factors help to balance Nb4 between its inhibitor versus substrate behavior, we next analyzed the hydrolysis of three Nb4 alanine mutants, namely Nb4 D99A, Nb4 R111A, and Nb4 Y113A, by uPA. As mentioned previously, these mutations aberrated the inhibitory activity of Nb4 and were strategically chosen as they form an intra-loop interaction network in the CDR-H3 of Nb4, which interconnects the P- and P′-sides of the putative scissile bond (Fig. 4a). Our results revealed that the removal of the salt bridge between Asp-99 and Arg-111 by mutating either residue enhanced the substrate behavior of Nb4, as these mutants were completely hydrolyzed by uPA within 2 min (Fig. 5, c and d). Mutation of Tyr-113, which forms hydrogen bonds to the P2 Gly-109 and Ser-106, and mediates cation-π interactions with Arg-111, also enhanced the substrate behavior of Nb4 although not as efficiently as the Nb4 D99A and Nb4 R111A mutants (Fig. 5e).
FIGURE 5.

Intra-loop hydrogen interaction network is decisive for the inhibitor versus substrate behavior of Nb4. a, proteolytic cleavage of Nb4 (3 μg) by uPA (3 μg) visualized by non-reduced SDS-PAGE analysis at different time points. The N-terminal sequence of the cleavage products is indicated to the right. b, on the left y axis is shown the quantification by densitometry of intact Nb4 at the indicated time points after incubation with uPA. On the right y axis is shown the remaining amidolytic activity of uPA hydrolysis of CS-61(44) (47 μm) after incubation with Nb4 for the indicated time points. Error bars show the standard deviation from three experiments. c, proteolytic cleavage of Nb4 R111A (3 μg) by uPA. d, proteolytic cleavage of Nb4 D99A (3 μg) by uPA. e, proteolytic cleavage of Nb4 Y113A (3 μg) by uPA. The N-terminal sequence of the cleavage products is indicated to the right.
Binding Kinetics of Nb4 Is Regulated by the Intra-loop Interaction Network within the CDR-H3
The balance between inhibitor versus substrate behavior of Nb4 was further analyzed using surface plasmon resonance (SPR) to determine the association rate constant (kon), the dissociation rate constant (koff), and the equilibrium dissociation constant (KD) of Nb4, Nb4 D99A, Nb4 R111A, and Nb4 Y113A toward the active two-chain uPA and the catalytically inactive two-chain uPA S195A mutant (Table 4 and Fig. 6). The analysis showed that Nb4 formed a high affinity and tight complex with the two-chain uPA (KD = 54 pm). Such high affinity is mainly due to a very slow dissociation rate. On the contrary, Nb4 D99A and Nb4 R111A mutants failed to produce binding curves for the determination of the kinetic parameters toward the active two-chain uPA. This is in agreement with the results from the proteolysis experiment in which the mutant proteins behaved like substrates and were hydrolyzed by uPA and could therefore not bind the active protease. The SPR analysis further revealed that Nb4 bound to the catalytically inactive two-chain uPA S195A with an apparent 10-fold higher affinity than to the wild-type two-chain uPA. The higher affinity was mainly due to a 6-fold decrease in the dissociation rate. Because the uPA S195A mutant is inactive, Nb4 D99A, Nb4 R111A, and Nb4 Y113A mutants were able to bind the mutant protease, although with a 400–20,000-fold lower affinity than the wild-type Nb4 (Table 4 and Fig. 6).
TABLE 4.
SPR analysis of binding of wild-type Nb4 or variants to active uPA or catalytically inactive uPA S195A
Association rate constants (kon), dissociation rate constants (koff), and the equilibrium dissociation constants (KD) were determined by fitting the SPR data to a 1:1 binding model. Data are represented as mean ± S.D. for three experiments.
| kon | kon (WT)/kon (mut) | koff | koff (mut)/koff (WT) | KD | KD (mut)/KD (WT) | |
|---|---|---|---|---|---|---|
| μm−1·s−1 | s−1 × 10−5 | nm | ||||
| uPA | ||||||
| Nb4 | 1.0 ± 0.2 | 5.4 ± 0.2 | 0.054 ± 0.02 | |||
| uPA S195A | ||||||
| Nb4 | 1.8 ± 0.5 | 0.9 ± 0.4a | 0.006 ± 0.003† | |||
| Nb4 D99A | 0.026 ± 0.005b | 70 | 290 ± 10b | 322 | 110 ± 22b | 20000 |
| Nb4 R111A | 0.023 ± 0.002b | 78 | 180 ± 10b | 200 | 78 ± 4b | 14200 |
| Nb4 Y113A | 0.009 ± 0.002b | 200 | 2.0 ± 0.7 | 2 | 2.3 ± 0.3b | 420 |
a Data are significantly different from the value of wild-type Nb4 binding to uPA (p < 0.05 by Student's t test).
b Data are significantly different from the value of wild-type Nb4 binding to uPA S195A (p < 0.001 by Student's t test).
FIGURE 6.

Surface plasmon resonance sensorgrams. Determination of kinetics of binding of wild-type Nb4 to active uPA (a) and wild-type Nb4 (b), Nb4 D99A (c), Nb4 R111A (d), and Nb4 Y113A (e) to catalytically inactive uPA S195A. The experimental data (red curves) were fitted to a 1:1 binding model (black curves) by the BIAcore evaluation software. Sensorgrams are representative of three experiments, and the dissociation rate (koff) is indicated above the curves. f, SPR sensorgram for the binding of wild-type Nb4 (black), Nb4 D99A (orange), Nb4 R111A (red), and Nb4 Y113A (blue) to active uPA.
Discussion
Here, the availability of a Camelid-derived antibody fragment, which binds in a substrate-like manner to the active site of the trypsin-like serine protease uPA, allowed us to ask the following question. Which factors determine whether a serine protease inhibitor with a substrate-like binding mode is an inhibitor or a substrate?
