

Abstract
Forkhead box O1 (FoxO1) is a key molecule for the development and functions of peripheral T cells. However, the precise mechanisms regulating FoxO1 expression in peripheral T cells remain elusive. We previously reported that Zfatf/f-CD4Cre mice showed a marked decline in FoxO1 protein levels in peripheral T cells, partially through proteasomal degradation. Here we have identified the precise mechanisms, apart from proteasome-mediated degradation, of the decreased FoxO1 levels in Zfat-deficient T cells. First, we confirmed that tamoxifen-inducible deletion of Zfat in Zfatf/f-CreERT2 mice coincidently decreases FoxO1 protein levels in peripheral T cells, indicating that Zfat is essential for maintaining FoxO1 levels in these cells. Although the proteasome-specific inhibitors lactacystin and epoxomicin only moderately increase FoxO1 protein levels, the inhibitors of lysosomal proteolysis bafilomycin A1 and chloroquine restore the decreased FoxO1 levels in Zfat-deficient T cells to levels comparable with those in control cells. Furthermore, Zfat-deficient T cells show increased numbers of autophagosomes and decreased levels of p62 protein, together indicating that Zfat deficiency promotes lysosomal FoxO1 degradation through autophagy. In addition, Zfat deficiency increases the phosphorylation levels of Thr-308 and Ser-473 of Akt and the relative amounts of cytoplasmic to nuclear FoxO1 protein levels, indicating that Zfat deficiency causes Akt activation, leading to nuclear exclusion of FoxO1. Our findings have demonstrated a novel role of Zfat in maintaining FoxO1 protein levels in peripheral T cells by regulating the activities of autophagy and the Akt signaling pathway.
Keywords: Akt PKB, autophagy, FOXO, protein degradation, T cell, Zfat
Introduction
Forkhead box O 1 (FoxO1), a member of the FoxO subfamily, is a multifunctional transcription factor that has important roles in regulating diverse cellular processes, such as differentiation, proliferation, survival, and metabolism (1). In addition to a variety of posttranslational modifications, the activity of FoxO1 is mainly regulated by Akt-mediated phosphorylation on three conserved sites, leading to the translocation of FoxO1 from the nucleus to the cytosol and its subsequent degradation through the ubiquitin-proteasome pathway (2–4). Several recent studies have identified the key role played by FoxO1 in the development and function of peripheral T cells in the immune system (5, 6). T cell-specific FoxO1-deficient mouse models revealed that FoxO1 is required for the proper control of T cell quiescence and tolerance by regulating the expression of KLF2 and the α subunit of the interleukin 7 receptor (IL-7Rα)3 (7, 8). Furthermore, FoxO1-dependent transcriptional programs regulate the development and functions of regulatory T cells (9–12) and determine the functional differentiation of CD8+ T cells into effector and memory cell subsets (13, 14). Despite the identification of these important roles of FoxO1, the mechanisms regulating FoxO1 expression in peripheral T cells remain elusive.
The nuclear zinc-finger protein Zfat has important roles in the immune system (15–19). We have reported that Zfat-gene ablation in thymic T cells in Zfatf/f-LckCre mice results in a drastic decrease in the number of CD4+CD8+ double positive cells, accompanied by impaired positive selection and excessive apoptosis (20, 21). Furthermore, we have reported that Zfat deficiency in peripheral T cells in Zfatf/f-CD4Cre mice results in a decrease in the number of peripheral T cells, accompanied by the decreased surface expression of IL-7Rα and the impairment in the induction of IL-2 in response to T cell receptor stimulation (22). These studies have clearly demonstrated that Zfat is a key molecule for the development, survival, and proliferation of both thymic and peripheral T cells.
Recently, we reported that FoxO1 protein levels were diminished in splenic T cells in Zfatf/f-CD4Cre mice (23). Furthermore, epoxomicin, which is an inhibitor specific to proteasomes, increased FoxO1 protein levels in Zfat-deficient T cells, suggesting that the decreased FoxO1 levels are partially due to the dysregulation of proteasomal proteolysis (23). In this study, we have identified the precise cause underlying the decline in FoxO1 protein levels in Zfat-deficient cells using Zfat knockout mouse models and protease inhibitors. We found that Zfat-deficient T cells showed hyperactivation of autophagy and the Akt signaling pathway, leading to the nuclear exclusion of FoxO1 and its enhanced lysosomal degradation. Here we demonstrated a novel role of Zfat in maintaining FoxO1 protein levels in peripheral T cells by regulating the activities of autophagy and the Akt signaling pathway.
Results
Zfat Is Essentially Required for Maintaining FoxO1 Expression Specifically in Peripheral T Cells
To elucidate the precise cause of the decreased FoxO1 levels in Zfat-deficient T cells, we employed two lines of mouse models with T cell-specific deletion of the Zfat gene, Zfatf/f-CD4Cre and Zfatf/f-LckCre mice. Consistent with our previous study (23), FoxO1 protein levels were markedly decreased in CD4+ T and CD8+ T cells derived from either spleen or lymph nodes in Zfatf/f-CD4Cre mice compared with those from Zfatf/f mice (Fig. 1A). The decreased expression of FoxO1 was also observed in peripheral T cells from Zfatf/f-LckCre spleen (Fig. 1B). On the other hand, FoxO1 protein levels in thymocytes were comparable between Zfatf/f and Zfatf/f-LckCre mice despite an obvious decrease in Zfat protein (Fig. 1B), similar to our observations in Zfatf/f-CD4Cre mice (23). These results suggest that Zfat is involved in the control of FoxO1 expression, specifically in peripheral T cells.
FIGURE 1.

Decrease in FoxO1 protein in Zfat-deficient T cells. A–D, immunoblot analysis of the indicated proteins on peripheral CD4+ T, CD8+ T, and B220+ cells or thymocytes from the indicated genotype mice. Actin was used as a loading control. Levels of protein expression were quantified by densitometry and normalized to actin levels. C, Zfatf/w-CreERT2 and Zfatf/f-CreERT2 mice were treated with tamoxifen for 3 days. A–C, data are representative of two or three independent experiments.
