

Abstract
Improving potencies through concomitant blockage of multiple epitopes and avid binding by fusing multiple (different) monovalent Nanobody building blocks via linker sequences into one multivalent polypeptide chain is an elegant alternative to affinity maturation. We explored a large and random formatting library of bivalent (combinations of two identical) and biparatopic (combinations of two different) Nanobodies for functional blockade of Pseudomonas aeruginosa PcrV. PcrV is an essential part of the P. aeruginosa type III secretion system (T3SS), and its oligomeric nature allows for multiple complex binding and blocking options. The library screening yielded a large number of promising biparatopic lead candidates, revealing significant (and non-trivial) preferences in terms of Nanobody building block and epitope bin combinations and orientations. Excellent potencies were confirmed upon further characterization in two different P. aeruginosa T3SS-mediated cytotoxicity assays. Three biparatopic Nanobodies were evaluated in a lethal mouse P. aeruginosa challenge pneumonia model, conferring 100% survival upon prophylactic administration and reducing lung P. aeruginosa burden by up to 2 logs. At very low doses, they protected the mice from P. aeruginosa infection-related changes in lung histology, myeloperoxidase production, and lung weight. Importantly, the most potent Nanobody still conferred protection after therapeutic administration up to 24 h post-infection. The concept of screening such formatting libraries for potency improvement is applicable to other targets and biological therapeutic platforms.
Keywords: epitope mapping; high throughput screening (HTS); Pseudomonas aeruginosa (P. aeruginosa); single-domain antibody (sdAb, Nanobody); type III secretion system (T3SS); PcrV; avidity; biparatopic; bivalent; formatting
Introduction
Nanobodies are a novel class of therapeutic proteins based on immunoglobulin single variable domains (Nanobody® is a trademark of Ablynx N.V.) (1). The immunoglobulin single variable domains may be derived from the variable domains of heavy chain-only antibodies that naturally occur in camelids (2). Compared with heterotetrameric conventional antibodies, heavy chain-only antibodies are homodimeric heavy chains lacking the CH1 domain. Many reports detail the superior solubility, monomeric behavior, and biophysical stability of Nanobodies compared with fragments derived from conventional antibodies (2).
One of the most compelling yet often overlooked features of Nanobodies is that they are easily amenable to formatting (i.e. the genetic fusion of multiple (different) monovalent Nanobody building blocks by means of linker sequences into one multivalent polypeptide chain). Formatting represents an elegant way to improve potencies by avidity and concomitant blockage of multiple epitopes without the need for cumbersome affinity maturation. For homomultimeric targets, the combination of multiple identical (or different competing) Nanobody building blocks, appropriately spaced by linkers and each targeting a single protomer, may result in increased potencies driven by the avid binding of the multivalent Nanobody, compared with its monovalent counterparts (3–8). For monomeric or heteromultimeric targets, similar potency increases can be observed with multivalent Nanobodies combining different building blocks binding to non-overlapping epitopes on the same protomer or each binding to a different protomer, respectively (6, 8–10).
Pseudomonas aeruginosa is a versatile Gram-negative bacterium, noted for its ability to cause a wide spectrum of chronic and acute infections in humans. This opportunistic pathogen is the leading cause of nosocomial Gram-negative pneumonia, especially in mechanically ventilated patients, and is highly prevalent in other respiratory, bloodstream, urinary tract, and burn wound infections (11). The pathogen's innate resistance to several antibiotics and the emergence of multi-resistant strains underscore the need for alternative treatments (12). Among its large arsenal of virulence systems, the type III secretion system (T3SS)6 is a major determinant in the pathogenesis of acute infections (13).
The T3SS comprises three main subassemblies: a basal body spanning the periplasm, an extracellular needle, and a pore-forming translocon required for effector protein translocation across the host cell plasma membrane (13). It is activated upon sensing the presence of host cells and can mediate cell death by means of two distinct mechanisms. The best studied mechanism is through the secretion and translocation of exotoxins into the host cell cytoplasm. Four different P. aeruginosa exotoxins are described (ExoS, ExoY, ExoT, and ExoU), which act in different ways on the host cell to ultimately cause cell death. Different P. aeruginosa strains secrete different combinations of exotoxins, with strains expressing ExoU exhibiting the greatest virulence (13). Alternatively, in an exotoxin-independent way, the T3SS can mediate macrophage and neutrophil cytotoxicity, resulting in lung injury and inflammatory responses, a process called oncosis (14–16).
PcrV forms an oligomeric (most likely pentameric) ring at the top end of the needle structure (17–21) and is crucial for the correct functioning of the T3SS. Knock-out of the pcrV gene leads to aberrant leakage of the exotoxins to the bacterial growth medium and to impeded exotoxin translocation into the host cell cytoplasm, resulting in nearly complete abolition of in vitro and in vivo cytotoxicity (16, 22–26). More so, PcrV is essential for the correct assembly of the PopB/PopD T3SS translocon into the host cell membrane (23, 27).
Passive and active immunization studies confirm that PcrV is an attractive target for protection against T3SS-mediated (lethal) P. aeruginosa infection, irrespective of the strain's exotoxin genotype, in a range of P. aeruginosa infection models (21, 22, 28–32). Anti-PcrV antibodies and derived Fab fragments have been shown to block PopB/PopD translocon assembly in the host cell membrane and subsequent exotoxin translocation as well as T3SS-dependent oncosis (27, 29, 30).
Here we describe the identification of large numbers of highly potent PcrV Nanobodies by high throughput formatting for potency improvement. A large biparatopic/bivalent Nanobody library covering four different epitope bins was explored to identify the most potent/efficacious combinations functionally blocking the oligomeric PcrV ring of the P. aeruginosa T3SS needle. Many different avid binding options can be envisaged on the oligomeric target; however, we found that (i) combinations of members of different epitope bins were preferred over combinations of the same epitope bin, (ii) combinations of the most potent monovalent building blocks did not necessarily yield superior biparatopic Nanobodies, and (iii) weakly potent monovalent building blocks made excellent partners in biparatopic constructs. Several of these biparatopic Nanobodies showed encouraging therapeutic potential in an acute and lethal P. aeruginosa-induced mouse pneumonia model.
Experimental Procedures
Immunizations, Phage Display Selection, and Production of Nanobodies
Llamas were immunized essentially as described (10) with recombinant PcrV (rPcrV) protein. Immune blood samples were taken, and total RNA was extracted. Nanobody-encoding cDNA was amplified by RT-PCR and ligated into a phagemid vector to generate Nanobody phage libraries (size ∼107) where the phage particles display Nanobodies with C-terminal FLAG3 and HIS6 tags fused to pIII, as described previously (33).
Phage display selections were performed against immobilized rPcrV protein (10 or 15 μg/ml) or in solution against different concentrations of biotinylated rPcrV (50 to 0.005 nm). After 2–3 rounds of selection, the resulting phage outputs were transfected into Escherichia coli strain TG1, and individual colonies were grown in 96-deep-well plates. The expression of monoclonal Nanobodies was induced by isopropyl 1-thio-β-d-galactopyranoside. The periplasmic extracts containing the Nanobodies were prepared by freeze-thawing the bacterial pellets in PBS and subsequent centrifugation to remove cell debris.
Productions and purifications of monovalent, bivalent, and biparatopic Nanobodies were performed essentially as described elsewhere (10). The flexible Gly-Ser linkers used to combine the monovalent building blocks in the bivalent and biparatopic constructs are referred to as 40GS ((GGGGS)8).
Cytotoxicity Assays
A P. aeruginosa exotoxin-dependent cytotoxicity assay targeting P3-X63-Ag8 mouse myeloma cells (P3X63) was set up essentially as described before (30). 2 × 105 P3X63 cells/well were seeded in 96-well plates. Diluted periplasmic extracts containing Nanobodies or dilution series of purified Nanobodies were preincubated with P. aeruginosa strain PA103 (expressing both ExoU and ExoT exotoxins), grown in LB medium supplemented with 5 mm EGTA (34). Next, the mixtures of Nanobodies and bacteria were added to the P3X63 cells at an average multiplicity of infection of 8 bacteria to 1 myeloma cell (range 6:1 to 10:1). After an incubation step of 3 h at 37 °C, P3X63 cells were stained with propidium iodide (Sigma-Aldrich) and fixated with 2% formaldehyde (Sigma-Aldrich). P. aeruginosa-mediated cell death was quantified through the uptake of the propidium iodide by dead cells using a FACS Array (BD Biosciences) and FCS Express (De Novo Software, Glendale, CA). Values were normalized to calculate the percentage inhibition of cell death according to the formula, 100 × (Y − X)/(Y − Z), where X represents the percentage of dead cells of the evaluated data point, Y is the average percentage of dead cells of the PA103-treated wells incubated with irrelevant control Nanobody, and Z is the average percentage of dead cells of the untreated wells, incubated with irrelevant control Nanobody.