As evaluated by our x-ray crystal structure analysis, Nb4 binds to the active site of uPA and occludes the S4-S3′ substrate-binding pockets. Although we did not observe significant conformational changes in uPA upon binding to Nb4, there are small conformational changes in the CDR-H3-binding loop of Nb4 (Fig. 7). To contrast the differences in conformational changes, we calculated the main-chain torsion angles (ϕ,ψ) of the P3-P3′ peptide segment in both free and uPA-bound Nb4 (Table 5). The comparison demonstrated that the conformational change mainly occurs at the P3-P1 segment, whereas the P1′-P3′ segment remains largely the same upon complex formation with uPA (Table 5 and Fig. 7). However, once bound, Nb4 remains tightly associated with uPA, as determined by a slow dissociation rate (koff). It allows Nb4 to competitively inhibit the interaction of uPA with its physiological substrate plasminogen, as well as hydrolysis of small chromogenic substrates in our studies.
FIGURE 7.

Conformational changes associated with complex formation between uPA and Nb4. a, crystal structure of Nb4 in its free form (green) was superimposed (root mean square of all atoms is 1.377Å2) to Nb4 in its uPA-bound form (orange). The small conformational change in the P3-P1 segment of the CDR-H3 is highlighted. b, uPA (green) from the crystal structure of uPA in complex with Nb4 was superimposed (root mean square of all atoms 1.282 Å2) to uPA (blue) in its free form (PDB code 4DVA). The catalytic triad (His-57, Asp-102, and Ser-195), Asp-189, Asp-194, and Ile-16, is shown as sticks.
TABLE 5.
Comparison of the main-chain torsion angles of the P3-P3′ peptide segment of Nb4 with BPTI, CI2, Eglin c and PAI-1
The numbers in parentheses indicate the values of the main-chain torsion angles of the free inhibitor. The values for BPTI, CI2, Eglin c, and PAI-1 were obtained from the atomic coordinate files in the Protein Data Bank using the following PDB codes: BPTI/trypsin (PDB code 4Y0Y); BPTIfree (PDB code 5PTI); CI2/subtilisin (PDB code 1LW6); CI2free (PDB code 2CI2); Eglin c/subtilisin (PDB code 1CSE); and PAI-1/uPA (PDB code 3PB1).
| P3 | P2 | P1 | P1′ | P2′ | P3′ | |
|---|---|---|---|---|---|---|
| Nb4 | Ser | Gly | Arg | Arg | Arg | Tyr |
| ϕ | −109.91 (−146.84) | −66.06 (−131.64) | −100.97 (−69.10) | −66.75 (−64.94) | −80.74 (−79.18) | −113.85 (−78.21) |
| ψ | −16.62 (112.10) | 138.85 (154.40) | 22.20 (−16.02) | 168.54 (150.02) | 138.64 (130.00) | 143.90 (143.62) |
| BPTI | Pro | Cys | Lys | Ala | Arg | Ile |
| ϕ | −88.28 (−82.42) | −73.44 (−84.35) | −109.28 (−108.13) | −75.82 (−76.04) | −125.154 (−128.87) | −106.28 (−108.77) |
| ψ | −18.31 (−3.42) | 158.34 (154.75) | 29.83 (21.39) | 168.10 (168.38) | 81.19 (86.21) | 128.13 (117.47) |
| CI2 | Val | Thr | Met | Glu | Tyr | Arg |
| ϕ | −133.38 (−102.63) | −60.48 (−77.11) | −103.67 (−63.17) | −84.10 (−90.69) | −107.63 (−93.15) | −114.16 (−125.05) |
| ψ | 162.40 (166.46) | 146.33 (134.44) | 30.75 (26.94) | 153.64 (127.71) | 112.97 (116.84) | 108.19 (114.65) |
| Eglin c | Val | Thr | Leu | Asp | Leu | Arg |
| ϕ | −138.53 | −62.17 | −115.42 | −96.72 | −117.414 | −121.08 |
| ψ | 167.98 | 143.06 | 44.67 | 168.83 | 109.86 | 111.94 |
| PAI-1 | Ser | Ala | Arg | Met | Ala | Pro |
| ϕ | −85.34 | −55.10 | −98.95 | −62.81 | −70.64 | −68.52 |
| ψ | −29.16 | 134.90 | 12.74 | 164.71 | 115.74 | 139.41 |
Although the binding mode of Nb4 is similar to that observed in the Michaelis complex between uPA and PAI-1 and similar to the substrate-like binding mode of the classical standard mechanism inhibitor bovine pancreatic trypsin inhibitor (BPTI), it is different from previously reported active site binding anti-serine protease antibody fragments (Fig. 8). For instance, a series of antibody fragments against matriptase all insert their CDR-H3 loop in a non-substrate-like reversed C- to N-terminal orientation, which places the putative P1-P1′ scissile bond out of reach for a nucleophilic attack by Ser-195 (18, 19). Moreover, the P1 Arg residue forms a suboptimal interaction in which a water molecule is required to mediate the interaction between the P1 Arg residue and Asp-189 at the bottom of the S1 pocket. In contrast, the anti-plasma kallikrein antibody DX-2930 binds in the normal N- to C-terminal orientation, but a non-substrate-like conformation of the loop prevents it from binding the S1′-S3′ pockets and thereby hydrolysis of the putative scissile bond (21). The anti-HGFA Fab58 also binds to the active site region of the protease, occluding the S2 and S3 but not the S1 pocket (20). In contrast to the previously described anti-protease antibody fragments, Nb4 places the CDR-H3 loop into the active site of uPA in a substrate-like manner aligning the P1-P1′ putative scissile bond within 2.8 Å of the hydroxyl group of Ser-195, which is optimal for a nucleophilic attack.
FIGURE 8.