To further confirm that Zfat is essentially required for FoxO1 expression in peripheral T cells, we deleted the Zfat gene in peripheral T cells using CreERT2 transgenic mice. In cells expressing CreERT2, Cre recombinase is not active until tamoxifen is provided. We generated Zfatf/f-CreERT2 mice by crossing CreERT2 mice with Zfatf/f mice and employed this system to delete Zfat in peripheral T cells. Zfatf/w-CreERT2 and Zfatf/f-CreERT2 mice were injected daily with tamoxifen for 3 days and analyzed 24 h after the final administration. Tamoxifen treatment caused a decrease in Zfat protein in both CD4+ T cells and CD8+ T cells from Zfatf/f-CreERT2 mice compared with those from Zfatf/w-CreERT2 control mice (Fig. 1C). FoxO1 protein levels were diminished in splenic T cells, particularly in CD4+ T cells, from tamoxifen-treated Zfatf/f-CreERT2 mice compared with those from control mice (Fig. 1C). These results clearly indicated that Zfat is essentially important for maintaining FoxO1 protein levels in peripheral T cells and that the decreased FoxO1 levels are not due to defects in thymocyte differentiation, which might be caused by Zfat deficiency in the Zfatf/f-CD4Cre and Zfatf/f-LckCre thymuses.
FoxO1 belongs to the FoxO family, which consists of FoxO1, FoxO3, FoxO4, and FoxO6. FoxO family proteins share a high homology in amino acid sequence and have redundancy in their function and regulation (24). As FoxO6 is known to be predominantly expressed in the brain, we examined the expression levels of FoxO3 and FoxO4 in splenic CD4+ T cells from Zfatf/f and Zfatf/f-CD4Cre mice. Zfat deficiency caused a marked decrease in the protein levels of FoxO3 and FoxO4 in peripheral T cells (Fig. 1D), suggesting that Zfat is required for maintaining the levels of these FoxO proteins expressed in peripheral T cells.
Decreased FoxO1 Levels in Zfat-deficient T Cells Are Only Partially Dependent on Proteasome Activity
We previously reported that the FoxO1 mRNA levels were not affected by Zfat deficiency and that a proteasome-specific inhibitor, epoxomicin, increased FoxO1 protein levels in Zfatf/f-CD4Cre CD4+ T cells, suggesting that the dysregulation of its proteasomal degradation is involved in the decline in FoxO1 protein levels in Zfat-deficient T cells (23). We first examined the proteasome activity in CD4+ T cells, which showed no difference between Zfatf/f and Zfatf/f-CD4Cre mice (Fig. 2A). Next, we assessed the effects of MG132 on FoxO1 protein levels in peripheral T cells. MG132 is known to be a proteasome inhibitor, but it also inhibits several other proteases (25, 26). Surprisingly, MG132 elevated FoxO1 protein levels in Zfat-deficient T cells more markedly compared with those in cells treated with epoxomicin or lactacystin, both of which are proteasome-specific inhibitors (27, 28) (Fig. 2B). Lactacystin and epoxomicin increased the ubiquitinated proteins at levels comparable with those in the cells treated with MG132 (Fig. 2B), indicating that lactacystin and epoxomicin properly inhibit proteasome activity at the concentrations used in this study. These results indicated that the decreased FoxO1 levels in Zfat-deficient T cells are only partially dependent on proteasome activity.
FIGURE 2.

Effects of protease inhibitors on FoxO1 protein levels in Zfat-deficient T cells. A, proteasome activity in splenic CD4+ T cells from Zfatf/f and Zfatf/f-CD4Cre mice after treatment with or without 10 μm MG132 at 37 °C for 2 h. The data are mean ± S.D. (n = 3). n.s., not significant. RLU, relative light units. B and C, immunoblot analysis of Zfat, FoxO1, or ubiquitin on splenic CD4+ T cells from Zfatf/f and Zfatf/f-CD4Cre mice after treatment with protease inhibitors. CD4+ T cells prepared from Zfatf/f and Zfatf/f-CD4Cre mice were harvested immediately (control) or after incubation with 10 μm MG132, 10 μm Z-VLL-CHO, 10 μm lactacystin, 1 μm epoxomicin, 10 μm β-secretase inhibitor IV, 25 μm DAPT, 25 μm DAPM, or vehicle (DMSO) at 37 °C for 3 h. The cells were lysed and subjected to immunoblotting with the specific antibodies. Actin was used as a loading control. Levels of FoxO1 protein expression were quantified by densitometry and normalized to actin levels. A–C, data are representative of three independent experiments.
We compared the effect of MG132 and its structural analog N-benzyloxycarbonyl-Val-Leu-leucinal (Z-VLL-CHO) on FoxO1 protein levels. Both MG132 and Z-VLL-CHO elevated FoxO1 protein levels in splenic CD4+ T cells from Zfatf/f-CD4Cre mice, even at a concentration of 0.01 μm (Fig. 2C). Because these peptidyl aldehyde inhibitors are known to inhibit not only proteasome activity but also the activities of β- and γ-secretases (29–31), we examined the effects of inhibitors specific for these secretases on FoxO1 protein levels. However, β-secretase inhibitor IV, and DAPT and DAPM, which are inhibitors specific for γ-secretase, failed to increase FoxO1 protein levels (Fig. 2B). Taken together, these results suggest that there are MG132-sensitive proteases other than proteasomes and secretases that are responsible for decreased FoxO1 levels in Zfat-deficient T cells.
Inhibition of Lysosomal Proteolysis Restores Decreased FoxO1 Protein Levels in Zfat-deficient T Cells
MG132 is known to inhibit the activities of cathepsins as well as those of proteasomes and secretases (32). Cathepsins are lysosomal proteases that are optimally active under acidic conditions. We examined the effects of bafilomycin A1 and chloroquine, both of which inhibit the activities of lysosomal proteases by blocking acidification of the lysosome (33, 34). Surprisingly, bafilomycin A1 and chloroquine markedly increased FoxO1 protein levels in Zfat-deficient T cells (Fig. 3A). Next, we compared the effects of bafilomycin A1 with those of MG132. The effect of MG132 on FoxO1 protein levels was that they rapidly reached a maximum at 0.5 h, whereas bafilomycin A1 elevated the FoxO1 levels more slowly (Fig. 3B). Furthermore, co-treatment with MG132 and bafilomycin A1 was not additive in their effects on FoxO1 protein levels (Fig. 3C). Taken together, these results suggest the possibility that MG132 and bafilomycin A1 may increase FoxO1 protein levels by targeting different molecules on the same pathway or the same molecule with different kinetics.