Similarly, 8 × 104 A549 (ATCC CCL-185) cells/well were added to a 96-well E-plate (Acea Biosciences 05232368001) and placed in the xCELLigence RTCA W380 (Acea Biosciences) for 6–8 h at 37 °C. P. aeruginosa strain PA103, grown as described above, was preincubated for 1 h at 37 °C with dilution series of purified anti-PcrV Nanobodies and then added to the A549 cells at an average multiplicity of infection of 8 (range 6:1 to 10:1) and incubated for another 24 h at 37 °C. Data were analyzed at the time points where the reduction in cell index of the treated wells was between 75 and 90% of the cell index window. Data were normalized to the time point of inoculum addition.
Generation of rPcrV and PcrV/LcrV Chimeras
The genes encoding the full-length PcrV protein of P. aeruginosa reference strain PAO1 (amino acid residues 1–294) and seven PcrV/LcrV chimeric molecules were cloned into a pUC119-derived expression vector in frame with a C-terminal His6 tag. After transformation in E. coli (TG-1), expression cultures were grown, induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside, and allowed to continue for 4 h at 37 °C. Cells pellets were lysed by sonication, and cytosolic fractions were isolated by centrifugation. The recombinant proteins were purified from the crude extracts via immobilized metal affinity chromatography on HisTrap FF crude columns and buffer-exchanged using HiPrep 26/10 columns into Dulbecco's PBS, followed by ion exchange on a Source15Q column and finally gel filtration over a Superdex75 10/300 GL column (GE Healthcare). The purity and homogeneity of the expressed protein were confirmed by SDS-PAGE and analytical size exclusion chromatography.
Modeling
The homology model of PcrV was generated with Discovery Studio 3.0 (Accelrys Software Inc.). The sequence of PcrV was taken from Uniprot (ID O30527), and the structure of Yersinia pestis V-antigen was used as a structural template (Protein Data Bank entry 1R6F) (35). The sequence identity between the two proteins is 36%. The amino acid sequence of PcrV was aligned to the sequence of the structural template using BLOSUM62 matrix and further manually modified in order not to break secondary structural elements. 20 different models were generated using the standard “Build homology model” tool of Discovery Studio. The best model was chosen based on total energy value, visual inspection, and the Verify Protein MODELER tool and further minimized with 1000 steps of steepest descent with a root mean square gradient tolerance of 3, followed by conjugate gradient minimization. Protein stereochemistry evaluation was performed by several tools (Ramachandran plot, bumps monitor Verify Protein Profiles-3D tool) implemented in Discovery Studio.
The model of the P. aeruginosa T3SS needle tip was generated starting from a PcrV homology model, the crystal structure of the truncated C terminus of the Shigella flexneri needle subunit (Protein Data Bank entry 2CA5), and the 16 Å electron microscopy density map of the complete model assembly (Protein Data Bank entry 2V6L) for LcrV protein assembly in the formation of the Yersinia needle tip (36). Superimposition was carried out with Molsoft ICM (Molsoft LLC), considering the backbone atoms of the C-terminal helix residues Lys258–Asp289 of the PcrV model and the residues from Gln45 to Asp75 of the S. flexneri needle subunit molecule.
Seven different chimeric proteins were designed by replacing solvent-exposed portions of PcrV in the context of the pentameric model with corresponding counterparts of LcrV as follows: Chimera 1 (residues 1–26 of PcrV replaced by residues 1–47 of LcrV), Chimera 2 (57–91 replaced by 86–112), Chimera 3 (106–125 replaced by 127–147), Chimera 4 (156–177 replaced by 178–199), Chimera 5 (178–195 replaced by 200–216), Chimera 6 (194–211 replaced by 215–240), and Chimera 7 (209–249 by 238–278). Particular attention was paid to secondary structure elements to avoid disturbance of the protein structure integrity. Secondary structure and total percentage of solvent accessibility data were obtained from the crystal structure of LcrV (Protein Data Bank entry 1R6F) and the homology model of PcrV. The total accessible surface area was calculated with Discovery Studio version 3.0 using 240 grid points/atom and a probe radius of 1.4 Å.
Generation of Anti-PcrV Fab
The gene segments encoding VL and VH from Fab 1A8 (WO 2009/073631 (30)) were cloned into an in-house human IgG1/κ Fab expression vector that encodes a C-terminal HA and His6 tag in frame with the HC coding sequence. The Fab fragments were expressed in E. coli and purified under native, non-reducing conditions via immobilized metal affinity chromatography on a HisTrap FF crude 1-ml column (GE Healthcare), followed by affinity chromatography for the human Fabκ light chain (CaptureSelect LC-κ, Thermo Fisher Scientific) and size exclusion chromatography on a Superdex75 10/300 GL column (GE Healthcare) or desalting via Zeba spin columns (Pierce).
Binding ELISA
Periplasmic extracts were tested for binding to rPcrV by ELISA. 10 nm biotinylated PcrV protein was captured on NeutrAvidin (2 μg/ml)-coated 96-well MaxiSorp plates (Nunc, Wiesbaden, Germany). Wells were blocked with a casein solution (1%). After addition of a 10-fold dilution of the periplasmic extracts, Nanobody binding was detected using a mouse anti-FLAG-HRP conjugate (Sigma) and a subsequent enzymatic reaction in the presence of the substrate 3,3′,5,5′-tetramethylbenzidine (SDT, Brussels, Belgium). Clones showing ELISA signals >2-fold above background were considered to encode positive PcrV-binding Nanobodies.
Off-rate Determinations
Off-rate screening on periplasmic extracts containing monovalent Nanobodies was done with surface plasmon resonance on a ProteOn XPR36 instrument (Bio-Rad). To this end, rPcrV protein was covalently bound to a GLC ProteOn Sensor chip via amine coupling using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and N-hydroxysuccinimide. Periplasmic extracts containing anti-PcrV Nanobodies were diluted 10-fold and injected at a flow rate of 45 μl/min for 2 min for binding to the immobilized rPcrV. Between sample injections, the surface was regenerated with ProteOn phosphoric acid solution, 0.85%. Off-rates were determined by fitting a 1:1 interaction model (Langmuir model) to the individual dissociation curves. Off-rate analysis on purified monovalent and biparatopic Nanobodies was done with surface plasmon resonance on a Biacore T100 instrument. rPcrV was immobilized on a CM5 chip via amine coupling using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and N-hydroxysuccinimide. Nanobodies were injected for 2 min at three different concentrations (5, 20, and 50 nm) and allowed to dissociate for 50 min at a flow rate of 45 μl/min. Between sample injections, the surfaces were regenerated with 10 mm glycine, pH 1.5, and 100 mm HCl. HBS-N (Hepes buffer, pH 7.4) was used as running buffer. Off-rates were determined by fitting a 1:1 interaction model (Langmuir model) to the individual dissociation curves.
Epitope Binning Experiments
Different ELISAs were performed. In a first experiment, 1 μg/ml rPcrV protein was coated in a 96-well MaxiSorp plate (Nunc). Dilution series (concentration range 100 nm to 6.4 pm) of the purified Nanobodies in PBS buffer containing 0.1% casein and 0.05% Tween 20 (Sigma) were incubated in the presence of 1 nm Fab1A8. Residual binding of Fab1A8 to rPcrV was detected using mouse anti-HA IgG (Zymed Laboratories Inc., South San Francisco, CA) followed by HRP-conjugated rabbit anti-mouse IgG (Dako, Glostrup, Denmark) and a subsequent enzymatic reaction in the presence of the substrate 3,3′,5,5′-tetramethylbenzidine (SDT).
In a second experiment, 2 nm biotinylated rPcrV was captured on a 96-well MaxiSorp plate (Nunc) coated with 2 μg/ml NeutrAvidin. A mixture of periplasmic extracts and phage particles of different Nanobodies was incubated. Residual binding of the phages to biotinylated rPcrV was detected using a monoclonal anti-M13-HRP conjugate (GE Healthcare) followed by incubation of the substrate 3,3′,5,5′-tetramethylbenzidine (SDT).