CDR-H3 of Nb4 adopts a substrate-like binding mode. a, crystal structure of uPA in complex with PAI-1 (PDB code 3PB1) was superimposed to uPA-Nb4. Shown is the reactive loop of the serpin PAI-1 (yellow) and the CDR-H3 of Nb4 (orange). b, crystal structure of trypsin in complex with the standard mechanism inhibitor BPTI (PDB code 4Y0Y) was superimposed to uPA-Nb4. Shown is the reactive loop of BPTI (white) and the CDR-H3 of Nb4 (orange). c, comparison between the CDR-H3's of the anti-matriptase antibody E2 (magenta) (PDB code 3BN9) and Nb4 (orange). The crystal structure of E2 in complex with matriptase was superimposed to uPA-Nb4. In all panels the catalytic Ser-195 colored red, and the residues in uPA (green) that interact with Nb4 are highlighted in light blue. The N and C termini of the peptide segments are indicated in yellow for Nb4, PAI-1, and BPTI and in green for E2.
The previously reported anti-protease antibody fragments achieve exquisite selectivity toward their target protease by forming exosite interactions with a surface-exposed loop of the protease. For Nb4, it was evident from the crystal structure that its CDR-H1 loop interacts with the 37s loop of uPA, which is a non-conserved region that varies in sequence and length among trypsin-like serine proteases. Thus, specific interactions between Nb4 and this region, particularly at residues 35, 37, and 37d, may contribute to the specificity of Nb4 for uPA. Further supporting this notion, Nb4 efficiently inhibits the activity of uPA from dog and rabbit, as the Nb4-interacting residues Arg-35, His-37, and Ser-37d are completely conserved between uPA from human and from these species (Fig. 9). Interestingly, although Nb4 also inhibits the activity of murine uPA, although less efficiently, it has no effect on rat uPA. Comparison of the sequence shows that the 37s loop of murine uPA contains Gln-35, Asn-37, and Ser-37d at the Nb4-interacting positions, whereas rat uPA contains a hydrophobic leucine residue at position 35. Taken together, these observations suggest that the conservation of polar amino acids at all three positions is a prerequisite for the protease to be recognized by Nb4.
FIGURE 9.

Sequence alignment of the 37s loop of uPA to uPA-related trypsin-like serine proteases and uPA from other species. The Nb4 interacting residues are highlighted in cyan, and their conserved position in the other proteases are highlighted in yellow. The sequences were obtained from the UniProt database.
The substrate-like binding mode of Nb4 suggested that it may act as a poor uPA substrate with a presumably low kcat. In fact, we found that Nb4 was hydrolyzed by uPA. The hydrolysis was extremely slow, explaining the inhibitory activity of Nb4. Given the substrate-like binding mode of Nb4 to uPA, we expected a continuous accumulation of cleaved Nb4. Intriguingly, this was not observed, as the ratio between cleaved and intact Nb4 stayed largely constant over time, and ∼30–40% of Nb4 remained intact and inhibitory. This observation proposes that Nb4 prevents complete hydrolysis by uPA through a mechanism involving a cleavage resynthesis equilibrium between cleaved and intact inhibitor. The cleavage resynthesis equilibrium is reached after 3–6 h, which is in good agreement with an estimated half-life of the uPA-Nb4 complex at 3.6 h as determined by the dissociation rate from the SPR analysis (Table 4).
To address these puzzling observations, we hypothesized that the intra-loop interaction network involving Asp-99, Arg-111, and Tyr-113 is responsible for the cleavage resynthesis equilibrium between cleaved and intact inhibitors by balancing the inhibitor versus substrate behavior of Nb4. A similar intra-loop interaction network was not observed in the apo-Nb4 structure (Fig. 2b). In fact, electron density for Arg-111 in the apo-Nb4 structure was weak suggesting that Arg-111 is not involved in the CDR-H3 intra-loop interaction network (Fig. 2b). Thus, we believe that the intra-loop interaction network involving Asp-99, Arg-111, and Tyr-113 is first formed upon complex formation with uPA to ensure a rigid conformation that leads to a slow dissociation rate of the uPA-Nb4 complex. In agreement with our hypothesis, disruption of the intra-loop interaction network by mutations converted Nb4 from an inhibitor to a substrate. However, different mutations to Nb4 affected the binding kinetics differently, thus allowing us to identify decisive molecular factors for the inhibitor versus substrate behavior of Nb4. As evaluated by our findings, such factors seems to be confined to the Asp-99–Arg-111 salt bridge as the Nb4 D99A and Nb4 R111A mutants were cleaved instantly by uPA (Fig. 5). Accordingly, disruption of the salt bridge caused a 300-fold increase in dissociation rate toward the catalytically inactive uPA S195A variant as compared with wild-type uPA. In contrast, mutation of Tyr-113 to Ala only resulted in a modest 2-fold increase in dissociation rate (Table 4). This strongly suggests that the dissociation rate of the P1′-leaving group is the single most decisive factor, which determines the inhibitor versus substrate behavior of Nb4. In the Nb4 D99A and Nb4 R111A mutants, the interconnectivity between the P- and P′-site of the Arg-110–Arg-111 or Arg-110–Ala-111 scissile bond is significantly weakened, and the more flexible position of the P1′ residue may allow a fast dissociation of the P′-leaving group resulting in entry of the catalytic water to complete the catalytic cycle. Oppositely, following cleavage of the Arg-110–Arg-111 scissile bond in wild-type Nb4, the rigid position of the P1′ Arg-111, secured by Asp-99, ensures a slow dissociation. Following this hypothesis, the slow dissociation of cleaved Nb4 may allow resynthesis of the Arg-110–Arg-111 peptide bond, as the active site of uPA is inaccessible to the catalytic water. This hypothesis is in reasonable agreement with the observation of a cleavage-resynthesis equilibrium between cleaved and intact Nb4 and with the observation of an intact Arg-110–Arg-111 peptide bond in a planar configuration in the crystal structure of Nb4 in complex with uPA.
A substrate-like binding mode is also observed in a group of naturally occurring serine protease inhibitors termed “standard mechanism” or “Laskowski” inhibitors (11, 12). A number of publications have described the determinants of the proteolytic resistance of standard mechanism inhibitors. Early studies on chymotrypsin inhibitor 2 (CI2) and BPTI demonstrated that an intra-loop hydrogen interaction network within the protease-binding loop of CI2 was important for its resistance to proteolysis, whereas BPTI was found to be resistant to proteolysis due to a disulfide bond stabilizing the conformation of the protease-binding loop (25–27). However, the resistance to proteolysis may also be dependent on the scaffold of the inhibitor, which supports and confers rigidity to the protease-binding loop as shown in amyloid precursor protein and CI2 (28, 29). Similarly to CI2, our results demonstrate that the resistance to proteolysis of Nb4 depends heavily on the intra-loop interaction network within the CDR-H3.