FIGURE 3.

Inhibition of lysosomal proteolysis restores FoxO1 protein levels in Zfat-deficient T cells. A–D, immunoblot analysis of FoxO1 or phospho-S6 ribosomal protein (Ser-235/Ser-236) on splenic CD4+ T cells from Zfatf/f and Zfatf/f-CD4Cre mice after treatment with inhibitors. CD4+ T cells prepared from Zfatf/f and Zfatf/f-CD4Cre mice were harvested immediately (control) or after incubation with 10 μm MG132, 100 nm bafilomycin A1, 50 μm chloroquine, 100 nm rapamycin, 10 μm E-64, or vehicle (DMSO) at 37 °C for 3 h or the indicated time periods. The cells were lysed and subjected to immunoblotting with the specific antibodies. Actin was used as a loading control. Levels of FoxO1 protein expression were quantified by densitometry and normalized to actin levels. Data are representative of three independent experiments.
Lysosomal acidification is required not only for the optimal activities of acidic proteases within the lysosome but also for activation of the mammalian target of rapamycin complex 1 (mTORC1) (35). In addition, mTORC1 activity is known to be a requirement for the nuclear exclusion of FoxO1 in peripheral CD8+ T cells (14). To explore whether bafilomycin A1 increases FoxO1 protein levels by inhibiting mTORC1 activity, we examined the effect of rapamycin, which is an inhibitor specific for mTORC1 activity (36). Rapamycin had no effect on FoxO1 protein levels in Zfat-deficient T cells, whereas the phosphorylation of S6 ribosomal protein, which is a surrogate marker for the mTORC1 activity, was strikingly suppressed by rapamycin treatment (Fig. 3D), indicating that mTORC1 activity is not involved in the effect of bafilomycin A1 on FoxO1 levels. Furthermore, E-64, which directly inhibits cysteine proteases such as cathepsins (37), significantly increased FoxO1 protein levels in Zfat-deficient T cells (Fig. 3D). Together, these results indicated that bafilomycin A1 increases FoxO1 protein levels in Zfat-deficient T cells through the inhibition of protease activities within the lysosome, implying that Zfat deficiency promotes FoxO1 degradation through the lysosomal proteases.
Activation of Autophagy in Zfat-deficient T Cells
Autophagy is a unique digestion process by which cytoplasmic proteins and organelles are delivered to the lysosome for degradation (38). We hypothesized that the dysregulation of autophagy is related to the enhanced lysosomal degradation of FoxO1. First, we assessed autophagosome formation in splenic CD4+ T and B220+ cells via flow cytometry using an autophagosome-specific fluorescent probe. As shown in Fig. 4A, the abundance of autophagosomes was higher in CD4+ T cells from Zfatf/f-CD4Cre mice than those from Zfatf/f mice, whereas it was comparable in B220+ cells from Zfatf/f and Zfatf/f-CD4Cre mice. Furthermore, immunoblotting analysis revealed that the level of p62 protein, which is a well known autophagic marker protein, was significantly lower in CD4+ T cells from Zfatf/f-CD4Cre mice than in those from Zfatf/f mice (Fig. 4B). In contrast, by treatment with bafilomycin A1, p62 protein accumulated in Zfat-deficient T cells at higher levels than it did in control cells (Fig. 4B). All of these results indicate that Zfat deficiency causes the activation of autophagy in peripheral CD4+ T cells.
FIGURE 4.
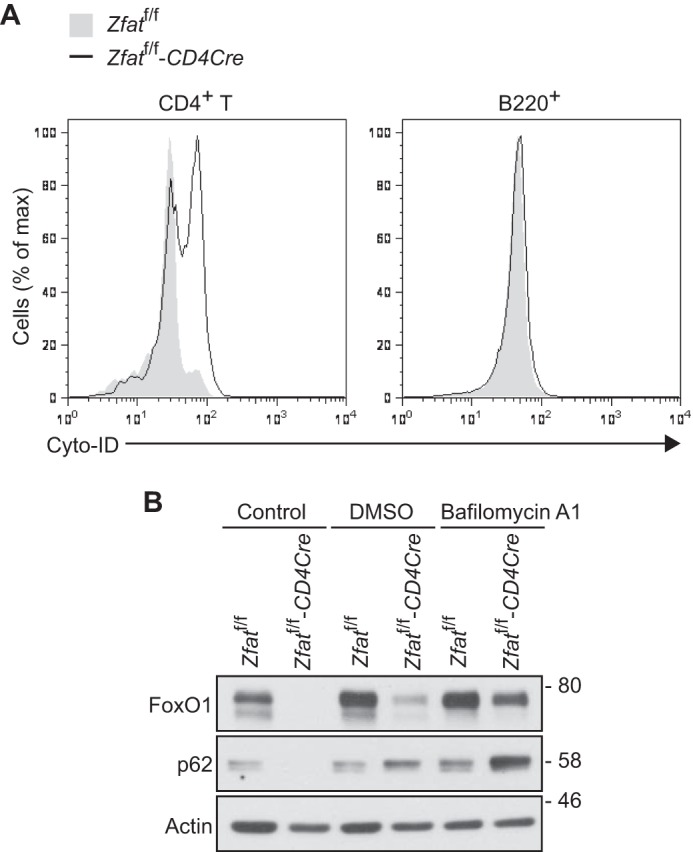
Zfat deficiency causes activation of autophagy. A, autophagosome formation in CD4+ T and B220+ cells. Total splenocytes from Zfatf/f and Zfatf/f-CD4Cre mice were stained with an autophagosome-specific fluorescent probe, Cyto-ID, and anti-CD4 or anti-B220 antibody for flow cytometry. B, Immunoblot analysis of FoxO1 and p62 on splenic CD4+ T cells from Zfatf/f and Zfatf/f-CD4Cre mice. CD4+ T cells prepared from Zfatf/f and Zfatf/f-CD4Cre mice were harvested immediately (control) or after incubation with 100 nm bafilomycin A1 or vehicle (DMSO) at 37 °C for 6 h. Actin was used as a loading control. Data are representative of three independent experiments.