Binding to the seven PcrV/LcrV chimeric molecules was tested in ELISA. Nanobodies at 3 μg/ml and Fab1A8 at 9 μg/ml were coated overnight at 4 °C in a 96-well MaxiSorp plate (Nunc). Wells were blocked with a casein solution (1% in PBS). PcrV/LcrV chimeras were applied (1:10), and binding was detected using a biotinylated mouse anti-Myc antibody (Serotec) followed by HRP-conjugated extravidin (Sigma) and a subsequent enzymatic reaction in the presence of the substrate 3,3′,5,5′-tetramethylbenzidine (SDT).
Other epitope binning experiments were performed with a Biacore T100 instrument. rPcrV was immobilized on a CM5 chip using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and N-hydroxysuccinimide chemistry. The surface was saturated with injection of a first Nanobody for 2 min at a flow rate of 45 μl/min. In a second injection, a mixture of the first Nanobody at a saturating concentration and a test Nanobody (500 nm) were evaluated for the presence (different epitope) or absence (same epitope) of additional binding. The surface was regenerated with 0.85% H3PO4 buffer.
Animals
Female C57BL/6 mice at 8–10 weeks of age were obtained from Janvier (Le-Genest-Saint-Isle, France) and were acclimatized for 1 week in the animal unit before the start of the study. They were randomized according to their body weights in cages of five mice with free access to water and regular chow. Manipulations of animals were conducted carefully to reduce stress to a minimum. All experiments were performed in compliance with the guidelines for laboratory animals of the French Ministry of Agriculture (law 87-848).
One day before infection, P. aeruginosa bacteria (PA2310.55 serotype O11) were grown at 37 °C overnight with shaking at 150 rpm in liquid (brain-heart infusion medium supplemented with 10 mm nitrilotriacetic acid (Sigma)). On the day of infection, the preculture was diluted and grown in liquid brain-heart infusion medium to an A600 nm of 0.4 (equivalent to 2 × 108 cfu/ml) and further diluted to 5 × 107 cfu/ml. 20 μl of this solution was mixed either with 20 μl of Nanobody for the prophylactic setting or with 20 μl of saline for the therapeutic setting (106 cfu in 40 μl).
Mice were anesthetized with a low dose of intravenous ketamine/xylazine (1.25 and 0.5 mg/ml, respectively) and infected intranasally in an upright position with 40 μl of the inoculum using an ultrafine pipette tip. Control noninfected mice received 40 μl of saline intranasally. After infection, the leftover inoculum was plated in brain-heart infusion-agar to verify the cfu. Mice were sacrificed at 24 h post-inoculation to assess lung bacterial burden, myeloperoxidase activity, relative lung weight, and histology of the lung. The remaining mice were followed up until 4 or 5 days, and survival was recorded three times daily.
Lung weights were recorded and then expressed as a percentage of the initial body weight at sacrifice. The right lung and the other lobes (except for the big lobe, which was used for histopathological purposes) of the left lung were weighed and homogenized in 1.5 ml of isotonic sterile saline solution using the sterile disposable homogenization system Dispomix (Medic Tools AG, 6300-Zug, Switzerland). 10-Fold serial dilutions of lung homogenates, made in isotonic saline solution, were plated on brain-heart infusion-agar plates to determine the number of viable bacteria in the lung.
Lung homogenates were centrifuged (1000 × g for 10 min at 4 °C), and the supernatant was discarded. The remaining pellet was then resuspended in 1 ml of PBS containing 0.5% HTAB and 5 mm EDTA. Following a second centrifugation, 100 μl of the supernatants were placed in test tubes with 200 μl of PBS-HTAB-EDTA, 1 ml of Hanks' balanced salt solution, 100 μl of o-dianisidine dihydrochloride (1.25 mg/ml), and 100 μl of H2O2 (0.05%). After 15 min of incubation at 37 °C in an agitator, the reaction was stopped with 100 μl of NaN3 1%. The myeloperoxidase activity was determined as absorbance at 460 nm against medium.
The big lobe of the left lung was fixed in 4% buffered formaldehyde, embedded in paraffin, cut into 3-μm-thick sections, and stained with hematoxylin and eosin. The slides were scored blindly by two independent observers for pathological changes. Edema, hemorrhage, and cellular recruitment were graded using a semiquantitative scoring system with increasing severity from 0 to 5 with some modifications as described previously (37). Hemorrhage was scored as follows: 0 = normal histomorphology; 1 = minor hemorrhage; 2 = minor hemorrhage localized in perivascular spaces; 3 = moderate hemorrhage, including peribronchiolar spaces; 4–5 = extensive hemorrhage, including lung parenchyma. Edema was graded from 0 to 5 according to the ratio between blood vessel and perivascular space diameter. Cellular recruitment was graded from 0 to 5 according to its intensity as follows: 0 = normal cell infiltration; 1 = minor cell infiltration; 2 = moderate cell infiltration; 3 = moderate cell infiltration with minor alveolar hemorrhage; 4 = massive cell infiltration; 5 = massive cell infiltration with increased hemorrhage. The histological score index was the sum of these three separate scores (maximum total score of 15).
Data Analysis
Library diversity estimations were calculated with the GLUE algorithm (38). Significance of changes in proportions of building blocks and epitope bins among the hits of the biparatopic/bivalent library screening campaign compared with the library design was assessed by χ2 tests (GraphPad). Significance of potency differences between Nanobodies and Fab1A8 was evaluated by two-way analysis of variance followed by Dunnett's multiple-comparison test (GraphPad).
Statistical evaluation of differences between experimental groups for the lung outcomes was conducted by a linear model, including the categorical variable “group” as fixed effect only. Assumptions underlying a linear model (normal distribution and homoscedastic residuals) were fulfilled by estimating a different variance within each experimental group and/or log transformation of the responses. Pairwise comparisons with the vehicle group were performed for each outcome variable and were followed by a Hommel multiple-testing correction to control the overall type I error at the prespecified significance level of 5%. Pairwise log-rank tests with the vehicle-treated group were used for survival analysis, but due to the low number of animals and events per group, it was decided to perform an exploratory analysis at a 5% significance level for survival only without correction for multiple testing. All tests were performed with the statistical software SAS version 9.4 (SAS Institute).
Results
An overview of the different steps described below is shown in Fig. 1.
FIGURE 1.

Schematic overview of the work flow detailed under “Results.” Gray ovals represent Nanobody building blocks.
Identification of Monovalent PcrV Nanobodies
Phage display selections on rPcrV, followed by screening of enriched clones as periplasmic extracts in binding ELISA (data not shown) to rPcrV and sequence analysis, resulted in the identification of 19 different anti-PcrV Nanobodies (supplemental Table S1). Periplasmic extracts of these displayed off-rates on rPcrV ranging from 5 × 10−3 to 5 × 10−5 s−1. The purified Nanobodies showed varying abilities to inhibit P. aeruginosa T3SS-mediated killing of murine myeloma P3X63 cells, with IC50 values ranging from >10−6 to 4 × 10−9 m. The panel of 19 anti-PcrV Nanobodies could thus be divided into 11 functional blockers and 8 poor blockers or non-blockers.
Nanobody versus Nanobody and Fab1A8 (30) competition experiments with purified Nanobody material revealed that they could be grouped into four distinct epitope bins (Fig. 2 and supplemental Table S1). A number of leads (3B11, 3E10, and 7C10) competed with representatives of bins 1 and 2, although 3B11, in contrast to other bin 2 competitors, did not compete with Fab1A8. Two bin 1 representatives (1E11 and 2G09, underlined in Fig. 2) only partially inhibited cell death over a complete dose-response curve (supplemental Table S1). As such, they can be distinguished from the weakly functional (3B11, 4C03, 10C05, 13F07, 14B010, and 14E10) or non-functional (7E09 and 12B02) Nanobodies (in italic type in Fig. 2) for which only incomplete or no dose-response curves were obtained in the cell death assay over the tested concentration range (detailed in supplemental Table S1).
FIGURE 2.

Epitope binning of 19 monovalent anti-PcrV Nanobodies. Nanobodies blocking each other's binding to PcrV are grouped by solid lines into four distinct epitope bins. Nanobodies competing with the binding of Fab1A8 to PcrV are grouped by a dashed line. Partially effective Nanobodies of bin 1 are underlined, and weakly functional or non-functional Nanobodies are shown in italic type (according to supplemental Table S1).