The protease-binding loop of standard mechanism inhibitors exhibits a highly conserved main-chain conformation of the P3-P3′ segment, as exemplified by CI2 and Eglin c (30). However, BPTI deviates from the conserved pattern, as it does not form a main-chain conformation of an anti-parallel β-strand at the P3 position. Similarly, we found that Nb4 also deviates from the conserved P3-P3′ pattern by not forming a main-chain conformation of an anti-parallel β-strand at the P3 position (Table 5). Moreover, we also observed a deviation at the P2′ position, at which the ϕ- and ψ-angles of Arg-112 in Nb4 is altered between 30–50° and 30–60°, respectively, as compared with the P2′ position in BPTI, CI2, and Eglin c (Fig. 8 and Table 5). Interestingly, we observed a similar alteration at the P2′ position in PAI-1 when compared with BPTI, CI2, and Eglin c, hence highlighting a small difference in the binding mode between Nb4 and the standard mechanism inhibitors. These observations further suggest that an altered main-chain conformation at the P2′ position is required to bind in a substrate-like mode to the active site of uPA. Notably, the conformational change in the P3-P1 segment of Nb4 upon binding to Nb4 is larger than those observed in the standard mechanism inhibitors, suggesting that the CDR-H3 of free Nb4 is more flexible than the protease-binding loops of standard mechanism inhibitors.
Originally it was believed that standard mechanism inhibitors act as poor substrates with low kcat values by forming a tight and rigid interaction with their target protease thereby preventing the formation of the first tetrahedral transition state during catalysis (30–32). However, several studies have demonstrated that the catalysis is blocked after a rapid formation of the acyl-enzyme in which the tight protease-inhibitor interaction prevents dissociation of the newly formed leaving-group, thus favoring reformation of the P1-P1′ peptide bond in the inhibitor by a nucleophilic attack of the P1′ amino group on the carbonyl group of the acyl-enzyme (27, 33–35). In contrast, we were unable to detect the acyl-enzyme in our studies, suggesting that either a fast reformation of the Arg-110–Arg-111 peptide bond occurs or that a relatively low concentration of the acyl-enzyme intermediate was presented at the cleavage-resynthesis equilibrium (25, 34). Nevertheless, the observations that the P1′ Arg-111 in Nb4 is held in place by Asp-99 illustrates its potential in mediating a nucleophilic attack on the acyl-enzyme intermediate, which favors reformation of the Arg-110–Arg-111 peptide bond over the deacylation of the acyl-enzyme intermediate.
Standard mechanism inhibitors display broad target specificity. Although much effort has been directed toward optimizing the protease-binding loops of standard mechanism inhibitors to engineer them with therapeutic applications (15, 17, 36), our study demonstrates that high affinity antibody fragments, which specifically bind the active site of a serine protease in a substrate-like manner, can be selected by combinatorial techniques without affinity maturation or other ex vivo modifications. Disulfide-constrained peptides are another group of inhibitors derived by combinatorial phage-display techniques. The peptidic inhibitor mupain-1 and its derivative peptide 10 (pkalin-3) inhibit the activity of mouse uPA and human plasma kallikrein, respectively (7, 13). The inhibitory mechanisms of both peptides were explained from crystal structure analysis that demonstrates small changes in bond distances around the P1 Arg residue, which places the catalytic Ser-195 3.9 Å away from the P1 carbonyl group for mupain-1 and 3.0 Å for pkalin-3 (Fig. 10). However, an alternative mechanism cannot be ruled out that mupain-1 and pkalin-3 may inhibit their target protease in a manner similar to that of Nb4 by a cleavage resynthesis equilibrium mechanism involving reformation of the P1-P1′ peptide bond, as the distance from the Ser-195 to the carbonyl group of Arg-110 in Nb4 is 2.8 Å. Thus, the results presented in this study may be considered as a general mechanism by which inhibitors with a substrate-like binding mode act as inhibitors and not substrates. Moreover, the development of a highly specific uPA inhibitor may be useful for non-invasive molecular imaging for detection of uPA activity in particular molecular events such as remodeling of the extracellular matrix in the progression of cancer (37).
FIGURE 10.

Comparison of Nb4 to mupain-1 and peptide 10. a, orientation of the CDR-H3 loop of Nb4 (orange) in the active site of uPA (green). b, orientation of the peptidic inhibitor mupain-1 (green) in the active site of murinized human uPA (green) (PDB code 4X1Q). c, orientation of the peptidic inhibitor peptide 10 (pkalin-3) (yellow) in the active site of human plasma kallikrein (green) (PDB code 4ZJ6). In all panels, the catalytic Ser-195 is colored red. The N- to C-terminal orientation of the inhibitors is indicated in yellow. d, distances from the catalytic Ser-195 to the carbonyl group of the P1 Arg residue in uPA-Nb4 (orange), uPAH99Y-mupain-1 (green), and kallikrein-pkalin-3 (yellow). Gly-193 is also shown to highlight differences in alignment of the P1 carbonyl oxygen atom of the inhibitors in the oxyanion hole (amide groups of Ser-195 and Gly-193).
In conclusion, our study demonstrates that the conformational rigidity of the peptide segment that will be inserted into the active site of a protease is an important factor that determines whether the peptide segment behaves like an inhibitor or substrate. We have also presented the first crystal structure of a nanobody in complex with a serine protease and the first ever crystal structure of an antibody fragment that targets the active site of a serine protease in a substrate-like manner, thus demonstrating a new mechanism for inhibition of a protease by an antibody fragment. Taken together, our findings shed new light on the inhibitory mechanism of inhibitors with a substrate-like binding mode and provide the basis for designing potent and specific protease inhibitors by optimizing the intra-loop interaction network within the inhibitory peptide segment.