Zfat Deficiency Results in PI3K-dependent Hyperactivation of Akt and Promotes Nuclear Exclusion of FoxO1
Akt is known to tightly regulate the localization and degradation of FoxO1 (2–4). Upon activation of PI3K, both phosphoinositide-dependent kinase 1 (PDK1) and Akt are recruited to the plasma membrane, which enables PDK1 to phosphorylate Thr-308 of Akt (39, 40). To be completely activated, Akt is further phosphorylated at Ser-473 by mTORC2 (41). To explore whether Akt is involved in the enhanced FoxO1 degradation in Zfat-deficient T cells, we assessed the phosphorylation levels of Akt in splenic CD4+ T cells by immunoblotting analysis. We observed that the phosphorylation levels of both Thr-308 and Ser-473 of Akt were significantly higher in CD4+ T cells from Zfatf/f-CD4Cre spleen than in those from Zfatf/f spleen, even without any stimulation (Fig. 5A), indicating that Zfat deficiency caused an elevation in basal Akt activity. As Akt activity is known to be negatively regulated by phosphatase and tensin homolog (PTEN) and protein phosphatase 2A (PP2A), we examined their protein levels in peripheral T cells by immunoblotting analysis. As shown in Fig. 5B, Zfat deficiency did not affect the expression levels of PTEN or PP2A in peripheral T cells. In contrast, LY294002, which is an inhibitor for PI3K, greatly decreased the amount of constitutively phosphorylated Akt in Zfat-deficient CD4+ T cells (Fig. 5C), suggesting that an aberrant activation of PI3K may be the underlying cause for the hyperactivation of Akt in Zfat-deficient T cells.
FIGURE 5.

Zfat deficiency enhances PI3K-dependent activation of Akt and nuclear exclusion of FoxO1. A–D, immunoblot analysis of the indicated proteins on splenic CD4+ T cells from Zfatf/f and Zfatf/f-CD4Cre mice. CD4+ T cells were harvested immediately (A and B) or after incubation with 10 μm LY294002 or vehicle (DMSO) for 20 min (C and D). The cells were lysed (A–C) or fractionated into nuclear (Nu)/cytoplasmic (Cy) fractions (D) and then subjected to immunoblotting with the specific antibodies. Actin was used as a loading control. Levels of FoxO1 protein expression were quantified by densitometry. Values below the panel in D represent the proportion of FoxO1 protein residing in each fraction. Data are representative of two or three independent experiments. E, model of the enhanced lysosomal degradation of FoxO1 in Zfat-deficient T cells. Zfat deficiency causes Akt activation, leading to the nuclear exclusion of FoxO1, after which the cytoplasmic FoxO1 is degraded by lysosomal proteases through autophagy, which is up-regulated by Zfat deficiency.
Finally, we examined the subcellular localization of FoxO1 in CD4+ T cells by subcellular fractionation, followed by immunoblotting analysis. Consistent with previous studies (14, 42), most of the FoxO1 protein was detected in the nuclear fraction in Zfatf/f CD4+ T cells (Fig. 5D). On the other hand, about half of the FoxO1 protein was detected in the cytosolic fraction in Zfatf/f-CD4Cre CD4+ T cells, indicating that Zfat deficiency promotes the translocation of FoxO1 from the nucleus to the cytosol, where FoxO1 is rapidly degraded (Fig. 5D). Furthermore, LY294002 increased FoxO1 protein levels in the nuclear fraction in Zfat-deficient T cells but not in control cells (Fig. 5D). Taken together, these results suggest that Zfat deficiency results in PI3K-dependent hyperactivation of Akt in peripheral T cells, leading to the nuclear exclusion of FoxO1.
Discussion
The important roles played by FoxO1 in the development and function of peripheral T cells are being increasingly recognized (6). Therefore, the identification of factors controlling FoxO1 expression in peripheral T cells is important for understanding immunoregulation and for treating immunological diseases. Our study has shown that Zfat maintains FoxO1 protein levels in peripheral T cells by regulating the activities of autophagy and the Akt signaling pathway.
We reported previously that the deletion of Zfat decreased FoxO1 protein levels in peripheral T cells and that epoxomicin increased FoxO1 protein levels in Zfat-deficient T cells, which suggested that the decreased FoxO1 protein levels resulted from the dysregulation of its proteasomal proteolysis (23). However, in this study, we have revealed that the decrease in FoxO1 protein levels in Zfat-deficient T cells was attributable to the enhanced lysosomal degradation of the FoxO1 protein. First, we showed that MG132 and Z-VLL-CHO, which are proteasome inhibitors with relatively low specificities, increased FoxO1 protein levels in Zfat-deficient T cells more markedly compared with those in cells treated with epoxomicin or lactacystin, which are inhibitors with a high specificity for proteasomes (Fig. 2). This result implies that proteases other than proteasomes are involved in the decline of FoxO1 levels. Next, we showed that the decreased FoxO1 protein levels in Zfat-deficient T cells were significantly restored by inhibitors for lysosomal acidification, bafilomycin A1 and chloroquine, and by E-64, which is an inhibitor specific for cysteine proteases such as cathepsins (Fig. 3). Finally, we showed that Zfat deficiency caused the activation of autophagy in peripheral T cells (Fig. 4). These results indicate that Zfat deficiency promotes lysosomal FoxO1 degradation through autophagy, although proteasomes are only partially involved in decreased FoxO1 levels. In addition, we showed that Zfat deficiency yielded an elevation in the phosphorylation levels of both Thr-308 and Ser-473 of Akt and in the relative amounts of cytoplasmic to nuclear FoxO1, indicating that Zfat deficiency causes Akt activation, leading to the nuclear exclusion of FoxO1 (Fig. 5). Taken together, these results revealed that, in Zfat-deficient peripheral T cells, hyperactivated Akt promotes the phosphorylation of FoxO1, leading to its translocation from the nucleus to the cytosol, after which the cytoplasmic FoxO1 is degraded by lysosomal proteases through autophagy, which is up-regulated by Zfat deficiency (Fig. 5D).