Screening of Biparatopic/Bivalent Nanobody Library Yields Large Number of Potent Lead Candidates
PcrV is assembled in an oligomeric, possibly pentameric ringlike structure on the top of the P. aeruginosa T3SS needle (17–21). A P. aeruginosa needle tip model was built by superimposing a homology model of PcrV (Fig. 3A), based on the crystal structure of its Y. pestis LcrV homolog (35), onto an S. flexneri needle subunit model (36). As observed before (36) for the LcrV needle tip model, no more than five PcrV monomers could be added to the needle tip ring structure (Fig. 3, B and C). To improve the potency of the Nanobodies in the cell death assay, formatting of the monovalent building blocks into bivalent (combinations of two identical) and biparatopic (combinations of two different) Nanobodies was envisaged. The number of binding/blocking options for bivalent or biparatopic Nanobodies covering four distinct epitopes is considerable, given the oligomeric, probably pentameric, nature of PcrV. At any time, a maximum of two identical bivalent Nanobodies could bind in an avid way (across two PcrV protomers each) together with a third identical bivalent Nanobody in a non-avid way to the pentameric ring (Fig. 3D). In contrast, up to five identical biparatopic Nanobodies could bind in an avid way to the pentameric ring, each within one (Fig. 3E) or across two PcrV protomers (Fig. 3F).
FIGURE 3.
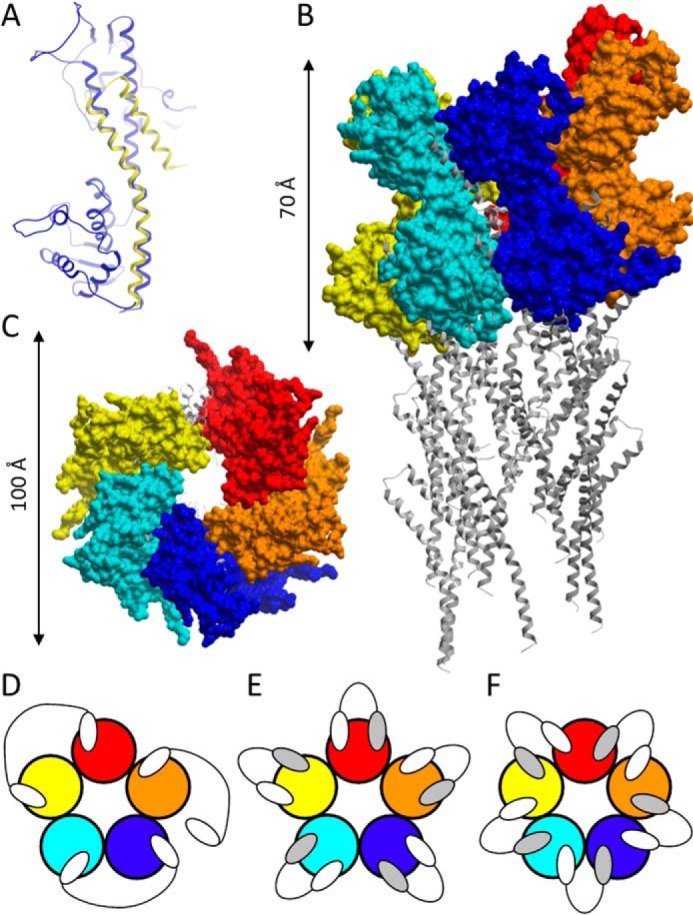
PcrV modeling and Nanobody binding modes. A, homology model of PcrV (blue), superimposed to the C-terminal helices of the S. flexneri needle subunit (yellow). Shown are side (B) and top (C) views (not drawn to the same scale) of the pentameric PcrV model complex (surface representation) on the tip of a T3SS needle (ribbon representation). Shown are theoretical binding modes of bivalent (D) and biparatopic (E and F) Nanobodies to a pentameric PcrV ring (not drawn to the same scale). Individual PcrV protomers are shown in different colors. White and gray ovals, Nanobody building blocks binding to distinct epitopes on a PcrV monomer.
Rather than evaluating a limited number of possible combinations, we opted to explore the full range of possibilities by screening a biparatopic/bivalent Nanobody library covering all 361 (19 × 19) combinations of the monovalent panel. The 19 building blocks were randomly combined with each other in an E. coli expression vector, separated by a long 40GS linker, allowing for maximal conformational flexibility. 80 randomly selected library clones were sequenced as a quality control, and results revealed that the library was of high quality; 65 of 80 (81%) of the clones had intact and correct biparatopic/bivalent sequences, representing 63 of 361 possible combinations. All 19 building blocks were captured in both N- and C-terminal positions with comparable frequencies matching the library design (Fig. 4 and supplemental Table S2). 960 clones were then produced as crude periplasmic extracts, representing an adjusted (960 × 81% = 778 clones) expected completeness of 88.4% for a library of 361 equiprobable variants as calculated by GLUE (38). Because the concentration of biparatopic/bivalent Nanobody in these crude periplasmic extracts is unknown, a pilot experiment (not shown) was performed to determine the optimal dilution factor allowing for a maximal spread of the data points. The crude periplasmic extracts were thus diluted 1:150 and screened in singlicate in the P3X63 cell death assay. The screening results of the 65 most potent clones (inhibiting P. aeruginosa-mediated cell death ≥55%) are summarized in Fig. 4 and supplemental Table S2. Sequence analysis revealed that they represent 50 unique combinations for which the following observations could be made. Combinations of building blocks from different epitope bins were preferred over combinations of the same bin (p = 0.001), in line with the prediction of more efficient blocking by biparatopic over bivalent Nanobodies (Fig. 3, D–F). Weakly blocking or non-blocking monovalents could be excellent biparatopic partners to a range of other building blocks, as illustrated by the significant enrichment of 13F07 (p = 0.0001). Significant preferences could be seen between building blocks of the same bin with comparable potencies (supplemental Table S1) (e.g. enrichment of 1E11 (p = 0.0012) compared with 2G09 (p = 0.3027) or enrichment of 7C10 (p = 0.0036) compared with 3E10 (p = 0.8019)). As a group, bin 1 members were less preferred (p = 0.0063), particularly in the N-terminal position (p = 0.0001). Bin 2 representatives were significantly (p < 0.0001) enriched in the most potent combinations, both in the N- and C-terminal positions (p = 0.0125 and 0.0048, respectively). Bin 3 members displayed a significant preference for the N-terminal position (p = 0.0125). Building block 12B02 (bin 4) was not represented, and neither were 3B11 (competing with bins 1 and 2 but not Fab1A8) and 4C03 (bin 1) (p = 0.0069).
FIGURE 4.

Screening of biparatopic/bivalent Nanobody library in the P3X63 cell death assay. Shown are the inhibition percentages of P. aeruginosa-mediated cell death of the 65 most potent clones (of 960 clones screened) in a 1:150 dilution of crude periplasmic extract. Inhibition percentages of P. aeruginosa-mediated cell death are expressed relative to the assay window spanned by the condition in the absence of P. aeruginosa (no P. aeruginosa-mediated cell death) and the condition in the presence of P. aeruginosa without blocking Nanobody (no inhibition of P. aeruginosa-mediated cell death). Values boxed in white, light gray, and dark gray are means of one, two, or three clones, respectively. The building blocks of the biparatopic/bivalent Nanobodies are grouped per epitope bin (according to Fig. 2). freq hits, proportion of a given building block or epitope bin across all 65 hits. freq lib QC, the proportion of a given building block or epitope bin across a random quality control sample of 65 library clones. Statistics are detailed in supplemental Table S2.
With unknown levels of biparatopic/bivalent Nanobody present in the crude periplasmic extracts, relative expression differences between library clones may influence the outcome of the screening. To this end, a comparison of the concentrations of 10 representative Nanobodies in the crude periplasmic extracts was performed (Fig. 5). At most, a 2.1-fold difference in concentration was observed (13F07-14E10 versus 1E11-5H01). Although this does not exclude the possibility of larger expression differences for other library clones, the impact of this factor on the outcome of the screening appears to be minor.
FIGURE 5.