Experimental Procedures
Generation of Anti-uPA Nanobodies
The immunization and construction of the nanobody phage library were conducted as described previously using single-chain zymogen uPA as antigen (38). Briefly, selection of anti-human uPA nanobodies was performed by immobilizing two-chain active uPA (ProSpec-Tany TechnoGene) (100 μg/ml) in 96-well MaxiSorp immunoplates (Nunc). After adding the nanobody phage library, bound phages were eluted with triethylamine (100 mm) and neutralized with 1 m Tris, pH 8.2. Recovered phages were amplified in Escherichia coli TG1 cells. During three subsequent selection rounds, the stringency of plate washing was gradually increased. uPA-binding nanobodies were identified by a polyclonal phage ELISA by randomly picking single colonies. Positive clones were sequenced, and unique clones were transformed into E. coli WK6 (su−) cells, produced and purified as described previously (39). Alanine mutants of Nb4 were prepared by standard QuickChange site-directed mutagenesis and expressed in E. coli WK6 (su−) cells.
Generation of Alanine Mutants and the Catalytic Domain of uPA
Full-length human uPA and uPA site-directed mutants were cloned into the pCDNA 3.1 vector. cDNAs were transfected into HEK293 6E suspension cells and cultured in a humidified 5% CO2 incubator at 37 °C. The media used was Freestyle F17 (Gibco) containing 4 mm l-glutamine, 0.1% Pluronic F68 (Gibco), 1% penicillin/streptomycin (Invitrogen), 25 μg/ml G418 (Geneticin, Thermo Fisher Scientific). Linear polyethyleneimine (2.2 mg) and cDNA (1.1 mg) were mixed in PBS for 15 min and added to 1 liter of cell cultures at a density of 1 × 106 cells/ml. Tryptone N1 (0.5%) was added to the culture 24 h post-transfection. The cultures were continued for 6 days before harvesting the conditioned media. The catalytic domain of uPA was recombinant expressed in Pichia pastoris as described previously (40).
Enzymatic Assays
All enzymatic assays were performed in HEPES-buffered saline (HBS: 10 mm HEPES, pH 7.4, 140 mm NaCl) with 0.1% bovine serum albumin (BSA), and the initial velocities were monitored at an absorbance of 405 nm for 1 h at 37 °C in a kinetic microplate reader (Multiscan Go, Thermo Scientific).
For the plasminogen activation assay, various concentrations of Nb4 (120 to 0 nm) were preincubated with uPA (0.25 nm) for 15 min at 37 °C. Next, the reaction was initiated by adding human plasminogen (100 nm), purified from plasma, and plasmin substrate H-d-Val-Leu-Lys-p-nitroanilide (0.5 mm) (S-2251, Chromogenix). To determine the velocity of plasminogen activation, the data were transformed to plot ΔA405/Δtime on the ordinate and time on the abscissa (41). Velocities were calculated from the time interval 5–20 min of these plots and used for calculation of IC50 values.
For inhibition of uPA hydrolysis, PyroGlu-Gly-Arg-p-Na-HCl (CS-61(44), Hyphen Biomed), various concentration of wild-type Nb4 or Nb4 mutants were pre-incubated with wild-type uPA (2 nm) or with various uPA alanine mutants in conditioned media for 15 min at 37 °C before adding CS-61(44) (47 μm) to initiate the reaction. The apparent inhibitory constant KDapp was calculated as described previously (42), and Ki values were determined by Equation 1.
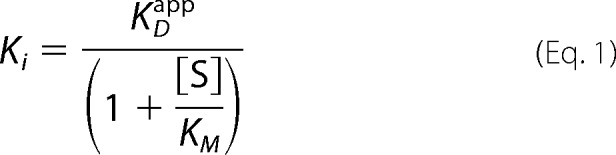 |
KM values were determined by measuring initial velocities of hydrolysis of CS-61(44) (2–0 mm) by wild-type uPA (2 nm) or by various uPA alanine mutants in conditioned media. The KM values were calculated by fitting the data to standard Michaelis-Menten kinetics.
To evaluate the specificity of Nb4, we incubated Nb4 (4 μm) with human plasma kallikrein (2 nm, 300 μm S-2302), human coagulation factor VIIa (20 nm, 1000 μm S-2288), human plasmin (5 nm, 750 μm S-2288), human matriptase (2 nm, 100 μm S-2288), human-activated protein C (8 nm, 375 μm S-2366), human hepsin (4 nm, 170 μm S-2366), human tissue-type plasminogen activator (2 nm, 500 μm S-2288), murine uPA (2 nm, 750 μm CS-61(44)), dog uPA (4 nm, 750 μm CS-61(44)), rat uPA (4 nm, 750 μm CS-61(44)), rabbit uPA (4 nm, 750 μm CS-61(44)), or human uPA (2 nm, 47 μm CS-61(44)) for 15 min at 37 °C before adding the chromogenic substrates at concentrations that equal half the experimentally determined KM value for each enzyme/substrate pair (data not shown). The chromogenic substrates S-2288 and S-2366 were purchased from Chromogenix, and the proteases were from Molecular Innovations, except for tissue-type plasminogen activator, which was purchased from Boehringer Ingelheim, and matriptase, hepsin, and mouse uPA, which were produced recombinant in HEK293 E6 cells. For measuring the activity of factor VIIa, 5 mm Ca2Cl was added to the buffer.