Consistent with the fact that FoxO family proteins share the mechanisms for Akt-mediated phosphorylation and subsequent nuclear exclusion (43), Zfat deficiency led to a marked decrease in protein levels of FoxO1, FoxO3, and FoxO4. Hyperactivation of Akt, which is caused by Zfat deficiency, will promote the phosphorylation of FoxO1, FoxO3, and FoxO4, leading to their translocation from the nucleus to the cytosol and, subsequently, their degradation. In addition to their specific roles, FoxO family proteins function in a redundant manner to regulate the development and function of T cells. In fact, mice with a deletion of both FoxO1 and FoxO3 in T cells showed more a severe inflammatory disorder phenotype than mice with a deletion of individual genes (11), indicating that Zfat is a critical molecule in immunoregulation.
The lysosome is a membrane-enclosed organelle containing various hydrolases responsible for the degradation of intracellular components and extracellular materials. Lysosomal dysfunction is known to be associated with several human diseases and the process of aging (44). Zfat deficiency promoted the lysosomal degradation of FoxO1. However, it remains unknown whether protein degradation in the lysosome is generally increased in Zfat-deficient T cells. Further studies will be required to elucidate the roles of Zfat in lysosomal protein degradation. As Zfat is considered to be a transcriptional regulator, identification of Zfat target genes in T cells will lead to a better understanding of the function of Zfat in lysosomal protein degradation. Zfat might regulate the expression of genes involved in lysosomal biogenesis, leading to increased lysosomal protein degradation.
Zfat deficiency did not affect FoxO1 protein levels in thymocytes (Fig. 1B), indicating that Zfat regulates FoxO1 levels in a cell type-specific manner. Zfat is required for maintaining FoxO1 protein levels in peripheral T cells but not in thymocytes. We showed previously that Zfat deficiency in peripheral T cells resulted in decreased IL-7Rα expression (22). However, the precise mechanisms by which Zfat deficiency lowered IL-7Rα expression remained unknown. In this study, using three lines of Zfat knockout mouse models, we showed that Zfat was essentially required for maintaining FoxO1 expression specifically in peripheral T cells (Fig. 1). Given that FoxO1 transcriptionally regulates IL-7Rα expression in peripheral T cells (7, 8), the lower IL-7Rα expression in Zfat-deficient T cells could be attributed to the decreased expression of FoxO1 protein. The homeostasis of peripheral T cells is largely maintained by signaling from both IL-7R and T cell receptor (45, 46). Accordingly, Zfat is considered to play an important role in peripheral T cell homeostasis by controlling FoxO1 protein levels.
Mice with T cell-specific deletion of the FoxO1 gene (FoxO1f/f-CD4Cre mice) showed similar immunological phenotypes as observed in Zfatf/f-CD4Cre mice. For example, both Zfatf/f-CD4Cre mice and FoxO1f/f-CD4Cre mice showed defects in survival and proliferation in peripheral T cells (7). Interestingly, FoxO1f/f-CD4Cre mice exhibited an autoimmune phenotype, such as multiorgan lymphocyte infiltration and autoantibody production (7). Indeed, FoxO1 deficiency in T cells resulted in defects in development and function in regulatory T (Treg) cells as well as an increase in the number of Th17 cells (9–12, 47). Although Zfat was identified as a candidate susceptibility gene for autoimmune thyroid disease (19), and genetic variants of Zfat were reported to be associated with the severity of Hashimoto disease and with interferon-β responsiveness in multiple sclerosis (48, 49), it remains unknown whether Zfat deficiency in T cells leads to the autoimmune phenotype. Further studies should be needed to elucidate the roles of Zfat in autoimmunity.
Autophagy is a fundamental catabolic process in which intracellular proteins and organelles are degraded via the lysosome, and it is up-regulated in response to extra- or intracellular stress and signals such as starvation, growth factor deprivation, and pathogen infection (50, 51). Furthermore, autophagy has emerged as a key process regulating many aspects of T cell function, including their development, survival, and homeostasis (52–55). Here we found that Zfat deficiency caused the activation of autophagy in peripheral T cells. Therefore, it is important to elucidate the mechanism by which Zfat influences the activity of autophagy in peripheral T cells. Given that Zfat is expected to be a transcriptional regulator in the nucleus, Zfat might affect the expression of the genes involved in autophagy regulation; this requires further investigation.
In this study, we found that both Thr-308 and Ser-473 of Akt were hyperphosphorylated in Zfat-deficient T cells. Akt plays a central role in the regulation of a variety of cellular processes, including cell survival and cell cycle progression, downstream of PI3K. Activated PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) to form phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3), leading to the recruitment of Akt to the plasma membrane, where PDK1 phosphorylates Thr-308 of Akt (39, 40). Akt is further phosphorylated at Ser-473 by mTORC2 to gain its full activation capacity (41). On the other hand, particular phosphatases, including PTEN and PP2A, negatively regulate Akt activity through distinct mechanisms (56–58). Zfat deficiency did not affect the expression levels of PTEN or PP2A in peripheral T cells, whereas the PI3K inhibitor LY294002 decreased the amount of constitutively phosphorylated Akt, suggesting that Akt hyperactivation caused by Zfat deficiency could be attributed to aberrant activation of PI3K. In T cells, PI3K is activated in response to stimulation through the T cell receptor as well as co-stimulatory, cytokine, and chemokine receptors (59–61). Zfat might regulate the expression levels of the molecules downstream of these receptors, which are involved in the control of PI3K activity. Further studies are required to elucidate the mechanisms by which Zfat regulates the PI3K/Akt signaling pathway in peripheral T cells.
In summary, we demonstrated that Zfat deficiency in peripheral T cells results in activation of autophagy and the Akt signaling pathway, leading to enhanced lysosomal degradation of the FoxO1 protein. Further studies will provide additional insights into the roles of Zfat in the development and function of T cells.