Expression comparison of selected biparatopic Nanobody library clones. Shown is a Coomassie-stained non-reduced SDS-polyacrylamide gel loaded per lane with 10 μl of undiluted crude periplasmic extract from 10 different library clones in duplicate (each representing an independent expression culture started from different single colony). Biparatopic Nanobody bands with molecular masses ranging from 31.5 to 33.3 kDa are marked by an asterisk. Quantification of the Nanobody bands (mean ± S.D. of the duplicates, expressed in ng) was done relative to the 100-ng control sample in the first lane, using Quantity One 4.6.1 software (Bio-Rad). Protein standard molecular masses are shown on the right in kDa.
Epitope Mapping of Functional Nanobodies
A chimeric molecule-based strategy was used to map the epitopes of selected purified monovalent Nanobodies representing the three functional bins. To this end, discrete surface-exposed parts of PcrV in the context of the pentameric model were replaced by their Y. pestis LcrV counterparts (detailed under “Experimental Procedures”). Three major binding patterns of the Nanobodies to the seven PcrV/LcrV chimeras could be distinguished (Fig. 6A), nicely correlating with the above described epitope binning experiments (Fig. 2). The epitope of bin 1 is covered by amino acids 156–177 and 194–211 and is situated in the center on the top side of the oligomeric ring (Fig. 6B). The bin 2 epitope is situated on the outer rim of the top side of the oligomeric ring and is encompassed by the 209–249 region (Fig. 6C). The close proximity of the bin 1 and 2 epitopes is in line with the existence of Nanobodies (3B11, 3E10, and 7C10) that compete with members of both bins (Fig. 2). Finally, the epitope of bin 3 resides in the 57–91 region, situated at the basolateral side of the pentameric ring (Fig. 6D). In contrast to most of the bin 1 and bin 2 Nanobodies, the two representatives of bin 3 are not active or at best only marginally active as monovalents in the P3X63 cell death assay (supplemental Table S1). This suggests that efficacious blocking of P. aeruginosa T3SS-mediated toxicity happens most efficiently through the top and not the basolateral side of PcrV. On the other hand, basolateral binders, such as 13F07, were identified as good partners in biparatopic constructs.
FIGURE 6.

Epitope mapping of selected functional monovalent Nanobodies. A, binding ELISA signals of Nanobodies and Fab1A8 to full-length rPcrV and the seven PcrV/LcrV chimeras (see “Experimental Procedures” for detailed information on the chimeras). Bin 1 representatives are depicted in shades of red, the bin 2 representative in blue, and bin 3 representatives in shades of green. The surface-exposed epitopes of bin 1 (B), bin 2 (C), and bin 3 (D) are highlighted in red, blue, and green, respectively, on one PcrV monomer (white surface representation) in the context of the pentameric model, with the two adjacent PcrV monomers shown in a ribbon representation. The exposed epitopes on a given PcrV monomer in the pentameric model are shown in side view (top panels) and top view (bottom panels).
In Vitro Characterization of Biparatopic PcrV Nanobodies
Because the library screening revealed a significant preference for combinations of different bins over combinations of the same bin, we decided to fully focus further characterization efforts on biparatopic formats. Ten purified biparatopic Nanobodies (a representative cross-section of the screening hits) were repeatedly assayed in a dose-response analysis in both P3X63 and A549 cell death assays (Table 1). They displayed excellent potencies in both assays, many of them significantly outperforming Fab1A8. All biparatopic Nanobodies were fully efficacious in the P3X63 assay (Table 1), which at first glance is at odds with the cell death inhibition percentages of these combinations ranging from 55 to 85% in the screening assay (Fig. 4). Most likely, the 1:150 dilution of the crude periplasmic extracts represented a Nanobody concentration <IC100, resulting in less than maximal inhibition of P. aeruginosa-mediated cell death (Fig. 4), whereas in the dose-response analysis, the maximal inhibitory concentrations were reached (Table 1 and Fig. 7A).
TABLE 1.
In vitro characterization of purified biparatopic Nanobodies in A549 and P3X63 cell death assays
| Nanobodya | P3X63 |
A549 |
||
|---|---|---|---|---|
| pIC50 (n)b | Efficacyc | pIC50 (n)b | Efficacyc | |
| m | % | m | % | |
| 1E11-5H01*** | 10.0 ± 0.3 (5) | 96 ± 3 | 8.8 ± 0.3 (5) | 91 ± 2 |
| 2G09-5H01ns | 8.3 ± 0.2 (2) | 91 ± 6 | 7.8 ± 0.1 (3) | 87 ± 0 |
| 5H01-11B09* | 10.4 ± 0.2 (3) | 104 ± 7 | 8.4 ± 0.5 (5) | 85 ± 1 |
| 6B05-1E11ns | 9.4 ± 0.2 (3) | 97 ± 2 | 7.7 ± 0.3 (6) | 66 ± 8 |
| 7C10-14E10ns | 9.3 ± 0.1 (2) | 98 ± 2 | 8.1 ± 0.1 (5) | 87 ± 1 |
| 13F07-2B02ns | 9.7 ± 0.3 (3) | 93 ± 3 | 7.7 ± 0.0 (3) | 74 ± 2 |
| 13F07-2B10** | 9.8 ± 0.2 (4) | 96 ± 1 | 8.9 ± 0.4 (5) | 83 ± 3 |
| 13F07-6B05** | 9.8 ± 0.2 (4) | 98 ± 2 | 8.9 ± 0.4 (4) | 79 ± 6 |
| 13F07-5H01**** | 9.8 ± 0.1 (5) | 96 ± 2 | 9.3 ± 0.2 (5) | 84 ± 3 |
| 13F07-14E10* | 9.6 ± 0.1 (5) | 96 ± 2 | 8.5 ± 0.2 (5) | 82 ± 3 |
| Fab1A8 | 8.9 ± 0.1 (10) | 96 ± 2 | 7.9 ± 0.2 (10) | 92 ± 2 |
a Data analysis (two-way analysis of variance) revealed significance (p < 0.0001) for the factors “assay type” and “Nanobody” but not for interaction between factors. Significance in potency differences of Nanobodies compared with Fab1A8 was assessed using Dunnett's multiple-comparison test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, not significant).
b Mean ± S.E. pIC50 is shown (number of independent data points in parentheses).
c Mean ± S.E. efficacy (percentage inhibition of P. aeruginosa-mediated cell death) is expressed relative to the assay window spanned by the condition in absence of P. aeruginosa (no P. aeruginosa-mediated cell death) and the condition in the presence of P. aeruginosa without blocking compound (Nanobody or Fab) (no inhibition of P. aeruginosa-mediated cell death).
FIGURE 7.
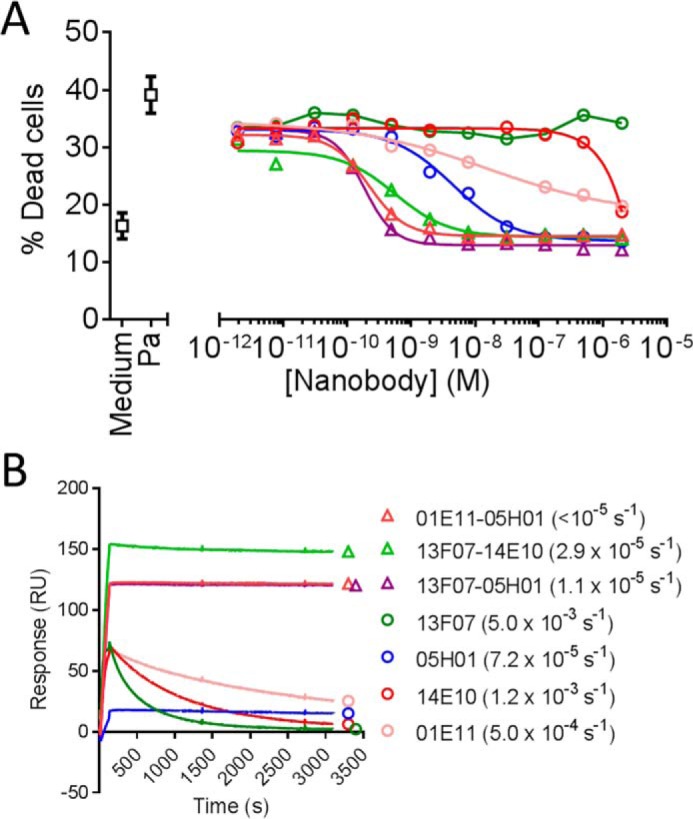
Comparison of selected biparatopic Nanobodies to their monovalent counterparts. A, inhibition of P. aeruginosa-mediated cell death of P3X63 cells by dilution series of purified Nanobodies. Data shown are of one representative experiment (see Table 1 for a summary of all experiments). Pa, top of the assay window in the presence of P. aeruginosa without blocking Nanobody (no inhibition of P. aeruginosa-mediated cell death). Medium, bottom of the assay window in the absence of P. aeruginosa (no P. aeruginosa-mediated cell death). B, surface plasmon resonance sensorgrams depicting binding of purified Nanobodies (5 nm) to rPcrV immobilized on a CM5 chip (see “Experimental Procedures” for more experimental details). Global koff values from three Nanobody concentrations tested (5, 20, and 50 nm) calculated with the Langmuir model are given in parentheses. The koff value of 1E11-5H01 was below the lower detection limit of the Biacore T100 instrument (<10−5 s−1).