Crystallography
Crystals of the free form of Nb4 were grown by sitting drop vapor diffusion by adding 2 μl of Nb4 (20 mg/ml) to 2 μl of mother liquid (0.1 m MES, 0.2 m ammonium sulfate, 30% w/v PEG5000, pH 6.5). Crystals grew within 3 days and were cryoprotected in mother liquid supplemented with 5% ethylene glycol and vitrified in liquid nitrogen. A native data set to 2.16 Å resolution was collected at 100 K at a wavelength of 0.98 Å on the BESSY beamline at the Berlin Electron Storage Ring Society for Synchrotron Radiation, Berlin, Germany. The diffraction data were indexed, integrated, and scaled in space group P43212 using the XDS program package (43). The initial phases were obtained by molecular replacement with PhenixMR (44) with PDB code 4JVP as the search model (45). Model building was performed in Coot (46); refinement and structure validation were performed using PHENIX (44). Molecular graphics were prepared in PyMOL (47). For each chain (A, B, C, or D), residues not having sufficient electron density were omitted from the model or modeled as alanine. In chain A, Ser-127 and Ala-128 in the C terminus were omitted. In chain B, Ser-127 was modeled as an Ala and Ala-128 was omitted. In chain C, Glu-107, Ser-108, and Gly-109 were omitted, and Arg-110 was modeled as an Ala. In chain D, Glu-107, Ser-108, and Gly-109 were omitted, and Arg-110 was modeled as an Ala; Ser-127 was modeled as an Ala; and Ala-128 was omitted.
Complexes were formed by incubating the catalytic domain of uPA with Nb4 at a molar ratio of 1:1.3 at 4 °C for 1 h. The complexes were then purified by gel filtration chromatography using a Superdex 75 10/300 GL (GE Healthcare) using phosphate-buffered saline (PBS: 137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.4) as running buffer. Crystals were grown using hanging drop vapor diffusion by adding 1 μl of uPA/Nb4 (3 mg/ml) to 1 μl of mother liquor (1.4 m ammonium sulfate, 0.1 m MES, pH 6.25, 0.1 m disodium phosphate dibasic and 0.5% (v/v) Tween 20) at 16 °C. Crystals were cryoprotected in mother liquor supplemented with 30% (v/v) glycerol and vitrified in liquid nitrogen. Crystals were tested and screened at 100 K using the Rigaku FRE+ x-ray source at the Centre for Protein Science and Crystallography, Chinese University of Hong Kong. The highest resolution data set at 1.97 Å was collected on the 13B1 beamline at the National Synchrotron Radiation Research Center (NSRRC), Taiwan. The diffraction data were indexed, integrated, and scaled in space group C121 using the HKL2000 program package (48). The initial phases were obtained by molecular replacement with PHASER of the CCP4 program suite (49) with the structures of uPA (PDB code 4DVA) and Nb4 as the search model. Model building was performed in Coot (46); refinement was performed using REFMAC (50); structure validation and conformational quality were assessed by PROCHECK (51).
Fluorescent Assay
uPA (0.4 μm) alone or mixed with Nb4 (500 nm), Nb4 R110A (500 nm), Nb4 R111A (500 nm), or Nb4 R112A (500 nm) were incubated for 15 min at 22 °C before adding p-aminobenzamidine (20 μm). Fluorescence emission spectra were recorded at 25 °C on a PTI QuantaMaster spectrofluorometer in a 2 × 10-mm semi-micro quartz cuvette. An emission scan of 340–400 nm using an excitation wavelength of 335 nm and an integration of 1–2 s over a 1.0-nm step resolution was used. The buffer used was HBS supplemented with 0.1% polyethyleneglycol 8000.
SDS-PAGE Analysis
Nb4 (3 μg), Nb4 D99A (3 μg), Nb4 R111A (3 μg), or Nb4 Y113A (3 μg) was incubated at 37 °C with active uPA (3 μg) in PBS, and the reactions were quenched by adding the protease inhibitor phenylmethylsulfonyl fluoride (PMSF, 1 mm, Sigma) and pre-warmed (96 °C) non-reducing sample buffer at the indicated time points. The samples were separated by non-reducing 18% SDS-PAGE, and the digestion products were N-terminally sequenced at the Department of Molecular Biology and Genetics, Aarhus University, Aarhus, Denmark, to determine the cleavage site. At each time point, 2 μl of each reaction was withdrawn and diluted 1000-fold in 10 mm HEPES, pH 7.4, 140 mm NaCl + 0.1% BSA before addition of the chromogenic substrate CS-61(44) (47 μm) to determine the initial rates of uPA hydrolysis of CS-61(44) by monitoring the absorbance at 405 nm for 1 h at 37 °C in the kinetic microplate reader. The densitometry analysis was performed using the GelEval software.
Surface Plasmon Resonance
The equilibrium dissociation constant KD, the association rate kon, and the dissociation rate koff of Nb4 binding to two-chain active uPA were determined by surface plasmin resonance on a BIAcore T100 (GE Healthcare). Nb4 was diluted to 0.5 μg/ml in immobilization buffer (10 mm sodium acetate, pH 5) and immobilized on a CM5 sensor chip (GE Healthcare) by amine coupling to ∼200 response units. Active uPA (1–0 nm) diluted in running buffer (HBS + 0.1% BSA) was subjected to a long association phase of 480 s to obtain curvature on the association curve. The dissociation was monitored for 3600 s to allow at least 10% of the analyte to dissociate before regenerating the surface. For binding of wild-type Nb4, Nb4 D99A, Nb4 R111A, and Nb4 Y113A to active two-chain uPA and catalytically inactive two-chain uPA S195A, the monoclonal anti-uPA clone 6 (52) was diluted to 75 μg/ml in immobilization buffer and immobilized on a CM5 sensor chip by amine coupling to ∼5000 response units. Active two-chain uPA (50 nm) was captured for 120 s before injection of wild-type Nb4 (150 nm), Nb4 D99A (150 nm), Nb4 R111A (150 nm), or Nb4 Y113A (150 nm) for 160 s. The dissociation was monitored for 60 s. For measuring binding kinetics toward two-chain uPA S195A from conditioned media, it was captured for 120 s before injection of wild-type Nb4 (5 to 0 nm), Nb4 D99A (300 to 0 nm), Nb4 R111A (300 to 0 nm), or Nb4 Y113A (300 to 0 nm) for 240 s. The dissociation was monitored for 600 s for Nb4 D99A, Nb4 R111A, and Nb4 Y113A, whereas it was monitored for 3000 s for wild-type Nb4. For wild-type Nb4 and Nb4 Y113A, the low dissociation rate made it experimentally impossible to monitor the dissociation long enough to allow 10% of the analyte to dissociate. In all experiments, the flow rate was 30 μl/min, and the surface was regenerated using 10 mm glycine, 0.5 m NaCl, pH 2.5. The kinetic constants were determined at 25 °C using the BIAcore evaluation software. The experimental curves fit perfectly to a 1:1 binding model, despite the long dissociation phase, confirming that there was little heterogeneity in the experimental samples.