Experimental Procedures
General Reagents and Antibodies
MG132, Z-VLL-CHO, lactacystin, epoxomicin, β-secretase inhibitor IV, N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester (DAPT), N-[N-3,5-difluorophenacetyl]-l-alanyl-S-phenylglycine methyl ester (DAPM), and LY294002 were purchased from Calbiochem. Bafilomycin A1 was from Wako Pure Chemical Industries. E-64, chloroquine diphosphate salt, tamoxifen, and anti-actin antibody (A2066) were from Sigma-Aldrich. Rapamycin and anti-FoxO1 (2880), anti-FoxO3a (2497), anti-FoxO4 (9472), anti-S6 ribosomal protein (2212), anti-phospho-S6 ribosomal protein (Ser-235/236, 2211), anti-Akt (9272), anti-phospho-Akt (Thr-308, 2965), anti-phospho-Akt (Ser-473, 4060), anti-PP2A C subunit (2259), and anti-PTEN (9552) antibodies were from Cell Signaling Technologies. The anti-ubiquitin antibody (P4D1) was from Santa Cruz Biotechnology. The Proteasome-Glo assay kit was from Promega. The anti-Zfat antibody was prepared as described previously (62).
Mice
Zfatf/f, Zfatf/f-CD4Cre, and Zfatf/f-LckCre mice were generated as described previously (21, 22). Zfatf/f mice were crossed to CreERT2 mice to generate tamoxifen-inducible Zfat knockout (Zfatf/f-CreERT2) mice in the C57BL/6 background. Zfatf/w-CreERT2 and Zfatf/f-CreERT2 mice were intraperitoneally injected daily with tamoxifen (2 mg/40 g of body weight) for 3 consecutive days and sacrificed 24 h after the final administration. All animal experiments were performed under Institutional Animal Care and Use Committee of Fukuoka University-approved guidelines in accordance with approved protocols.
Cell Isolation and Culture
Spleens, mesenteric lymph nodes, or thymuses from 8- to 12-week-old mice were processed into a single-cell suspension. CD4+ T, CD8+ T, or B220+ cells were isolated via positive selection using MACS (Miltenyi Biotech) following the protocol of the manufacturer. In experiments using inhibitors, purified T cells were incubated in RPMI1640 medium (Wako Pure Chemical Industries) supplemented with 10% FBS (Gibco), 50 μm β-mercaptoethanol (Sigma-Aldrich), penicillin (Life Technologies), and streptomycin (Life Technologies).
Nuclear/Cytoplasmic Fractionation, Cell Lysis, and Immunoblotting
Splenic CD4+ T cells were fractionated into their nuclear and cytoplasmic fractions using the NE-PER nuclear and cytoplasmic kit (Thermo Scientific) following the protocol of the manufacturer. Whole cell extracts were prepared by incubating cells in radioimmune precipitation assay (RIPA) buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1% Triton X-100) supplemented with Complete EDTA-free protease inhibitor and PhosSTOP phosphatase inhibitor (both from Roche) for 30 min at 4 °C. Cell pellets were removed by centrifugation, and then the supernatants were mixed with Laemmli sample buffer. Equal amounts of protein were resolved via SDS-PAGE and transferred to a nitrocellulose membrane (GE Healthcare). The membranes were blocked in TBST (10 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 0.1% Tween 20) containing 5% nonfat dry milk and then incubated overnight at 4 °C with primary antibodies diluted in TBST containing 1% BSA. Horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) and SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) were used for the detection. Quantitative analysis of the immunoblotting was performed using ImageJ software (National Institutes of Health). The amounts of total protein loaded on SDS-PAGE were determined by staining gels with CBB Stain One (Nacalai Tesque). Neither Zfat deficiency nor treatment with the inhibitors used in this study affected actin protein levels.
Autophagy Measurement by Flow Cytometry
The formation of autophagosomes was determined using the Cyto-ID autophagy detection kit (Enzo Life Science) according to the protocol of the manufacturer. In brief, splenic cells were depleted of erythrocytes by hypotonic lysis. After washing twice with PBS, the cells were incubated with Cyto-ID for 30 min at 37 °C in the dark. After washing with PBS containing 5% FBS, the cells were stained with anti-mouse CD4 APC-conjugated or anti-mouse B220 APC/Cy7-conjugated antibody (both from BioLegend). Data were collected using FACSAria II (BD Biosciences) and analyzed using FlowJo software (Tomy Digital Biology).
Author Contributions
S. I. and Y. I. designed, performed, and analyzed the experiments and wrote the paper. Y. T., K. D., M. K., H. L., K. N., and T. T. provided technical assistance for the experiments. T. O. provided Zfat transgenic mice. S. S. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by a Grant-in-Aid for Scientific Research B (to S. S.) from the JSPS and a Grant-in-Aid for the FCAM from the MEXT-supported Program for the Strategic Research Foundation at Private Universities. The authors declare that they have no conflicts of interest with the contents of this article.
- IL-7Rα
- subunit α of the IL-7 receptor
- DAPT
- N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester
- DAPM
- N-[N-3,5-difluorophenacetyl]-l-alanyl-S-phenylglycine methyl ester
- mTORC
- mammalian target of rapamycin complex
- PTEN
- phosphatase and tensin homolog
- PP2A
- protein phosphatase 2A
- Z-VLL-CHO
- N-benzyloxycarbonyl-Val-Leu-leucinal.