Biparatopic combinations with 1E11 or 2G09 were 100% efficacious in the P3X63 assay, although as monovalents, these building blocks inhibited P. aeruginosa-mediated cell death only partially (Table 1, supplemental Table S1, and Fig. 7A). Dramatic potency improvements, ranging from 10- to >10,000-fold, were observed for all biparatopic Nanobodies versus their monovalent counterparts (compare Table 1 with supplemental Table S1). This is, for instance, illustrated by the highly potent biparatopic 13F07-14E10, of which the monovalent building blocks were not functional or were only marginally functional (Fig. 7A). Another observation from the library screening, a preference for 1E11 over 2G09 as a partner despite a 5-fold weaker potency of the former monovalent, was confirmed; 1E11-5H01 was >10 times more potent than 2G09-5H01 (Table 1).
The off-rates to rPcrV of a further subselection of purified biparatopic Nanobodies and their monovalent counterparts were determined by means of surface plasmon resonance (Fig. 7B). Concomitant binding of the biparatopic Nanobodies to the epitopes of bins 1 and 2 (1E11-5H01), bins 3 and 2 (13F07-5H01), or bins 3 and 1 (13F07-14E10) brought about marked improvements in off-rate; 7- to >450-fold slower koff values were observed compared with the monovalent building blocks.
The combined data of Figs. 2 and 6 suggest that members of bins 1, 2, and 3 can concomitantly bind to their non-overlapping surface-exposed epitopes on a single PcrV protomer in the context of a pentameric ring, as illustrated by Fig. 3E. Moreover, the two monovalent building blocks of a biparatopic Nanobody are connected by a highly flexible 40GS linker, which would separate them by ±144 Å when fully stretched. This brings ample conformational opportunities to the two monovalent building blocks to bind to two adjacent (or non-adjacent) PcrV protomers in the context of a pentameric ring, with a height of ±70 Å (Fig. 3B) and diameter of ±100 Å (Fig. 3C), as illustrated by Fig. 3F. Both inter- and intra-PcrV protomer binding scenarios and combinations thereof can be envisaged for biparatopic Nanobodies targeting the epitopes of bins 1 and 2 (1E11-5H01), bins 3 and 2 (13F07-5H01), or bins 3 and 1 (13F07-14E10), but different scenarios are of course possible for other biparatopic Nanobodies. Certain epitope combinations will result in a more efficient functional block of PcrV than others, reflected by the trends observed in the library screening (Fig. 4). The above mentioned potency improvements of the biparatopic Nanobodies in the cell death assays are thus probably explained by their avid binding to and concomitant blocking of two different surface-exposed epitopes on PcrV.
Biparatopic PcrV Nanobodies Potently Inhibit P. aeruginosa in Lethal Acute Pneumonia Model in Therapeutic Setting
Three biparatopic Nanobodies (13F07-5H01, 13F07-14E10, and 1E11-5H01) were evaluated in an acute and lethal P. aeruginosa lung infection mouse model (37).
In a first prophylactic study, groups of mice were treated at the time of infection with Nanobodies (10 μg/mouse) and compared with Fab1A8 as positive control (30). All Nanobodies completely protected the mice from P. aeruginosa infection-related lethality similarly to those treated with high dose Fab1A8 (Fig. 8A). The irrelevant Nanobody- and vehicle-treated animals all died within the first 48 h post-infection.
FIGURE 8.

In vivo characterization of biparatopic Nanobodies in a lethal acute P. aeruginosa-induced pneumonia model; prophylactic setting. A, survival comparison of C57BL/6 mice (7 mice/group) intranasally treated with a premix of anti-PcrV Nanobody (10 μg/mouse), anti-PcrV Fab1A8 (10 μg/mouse), irrelevant control Nanobody (IRR; 10 μg/mouse), or vehicle with 106 cfu of P. aeruginosa strain 2310.55 at the time of infection. B, survival comparison of C57BL/6 mice (7–8 mice/group) intranasally treated with a premix of anti-PcrV Nanobody (0.028 μg/mouse), anti-PcrV Fab1A8 (0.05 μg/mouse), or vehicle with 106 cfu of P. aeruginosa strain 2310.55 at the time of infection. Animals treated as in B (3–5 mice/group) were sacrificed at 24 h after P. aeruginosa challenge, after which the lung bacterial burden (C), lung weight relative to total body weight (D), lung histological score (E), and myeloperoxidase (MPO) activity (F) were determined. Each animal is represented as an open symbol, whereas the group means are shown as horizontal lines. IRR, an irrelevant control Nanobody (0.028 μg/mouse). Statistically significant differences between the experimental groups and the vehicle-treated group were determined using a log-rank test for the survival experiments or a linear model followed by a Hommel multiple testing correction (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, not significant).
In a second prophylactic study at a >350-fold lower dose of 0.028 μg/mouse (equimolar to 0.05 μg/mouse of Fab1A8), all three Nanobodies significantly provided complete (13F07-5H01) or nearly complete protection (01E11-05H01, 13F07-14E10, and Fab1A8) (Fig. 8B) compared with the vehicle-treated group. Additional similarly treated mice were included in this study and sacrificed at 24 h post-infection for assessment of lung bacterial load and lung inflammation as assessed by myeloperoxidase activity in the lung, lung weight, and histology. The prolonged survival of the infected anti-PcrV Nanobody- or Fab1A8-treated mice versus the infected vehicle- or irrelevant Nanobody-treated controls was paralleled by significant decreases in mean lung bacterial burden of up to 500-fold (Fig. 8C). In addition, lung inflammation, as measured by myeloperoxidase activity in the lung homogenates, changes in relative lung weights and histological scores, was variably reduced by all of the Nanobodies (Fig. 8, D–F). Conversely, the irrelevant control Nanobody had no effect on these parameters, indicating that the observed effects could be attributed to the anti-PcrV activity of the Nanobodies.
Next, the therapeutic potential of 13F07-5H01 was evaluated on survival. 13F07-5H01 was administered intranasally at 10 μg/mouse at different time points ranging from 3 to 24 h post-infection. When administered at 3, 8, 12, and 18 h post-infection, 13F07-5H01 significantly increased the survival rate in comparison with the vehicle-treated groups (Fig. 9). A delay in mortality was still seen when treatment occurred as late as 24 h post-infection, although at this late time point, the effect was not significant (Fig. 9B).
FIGURE 9.