Author Contributions
T. K. H., J. N., and P. A. A. designed the research; T. K. H., E. O., and K. Y. performed the research; T. K. H., G. G., S. M., P. D., M. H., P. A. A., and J. N. analyzed the data; and T. K. H., P. A. A., and J. N. wrote the paper.
This work was supported in part by Danish National Research Foundation Grant 26-331-6 (to P. A. A.), Natural Science Foundation of China Grants 31161130356, 31170707, and 31370737 (to M. H.), Lundbeck Foundation Grant R83-A7826 (to P. A. A.), The Cancer Research Foundation of 1989 (to P. A. A.), Research Grants Council of Hong Kong SAR Grants GRF 473310 and CUHK1/CRF/13G, and CUHK One-off Funding for Joint Lab Project Number 3132980 (to J. C. K. N.) the Graduate School of Science and Technology, Aarhus University (to T. K. H.), Carlsberg Foundation Grant CF15-0814 (to T. K. H.). The authors declare that they have no conflicts of interest with the content of this article.
The atomic coordinates and structure factors (codes 5HGG and 5HDO) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CDR
- complementary determining region
- uPA
- urokinase-type plasminogen activator
- BPTI
- bovine pancreatic trypsin inhibitor
- PDB
- Protein Data Bank
- SPR
- surface plasmon resonance.
References
- 1. Puente X, O., and López-Otín C. (2008) in The Cancer Degradome (Edwards D., Høyer-Hansen G., Blasi F., and Sloane B. F., eds) pp. 3–15, Springer, New York [Google Scholar]
- 2. Turk B. (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799 [DOI] [PubMed] [Google Scholar]
- 3. Cudic M., and Fields G. B. (2009) Extracellular proteases as targets for drug development. Curr. Protein Pept. Sci. 10, 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drag M., and Salvesen G. S. (2010) Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 9, 690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schechter I., and Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 6. Hedstrom L. (2002) Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524 [DOI] [PubMed] [Google Scholar]
- 7. Zhao B., Xu P., Jiang L., Paaske B., Kromann-Hansen T., Jensen J. K., Sørensen H. P., Liu Z., Nielsen J. T., Christensen A., Hosseini M., Sørensen K. K., Nielsen N. C., Jensen K. J., Huang M., and Andreasen P. A. (2014) A cyclic peptidic serine protease inhibitor: increasing affinity by increasing peptide flexibility. PLoS One 9, e115872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luckett S., Garcia R. S., Barker J. J., Konarev A. V., Shewry P. R., Clarke A. R., and Brady R. L. (1999) High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 290, 525–533 [DOI] [PubMed] [Google Scholar]
- 9. Felizmenio-Quimio M. E., Daly N. L., and Craik D. J. (2001) Circular proteins in plants: solution structure of a novel macrocyclic trypsin inhibitor from Momordica cochinchinensis. J. Biol. Chem. 276, 22875–22882 [DOI] [PubMed] [Google Scholar]
- 10. Farady C. J., and Craik C. S. (2010) Mechanisms of macromolecular protease inhibitors. Chembiochem 11, 2341–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laskowski M. Jr., and Kato I. (1980) Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626 [DOI] [PubMed] [Google Scholar]
- 12. Laskowski M., and Qasim M. A. (2000) What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes? Biochim. Biophys. Acta 1477, 324–337 [DOI] [PubMed] [Google Scholar]
- 13. Xu P., Xu M., Jiang L., Yang Q., Luo Z., Dauter Z., Huang M., and Andreasen P. A. (2015) Design of specific serine protease inhibitors based on a versatile peptide scaffold: conversion of a urokinase inhibitor to a plasma kallikrein inhibitor. J. Med. Chem. 58, 8868–8876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stoop A. A., and Craik C. S. (2003) Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nat. Biotechnol. 21, 1063–1068 [DOI] [PubMed] [Google Scholar]
- 15. de Veer S. J., Swedberg J. E., Akcan M., Rosengren K. J., Brattsand M., Craik D. J., and Harris J. M. (2015) Engineered protease inhibitors based on sunflower trypsin inhibitor-1 (SFTI-1) provide insights into the role of sequence and conformation in Laskowski mechanism inhibition. Biochem. J. 469, 243–253 [DOI] [PubMed] [Google Scholar]
- 16. Quimbar P., Malik U., Sommerhoff C. P., Kaas Q., Chan L. Y., Huang Y. H., Grundhuber M., Dunse K., Craik D. J., Anderson M. A., and Daly N. L. (2013) High-affinity cyclic peptide matriptase inhibitors. J. Biol. Chem. 288, 13885–13896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swedberg J. E., de Veer S. J., Sit K. C., Reboul C. F., Buckle A. M., and Harris J. M. (2011) Mastering the canonical loop of serine protease inhibitors: enhancing potency by optimising the internal hydrogen bond network. PLoS One 6, e19302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneider E. L., Lee M. S., Baharuddin A., Goetz D. H., Farady C. J., Ward M., Wang C. I., and Craik C. S. (2012) A reverse binding motif that contributes to specific protease inhibition by antibodies. J. Mol. Biol. 415, 699–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farady C. J., Egea P. F., Schneider E. L., Darragh M. R., and Craik C. S. (2008) Structure of an Fab-protease complex reveals a highly specific non-canonical mechanism of inhibition. J. Mol. Biol. 380, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu Y., Eigenbrot C., Liang W. C., Stawicki S., Shia S., Fan B., Ganesan R., Lipari M. T., and Kirchhofer D. (2007) Structural insight into distinct mechanisms of protease inhibition by antibodies. Proc. Natl. Acad. Sci. U.S.A. 104, 19784–19789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kenniston J. A., Faucette R. R., Martik D., Comeau S. R., Lindberg A. P., Kopacz K. J., Conley G. P., Chen J., Viswanathan M., Kastrapeli N., Cosic J., Mason S., DiLeo M., Abendroth J., Kuzmic P., et al. (2014) Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J. Biol. Chem. 289, 23596–23608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., and Hamers R. (1993) Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 [DOI] [PubMed] [Google Scholar]