References
- 1. Eijkelenboom A., and Burgering B. M. (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 [DOI] [PubMed] [Google Scholar]
- 2. Huang H., Regan K. M., Wang F., Wang D., Smith D. I., van Deursen J. M., and Tindall D. J. (2005) Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. U.S.A. 102, 1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aoki M., Jiang H., and Vogt P. K. (2004) Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 13613–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matsuzaki H., Daitoku H., Hatta M., Tanaka K., and Fukamizu A. (2003) Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 100, 11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luo C. T., and Li M. O. (2013) Transcriptional control of regulatory T cell development and function. Trends Immunol. 34, 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hedrick S. M., Hess Michelini R., Doedens A. L., Goldrath A. W., and Stone E. L. (2012) FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol. 12, 649–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ouyang W., Beckett O., Flavell R. A., and Li M. O. (2009) An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity 30, 358–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kerdiles Y. M., Beisner D. R., Tinoco R., Dejean A. S., Castrillon D. H., DePinho R. A., and Hedrick S. M. (2009) Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat. Immunol. 10, 176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo C. T., Liao W., Dadi S., Toure A., and Li M. O. (2016) Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature 529, 532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ouyang W., Liao W., Luo C. T., Yin N., Huse M., Kim M. V., Peng M., Chan P., Ma Q., Mo Y., Meijer D., Zhao K., Rudensky A. Y., Atwal G., Zhang M. Q., and Li M. O. (2012) Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491, 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ouyang W., Beckett O., Ma Q., Paik J. H., DePinho R. A., and Li M. O. (2010) Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 11, 618–627 [DOI] [PubMed] [Google Scholar]
- 12. Kerdiles Y. M., Stone E. L., Beisner D. R., Beisner D. L., McGargill M. A., Ch'en I. L., Stockmann C., Katayama C. D., and Hedrick S. M. (2010) Foxo transcription factors control regulatory T cell development and function. Immunity 33, 890–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tejera M. M., Kim E. H., Sullivan J. A., Plisch E. H., and Suresh M. (2013) FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J. Immunol. 191, 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rao R. R., Li Q., Gubbels Bupp M. R., and Shrikant P. A. (2012) Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity 36, 374–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doi K., Ishikura S., and Shirasawa S. (2014) The roles of ZFAT in thymocyte differentiation and homeostasis of peripheral naive T-cells. Anticancer Res. 34, 4489–4495 [PubMed] [Google Scholar]
- 16. Tsunoda T., and Shirasawa S. (2013) Roles of ZFAT in haematopoiesis, angiogenesis and cancer development. Anticancer Res. 33, 2833–2837 [PubMed] [Google Scholar]
- 17. Tsunoda T., Takashima Y., Tanaka Y., Fujimoto T., Doi K., Hirose Y., Koyanagi M., Yoshida Y., Okamura T., Kuroki M., Sasazuki T., and Shirasawa S. (2010) Immune-related zinc finger gene ZFAT is an essential transcriptional regulator for hematopoietic differentiation in blood islands. Proc. Natl. Acad. Sci. U.S.A. 107, 14199–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujimoto T., Doi K., Koyanagi M., Tsunoda T., Takashima Y., Yoshida Y., Sasazuki T., and Shirasawa S. (2009) ZFAT is an antiapoptotic molecule and critical for cell survival in MOLT-4 cells. FEBS Lett. 583, 568–572 [DOI] [PubMed] [Google Scholar]
- 19. Shirasawa S., Harada H., Furugaki K., Akamizu T., Ishikawa N., Ito K., Ito K., Tamai H., Kuma K., Kubota S., Hiratani H., Tsuchiya T., Baba I., Ishikawa M., Tanaka M., et al. (2004) SNPs in the promoter of a B cell-specific antisense transcript, SAS-ZFAT, determine susceptibility to autoimmune thyroid disease. Hum. Mol. Genet 13, 2221–2231 [DOI] [PubMed] [Google Scholar]
- 20. Ishikura S., Ogawa M., Doi K., Matsuzaki H., Iwaihara Y., Tanaka Y., Tsunoda T., Hideshima H., Okamura T., and Shirasawa S. (2015) Zfat-deficient CD4+ CD8+ double-positive thymocytes are susceptible to apoptosis with deregulated activation of p38 and JNK. J. Cell. Biochem. 116, 149–157 [DOI] [PubMed] [Google Scholar]
- 21. Ogawa M., Okamura T., Ishikura S., Doi K., Matsuzaki H., Tanaka Y., Ota T., Hayakawa K., Suzuki H., Tsunoda T., Sasazuki T., and Shirasawa S. (2013) Zfat-deficiency results in a loss of CD3ζ phosphorylation with dysregulation of ERK and Egr activities leading to impaired positive selection. PLoS ONE 8, e76254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doi K., Fujimoto T., Okamura T., Ogawa M., Tanaka Y., Mototani Y., Goto M., Ota T., Matsuzaki H., Kuroki M., Tsunoda T., Sasazuki T., and Shirasawa S. (2012) ZFAT plays critical roles in peripheral T cell homeostasis and its T cell receptor-mediated response. Biochem. Biophys. Res. Commun. 425, 107–112 [DOI] [PubMed] [Google Scholar]
- 23. Iwaihara Y., Ishikura S., Doi K., Tsunoda T., Fujimoto T., Okamura T., and Shirasawa S. (2015) Marked reduction in FoxO1 protein by its enhanced proteasomal degradation in Zfat-deficient peripheral T-cells. Anticancer Res. 35, 4419–4423 [PubMed] [Google Scholar]
- 24. Arden K. C., and Biggs W. H. 3rd. (2002) Regulation of the FoxO family of transcription factors by phosphatidylinositol-3 kinase-activated signaling. Arch. Biochem. Biophys. 403, 292–298 [DOI] [PubMed] [Google Scholar]
- 25. Elliott P. J., Zollner T. M., and Boehncke W. H. (2003) Proteasome inhibition: a new anti-inflammatory strategy. J. Mol. Med. 81, 235–245 [DOI] [PubMed] [Google Scholar]
- 26. Hayashi M., Saito Y., and Kawashima S. (1992) Calpain activation is essential for membrane fusion of erythrocytes in the presence of exogenous Ca2+. Biochem. Biophys. Res. Commun. 182, 939–946 [DOI] [PubMed] [Google Scholar]
- 27. Fenteany G., Standaert R. F., Lane W. S., Choi S., Corey E. J., and Schreiber S. L. (1995) Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268, 726–731 [DOI] [PubMed] [Google Scholar]
- 28. Meng L., Mohan R., Kwok B. H., Elofsson M., Sin N., and Crews C. M. (1999) Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo anti-inflammatory activity. Proc. Natl. Acad. Sci. U.S.A. 96, 10403–10408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steinhilb M. L., Turner R. S., and Gaut J. R. (2001) The protease inhibitor, MG132, blocks maturation of the amyloid precursor protein Swedish mutant preventing cleavage by β-secretase. J. Biol. Chem. 276, 4476–4484 [DOI] [PubMed] [Google Scholar]