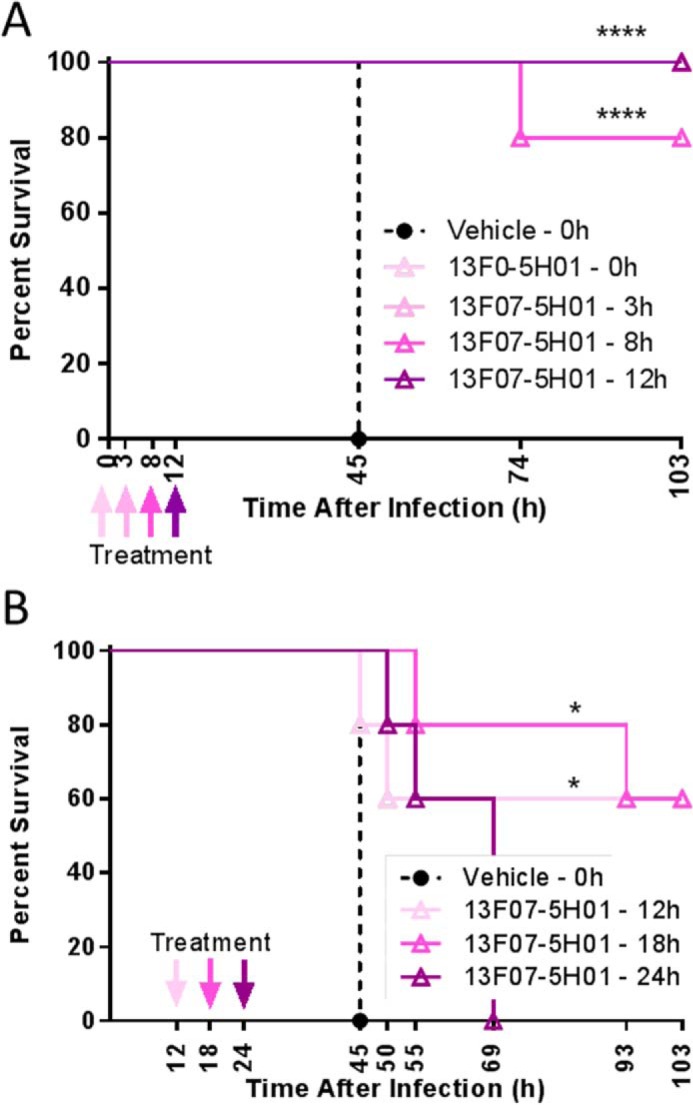
In vivo characterization of a biparatopic Nanobody in a lethal acute P. aeruginosa-induced pneumonia model; therapeutic setting. Shown is a survival comparison of C57BL/6 mice (5 mice/group) intranasally treated with vehicle or 13F07-5H01 (10 μg/mouse) at the indicated time points after infection with 106 cfu of P. aeruginosa strain 2310.55. A and B represent independent experiments. Statistically significant differences between the experimental groups and the vehicle-treated group were determined using a log-rank test (*, p < 0.05; ****, p < 0.0001).
Discussion
Large numbers of highly potent Nanobodies functionally inhibiting PcrV were identified by screening of a biparatopic/bivalent Nanobody library. The library was generated by randomly combining 19 monovalent anti-PcrV Nanobodies with each other, yielding 361 possible combinations separated by a long Gly-Ser linker, in an E. coli expression vector. The concept of potency improvement via avid binding and concomitant blockage of multiple epitopes by bivalent or biparatopic Nanobodies has already been demonstrated against a variety of targets (3–10) but to our knowledge has never been approached in depth, as was done here. The high throughput approach followed by a detailed analysis revealed clear preferences in terms of Nanobody building block and epitope bin combinations that would never have been observed using a more limited approach, typically exploring only a few predefined combinations of building blocks. Three arguments in favor of the high throughput approach can be brought forward and are demonstrated in this study: (i) a more limited approach probably would have excluded weakly functional or non-functional monovalent building blocks, which may turn out to be preferred biparatopic partners; (ii) in reverse, a more limited approach combining only the most potent monovalent building blocks would not necessarily result in the best biparatopic combinations; and (iii) an approach of randomly selecting a representative of a given epitope bin and potency would not necessarily yield the best biparatopic combinations covering this epitope. It is tempting to speculate on the underlying causes of the observed findings in combinatorial preferences, which probably include one or more of the following factors. The intrinsic potencies and efficacies of its monovalent building blocks very likely would impact on the behavior of a biparatopic combination. The same would apply to the steric context of the monovalent building blocks and the relative positions of their epitopes. Last, the expression level of the biparatopic combinations could have an impact because the library screening was performed using a fixed dilution of crude E. coli periplasmic extract containing an unknown concentration of biparatopic Nanobody. This would favor better expressing combinations, corroborated by the high fermentation expression yields obtained in yeast for the biparatopic Nanobodies that were evaluated in vivo, ranging from 1.5 to 4 g/liter.
When compared with traditional affinity maturation, the approach presented here consumes much less time and effort, has a higher likelihood (quadratic increase compared with the number of monovalent building blocks) of meeting prespecified potency criteria, and allows for concomitant blocking of multiple functionally relevant epitopes. In short, high throughput formatting for potency improvement more efficiently yields larger numbers of viable development candidates than does affinity maturation. This concept could be applied to other types of immunoglobulin-derived binding domains (e.g. scFvs) or other types of simple binding scaffolds but less efficiently so to more complex (multimeric) binders, such as Fabs and mAbs. It could even be extended to higher valencies or performed in the absence of prior knowledge regarding epitopes.
The library screening revealed a more efficient functional blockage of PcrV by Nanobodies combining two different epitope bins, compared with Nanobodies combining building blocks from the same epitope bin. Preferred were combinations of bin 2 members with either partial bin 1 blockers, bin 3 members, or blockers competing with both bins 1 and 2. The promise of the library screening was substantiated upon further characterization of a representative cross-section of biparatopic Nanobodies in two different P. aeruginosa cell death assays. A number of in vivo studies confirmed the potential of three of these Nanobodies, conferring 100% survival in a prophylactic setting. More importantly, one of the Nanobodies conferred a significant survival benefit in a therapeutic setting up to 18 h post-infection, and this trend continued up to 24 h post-infection (although not significantly). This strongly underscores the potential of these Nanobodies in comparison with other PcrV blockers for which therapeutic survival benefit in an acute P. aeruginosa pneumonia model was only demonstrated up to 4 h post-infection as monotherapy (39) or in combination with antibiotics (32).
Author Contributions
E. D. T., E. M., L. D., A. R., S. S., T. S., E. D., and B. D. designed the research. E. D. T., A. R., S. D. T., T. V., R. V. H., A. C., and E. M. performed the research. S. D. T., M. R., R. F., S. S., J. N., and W. V. d. V. contributed new reagents/analytical tools. E. D. T., A. R., E. M., L. D., T. S., and B. D. analyzed data. E. D. T., E. M., L. D., and B. D. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to the Ablynx CMC PcrV team for the production of the in vivo Nanobody batches, Ariella Van de Sompel for the statistical analysis of the in vivo data, and Tony De Fougerolles for a critical review of the manuscript. We also thank the anonymous reviewers for improving the manuscript with suggestions.
This work was supported by Ablynx N.V. All authors are or have been Ablynx employees.

This article contains supplemental Tables S1 and S2.
- T3SS
- type III secretion system
- rPcrV
- recombinant PcrV
- HTAB
- hexadecyltrimethyl ammonium bromide.