- 23. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., and Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Desmyter A., Transue T. R., Ghahroudi M. A., Thi M. H., Poortmans F., Hamers R., Muyldermans S., and Wyns L. (1996) Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Biol. 3, 803–811 [DOI] [PubMed] [Google Scholar]
- 25. Radisky E. S., Kwan G., Karen Lu C. J., and Koshland D. E. Jr. (2004) Binding, proteolytic, and crystallographic analyses of mutations at the protease-inhibitor interface of the subtilisin BPN′/chymotrypsin inhibitor 2 complex. Biochemistry 43, 13648–13656 [DOI] [PubMed] [Google Scholar]
- 26. Radisky E. S., Lu C. J., Kwan G., and Koshland D. E. Jr. (2005) Role of the intramolecular hydrogen bond network in the inhibitory power of chymotrypsin inhibitor 2. Biochemistry 44, 6823–6830 [DOI] [PubMed] [Google Scholar]
- 27. Zakharova E., Horvath M. P., and Goldenberg D. P. (2008) Functional and structural roles of the Cys-14-Cys38 disulfide of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 382, 998–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salameh M. A., Soares A. S., Navaneetham D., Sinha D., Walsh P. N., and Radisky E. S. (2010) Determinants of affinity and proteolytic stability in interactions of Kunitz family protease inhibitors with mesotrypsin. J. Biol. Chem. 285, 36884–36896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Radisky E. S., King D. S., Kwan G., and Koshland D. E. Jr. (2003) The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry 42, 6484–6492 [DOI] [PubMed] [Google Scholar]
- 30. Bode W., and Huber R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204, 433–451 [DOI] [PubMed] [Google Scholar]
- 31. Finkenstadt W. R., and Laskowski M. Jr. (1965) Peptide bond cleavage on trypsintrypsin inhibitor complex formation. J. Biol. Chem. 240, 962–963 [PubMed] [Google Scholar]
- 32. Bode W., and Huber R. (2000) Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta 1477, 241–252 [DOI] [PubMed] [Google Scholar]
- 33. Longstaff C., Campbell A. F., and Fersht A. R. (1990) Recombinant chymotrypsin inhibitor 2: expression, kinetic analysis of inhibition with α-chymotrypsin and wild-type and mutant subtilisin BPN′, and protein engineering to investigate inhibitory specificity and mechanism. Biochemistry 29, 7339–7347 [DOI] [PubMed] [Google Scholar]
- 34. Radisky E. S., and Koshland D. E. Jr. (2002) A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 10316–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Finkenstadt W. R., and Laskowski M. Jr. (1967) Resynthesis by trypsin of the cleaved peptide bond in modified soybean trypsin inhibitor. J. Biol. Chem. 242, 771–773 [PubMed] [Google Scholar]
- 36. Cohen I., Kayode O., Hockla A., Sankaran B., Radisky D. C., Radisky E. S., and Papo N. (2016) Combinatorial protein engineering of proteolytically resistant mesotrypsin inhibitors as candidates for cancer therapy. Biochem. J. 473, 1329–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andreasen P. A., Kjøller L., Christensen L., and Duffy M. J. (1997) The urokinase-type plasminogen activator system in cancer metastasis: a review. Int. J. Cancer 72, 1–22 [DOI] [PubMed] [Google Scholar]
- 38. Vincke C., Gutiérrez C., Wernery U., Devoogdt N., Hassanzadeh-Ghassabeh G., and Muyldermans S. (2012) Generation of single domain antibody fragments derived from camelids and generation of manifold constructs. Methods Mol. Biol. 907, 145–176 [DOI] [PubMed] [Google Scholar]
- 39. Hendrickx M. L., DE Winter A., Buelens K., Compernolle G., Hassanzadeh-Ghassabeh G., Muyldermans S., Gils A., and Declerck P. J. (2011) TAFIa inhibiting nanobodies as profibrinolytic tools and discovery of a new TAFIa conformation. J. Thromb. Haemost. 9, 2268–2277 [DOI] [PubMed] [Google Scholar]
- 40. Zhao G., Yuan C., Wind T., Huang Z., Andreasen P. A., and Huang M. (2007) Structural basis of specificity of a peptidyl urokinase inhibitor, upain-1. J. Struct. Biol. 160, 1–10 [DOI] [PubMed] [Google Scholar]
- 41. Petersen L. C., Brender J., and Suenson E. (1985) Zymogen-activation kinetics. Modulatory effects of trans-4-(aminomethyl)cyclohexane-1-carboxylic acid and poly-d-lysine on plasminogen activation. Biochem. J. 225, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kromann-Hansen T., Lund I. K., Liu Z., Andreasen P. A., Høyer-Hansen G., and Sørensen H. P. (2013) Allosteric inactivation of a trypsin-like serine protease by an antibody binding to the 37- and 70-loops. Biochemistry 52, 7114–7126 [DOI] [PubMed] [Google Scholar]
- 43. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tarr A. W., Lafaye P., Meredith L., Damier-Piolle L., Urbanowicz R. A., Meola A., Jestin J. L., Brown R. J., McKeating J. A., Rey F. A., Ball J. K., and Krey T. (2013) An alpaca nanobody inhibits hepatitis C virus entry and cell-to-cell transmission. Hepatology 58, 932–939 [DOI] [PubMed] [Google Scholar]
- 46. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.7.4.0, DeLano Scientific LLc, Palo Alto, CA [Google Scholar]
- 48. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 51. Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 52. Petersen H. H., Hansen M., Schousboe S. L., and Andreasen P. A. (2001) Localization of epitopes for monoclonal antibodies to urokinase-type plasminogen activator: relationship between epitope localization and effects of antibodies on molecular interactions of the enzyme. Eur. J. Biochem. 268, 4430–4439 [DOI] [PubMed] [Google Scholar]