- 30. Abbenante G., Kovacs D. M., Leung D. L., Craik D. J., Tanzi R. E., and Fairlie D. P. (2000) Inhibitors of β-amyloid formation based on the β-secretase cleavage site. Biochem. Biophys. Res. Commun. 268, 133–135 [DOI] [PubMed] [Google Scholar]
- 31. Pinnix I., Musunuru U., Tun H., Sridharan A., Golde T., Eckman C., Ziani-Cherif C., Onstead L., and Sambamurti K. (2001) A novel γ-secretase assay based on detection of the putative C-terminal fragment-γ of amyloid β protein precursor. J. Biol. Chem. 276, 481–487 [DOI] [PubMed] [Google Scholar]
- 32. Ito H., Watanabe M., Kim Y. T., and Takahashi K. (2009) Inhibition of rat liver cathepsins B and L by the peptide aldehyde benzyloxycarbonyl-leucyl-leucyl-leucinal and its analogues. J. Enzyme Inhib. Med. Chem. 24, 279–286 [DOI] [PubMed] [Google Scholar]
- 33. Yoshimori T., Yamamoto A., Moriyama Y., Futai M., and Tashiro Y. (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707–17712 [PubMed] [Google Scholar]
- 34. Wibo M., and Poole B. (1974) Protein degradation in cultured cells: II: the uptake of chloroquine by rat fibroblasts and the inhibition of cellular protein degradation and cathepsin B1. J. Cell Biol. 63, 430–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y., and Sabatini D. M. (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334, 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown E. J., Albers M. W., Shin T. B., Ichikawa K., Keith C. T., Lane W. S., and Schreiber S. L. (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758 [DOI] [PubMed] [Google Scholar]
- 37. Hashida S., Towatari T., Kominami E., and Katunuma N. (1980) Inhibitions by E-64 derivatives of rat liver cathepsin B and cathepsin L in vitro and in vivo. J. Biochem. 88, 1805–1811 [DOI] [PubMed] [Google Scholar]
- 38. Mizushima N., Ohsumi Y., and Yoshimori T. (2002) Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429 [DOI] [PubMed] [Google Scholar]
- 39. Stokoe D., Stephens L. R., Copeland T., Gaffney P. R., Reese C. B., Painter G. F., Holmes A. B., McCormick F., and Hawkins P. T. (1997) Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 277, 567–570 [DOI] [PubMed] [Google Scholar]
- 40. Currie R. A., Walker K. S., Gray A., Deak M., Casamayor A., Downes C. P., Cohen P., Alessi D. R., and Lucocq J. (1999) Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 337, 575–583 [PMC free article] [PubMed] [Google Scholar]
- 41. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 42. Hawse W. F., Sheehan R. P., Miskov-Zivanov N., Menk A. V., Kane L. P., Faeder J. R., and Morel P. A. (2015) Cutting edge: differential regulation of PTEN by TCR, Akt, and FoxO1 controls CD4+ T cell fate decisions. J. Immunol. 194, 4615–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tzivion G., Dobson M., and Ramakrishnan G. (2011) FoxO transcription factors: regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945 [DOI] [PubMed] [Google Scholar]
- 44. Schultz M. L., Tecedor L., Chang M., and Davidson B. L. (2011) Clarifying lysosomal storage diseases. Trends Neurosci. 34, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sprent J., and Surh C. D. (2011) Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat. Immunol. 12, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Surh C. D., and Sprent J. (2008) Homeostasis of naive and memory T cells. Immunity 29, 848–862 [DOI] [PubMed] [Google Scholar]
- 47. Lainé A., Martin B., Luka M., Mir L., Auffray C., Lucas B., Bismuth G., and Charvet C. (2015) Foxo1 is a T cell-intrinsic inhibitor of the RORγt-Th17 program. J. Immunol. 195, 1791–1803 [DOI] [PubMed] [Google Scholar]
- 48. Inoue N., Watanabe M., Yamada H., Takemura K., Hayashi F., Yamakawa N., Akahane M., Shimizuishi Y., Hidaka Y., and Iwatani Y. (2012) Associations between autoimmune thyroid disease prognosis and functional polymorphisms of susceptibility genes, CTLA4, PTPN22, CD40, FCRL3, and ZFAT, previously revealed in genome-wide association studies. J. Clin. Immunol. 32, 1243–1252 [DOI] [PubMed] [Google Scholar]
- 49. Comabella M., Craig D. W., Morcillo-Suárez C., Río J., Navarro A., Fernández M., Martin R., and Montalban X. (2009) Genome-wide scan of 500,000 single-nucleotide polymorphisms among responders and nonresponders to interferon β therapy in multiple sclerosis. Arch. Neurol. 66, 972–978 [DOI] [PubMed] [Google Scholar]
- 50. Nakatogawa H., Suzuki K., Kamada Y., and Ohsumi Y. (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467 [DOI] [PubMed] [Google Scholar]
- 51. He C., and Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pua H. H., Dzhagalov I., Chuck M., Mizushima N., and He Y. W. (2007) A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jia W., and He Y. W. (2011) Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J. Immunol. 186, 5313–5322 [DOI] [PubMed] [Google Scholar]
- 54. Stephenson L. M., Miller B. C., Ng A., Eisenberg J., Zhao Z., Cadwell K., Graham D. B., Mizushima N. N., Xavier R., Virgin H. W., and Swat W. (2009) Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 5, 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McLeod I. X., Zhou X., Li Q. J., Wang F., and He Y. W. (2011) The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Rα surface expression. J. Immunol. 187, 5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Newton R. H., and Turka L. A. (2012) Regulation of T cell homeostasis and responses by pten. Front. Immunol. 3, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuo Y. C., Huang K. Y., Yang C. H., Yang Y. S., Lee W. Y., and Chiang C. W. (2008) Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55α regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 283, 1882–1892 [DOI] [PubMed] [Google Scholar]
- 58. Rocher G., Letourneux C., Lenormand P., and Porteu F. (2007) Inhibition of B56-containing protein phosphatase 2As by the early response gene IEX-1 leads to control of Akt activity. J. Biol. Chem. 282, 5468–5477 [DOI] [PubMed] [Google Scholar]
- 59. Engelman J. A., Luo J., and Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 [DOI] [PubMed] [Google Scholar]
- 60. Huang Y. H., and Sauer K. (2010) Lipid signaling in T-cell development and function. Cold Spring Harb. Perspect. Biol. 2, a002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. So L., and Fruman D. A. (2012) PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem. J. 442, 465–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Koyanagi M., Nakabayashi K., Fujimoto T., Gu N., Baba I., Takashima Y., Doi K., Harada H., Kato N., Sasazuki T., and Shirasawa S. (2008) ZFAT expression in B and T lymphocytes and identification of ZFAT-regulated genes. Genomics 91, 451–457 [DOI] [PubMed] [Google Scholar]