References
- 1. Kolkman J. A., and Law D. A. (2010) Nanobodies: from llamas to therapeutic proteins. Drug Discov. Today Technol. 7, e139–e146 [DOI] [PubMed] [Google Scholar]
- 2. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 [DOI] [PubMed] [Google Scholar]
- 3. Coppieters K., Dreier T., Silence K., de Haard H., Lauwereys M., Casteels P., Beirnaert E., Jonckheere H., Van de Wiele C., Staelens L., Hostens J., Revets H., Remaut E., Elewaut D., and Rottiers P. (2006) Formatted anti-tumor necrosis factor α VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 54, 1856–1866 [DOI] [PubMed] [Google Scholar]
- 4. Jähnichen S., Blanchetot C., Maussang D., Gonzalez-Pajuelo M., Chow K. Y., Bosch L., De Vrieze S., Serruys B., Ulrichts H., Vandevelde W., Saunders M., De Haard H. J., Schols D., Leurs R., Vanlandschoot P., Verrips T., and Smit M. J. (2010) CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 20565–20570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hultberg A., Temperton N. J., Rosseels V., Koenders M., Gonzalez-Pajuelo M., Schepens B., Ibañez L. I., Vanlandschoot P., Schillemans J., Saunders M., Weiss R. A., Saelens X., Melero J. A., Verrips C. T., Van Gucht S., and de Haard H. J. (2011) Llama-derived single domain antibodies to build multivalent, superpotent and broadened neutralizing anti-viral molecules. PLoS One 6, e17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Terryn S., Francart A., Lamoral S., Hultberg A., Rommelaere H., Wittelsberger A., Callewaert F., Stohr T., Meerschaert K., Ottevaere I., Stortelers C., Vanlandschoot P., Kalai M., and Van Gucht S. (2014) Protective effect of different anti-rabies virus VHH constructs against rabies disease in mice. PLoS One 9, e109367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huet H. A., Growney J. D., Johnson J. A., Li J., Bilic S., Ostrom L., Zafari M., Kowal C., Yang G., Royo A., Jensen M., Dombrecht B., Meerschaert K. R. A., Kolkman J. A., Cromie K. D., et al. (2014) Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. mAbs 6, 1560–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bradley M. E., Dombrecht B., Manini J., Willis J., Vlerick D., De Taeye S., Van den Heede K., Roobrouck A., Grot E., Kent T. C., Laeremans T., Steffensen S., Van Heeke G., Brown Z., Charlton S. J., and Cromie K. D. (2015) Potent and efficacious inhibition of CXCR2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol. Pharmacol. 87, 251–262 [DOI] [PubMed] [Google Scholar]
- 9. Roovers R. C., Vosjan M. J. W. D., Laeremans T., el Khoulati R., de Bruin R. C. G., Ferguson K. M., Verkleij A. J., van Dongen G. A. M. S., and van Bergen en Henegouwen P. M. P. (2011) A biparatopic anti-EGFR Nanobody efficiently inhibits solid tumour growth. Int. J. Cancer 129, 2013–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maussang D., Mujić-Delić A., Descamps F. J., Stortelers C., Vanlandschoot P., Stigter-van Walsum M., Vischer H. F., van Roy M., Vosjan M., Gonzalez-Pajuelo M., van Dongen G. A. M. S., Merchiers P., van Rompaey P., and Smit M. J. (2013) Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J. Biol. Chem. 288, 29562–29572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stryjewski M. E., and Sexton D. J. (2003) Pseudomonas aeruginosa infections in specific types of patients and clinical settings. in Severe Infections Caused by Pseudomonas Aeruginosa (Hauser A. R., and Rello J., eds) pp. 1–15, Springer, New York [Google Scholar]
- 12. Potron A., Poirel L., and Nordmann P. (2015) Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int. J. Antimicrob. Agents 45, 568–585 [DOI] [PubMed] [Google Scholar]
- 13. Hauser A. R. (2009) The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7, 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dacheux D., Toussaint B., Richard M., Brochier G., Croize J., and Attree I. (2000) Pseudomonas aeruginosa cystic fibrosis isolates induce rapid, type III secretion-dependent, but ExoU-independent, oncosis of macrophages and polymorphonuclear neutrophils. Infect. Immun. 68, 2916–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dacheux D., Goure J., Chabert J., Usson Y., and Attree I. (2001) Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol. Microbiol. 40, 76–85 [DOI] [PubMed] [Google Scholar]
- 16. Audia J. P., Lindsey A. S., Housley N. A., Ochoa C. R., Zhou C., Toba M., Oka M., Annamdevula N. S., Fitzgerald M. S., Frank D. W., and Alvarez D. F. (2013) In the absence of effector proteins, the Pseudomonas aeruginosa type three secretion system needle tip complex contributes to lung injury and systemic inflammatory responses. PLoS One 8, e81792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gébus C., Caroline G., Faudry E., Eric F., Bohn Y.-S. T., Elsen S., Sylvie E., and Attree I. (2008) Oligomerization of PcrV and LcrV, protective antigens of Pseudomonas aeruginosa and Yersinia pestis. J. Biol. Chem. 283, 23940–23949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lunelli M., Hurwitz R., Lambers J., and Kolbe M. (2011) Crystal structure of PrgI-SipD: insight into a secretion competent state of the type three secretion system needle tip and its interaction with host ligands. PLoS Pathog. 7, e1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato H., and Frank D. W. (2011) Multi-functional characteristics of the Pseudomonas aeruginosa type III needle-tip protein, PcrV: comparison to orthologs in other Gram-negative bacteria. Front. Microbiol. 2, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Büttner D. (2012) Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 76, 262–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sawa T., Katoh H., and Yasumoto H. (2014) V-antigen homologs in pathogenic Gram-negative bacteria. Microbiol. Immunol. 58, 267–285 [DOI] [PubMed] [Google Scholar]
- 22. Sawa T., Yahr T. L., Ohara M., Kurahashi K., Gropper M. A., Wiener-Kronish J. P., and Frank D. W. (1999) Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 5, 392–398 [DOI] [PubMed] [Google Scholar]
- 23. Goure J., Pastor A., Faudry E., Chabert J., Dessen A., and Attree I. (2004) The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 72, 4741–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sundin C., Thelaus J., Bröms J. E., and Forsberg A. (2004) Polarisation of type III translocation by Pseudomonas aeruginosa requires PcrG, PcrV and PopN. Microb Pathog. 37, 313–322 [DOI] [PubMed] [Google Scholar]
- 25. Lee P.-C., Stopford C. M., Svenson A. G., and Rietsch A. (2010) Control of effector export by the Pseudomonas aeruginosa type III secretion proteins PcrG and PcrV. Mol. Microbiol. 75, 924–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohgita T., Hayashi N., Hama S., Tsuchiya H., Gotoh N., and Kogure K. (2013) A novel effector secretion mechanism based on proton-motive force-dependent type III secretion apparatus rotation. FASEB J. 27, 2862–2872 [DOI] [PubMed] [Google Scholar]
- 27. Goure J., Broz P., Attree O., Cornelis G. R., and Attree I. (2005) Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J. Infect. Dis. 192, 218–225 [DOI] [PubMed] [Google Scholar]
- 28. Holder I. A., Neely A. N., and Frank D. W. (2001) PcrV immunization enhances survival of burned Pseudomonas aeruginosa-infected mice. Infect. Immun. 69, 5908–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moriyama K., Wiener-Kronish J. P., and Sawa T. (2009) Protective effects of affinity-purified antibody and truncated vaccines against Pseudomonas aeruginosa V-antigen in neutropenic mice. Microbiol. Immunol. 53, 587–594 [DOI] [PubMed] [Google Scholar]
- 30. Baer M., Sawa T., Flynn P., Luehrsen K., Martinez D., Wiener-Kronish J. P., Yarranton G., and Bebbington C. (2009) An engineered human antibody fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 77, 1083–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Warrener P., Varkey R., Bonnell J. C., DiGiandomenico A., Camara M., Cook K., Peng L., Zha J., Chowdury P., Sellman B., and Stover C. K. (2014) A novel anti-PcrV antibody providing enhanced protection against Pseudomonas aeruginosa in multiple animal infection models. Antimicrob. Agents Chemother. 58, 4384–4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DiGiandomenico A., Keller A. E., Gao C., Rainey G. J., Warrener P., Camara M. M., Bonnell J., Fleming R., Bezabeh B., Dimasi N., Sellman B. R., Hilliard J., Guenther C. M., Datta V., Zhao W., et al. (2014) A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci. Transl. Med. 6, 262ra155. [DOI] [PubMed] [Google Scholar]
- 33. Roovers R. C., Laeremans T., Huang L., De Taeye S., Verkleij A. J., Revets H., de Haard H. J., and van Bergen en Henegouwen P. M. P. (2007) Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR nanobodies. Cancer Immunol. Immunother. 56, 303–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim J., Ahn K., Min S., Jia J., Ha U., Wu D., and Jin S. (2005) Factors triggering type III secretion in Pseudomonas aeruginosa. Microbiology 151, 3575–3587 [DOI] [PubMed] [Google Scholar]
- 35. Derewenda U., Mateja A., Devedjiev Y., Routzahn K. M., Evdokimov A. G., Derewenda Z. S., and Waugh D. S. (2004) The structure of Yersinia pestis V-antigen, an essential virulence factor and mediator of immunity against plague. Structure 12, 301–306 [DOI] [PubMed] [Google Scholar]
- 36. Deane J. E., Roversi P., Cordes F. S., Johnson S., Kenjale R., Daniell S., Booy F., Picking W. D., Picking W. L., Blocker A. J., and Lea S. M. (2006) Molecular model of a type III secretion system needle: implications for host-cell sensing. Proc. Natl. Acad. Sci. U.S.A. 103, 12529–12533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Secher T., Fauconnier L., Szade A., Rutschi O., Fas S. C., Ryffel B., and Rudolf M. P. (2011) Anti-Pseudomonas aeruginosa serotype O11 LPS immunoglobulin M monoclonal antibody panobacumab (KBPA101) confers protection in a murine model of acute lung infection. J. Antimicrob. Chemother. 66, 1100–1109 [DOI] [PubMed] [Google Scholar]
- 38. Firth A. E., and Patrick W. M. (2005) Statistics of protein library construction. Bioinformatics 21, 3314–3315 [DOI] [PubMed] [Google Scholar]
- 39. Shime N., Sawa T., Fujimoto J., Faure K., Allmond L. R., Karaca T., Swanson B. L., Spack E. G., and Wiener-Kronish J. P. (2001) Therapeutic administration of anti-PcrV F(ab′)2 in sepsis associated with Pseudomonas aeruginosa. J. Immunol. 167, 5880–5886 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.