

Abstract
Modifier of transcription 1 (Mot1) is a conserved and essential Swi2/Snf2 ATPase that can remove TATA-binding protein (TBP) from DNA using ATP hydrolysis and in so doing exerts global effects on transcription. Spt16 is also essential and functions globally in transcriptional regulation as a component of the facilitates chromatin transcription (FACT) histone chaperone complex. Here we demonstrate that Mot1 and Spt16 regulate a largely overlapping set of genes in Saccharomyces cerevisiae. As expected, Mot1 was found to control TBP levels at co-regulated promoters. In contrast, Spt16 did not affect TBP recruitment. On a global scale, Spt16 was required for Mot1 promoter localization, and Mot1 also affected Spt16 localization to genes. Interestingly, we found that Mot1 has an unanticipated role in establishing or maintaining the occupancy and positioning of nucleosomes at the 5′ ends of genes. Spt16 has a broad role in regulating chromatin organization in gene bodies, including those nucleosomes affected by Mot1. These results suggest that the large scale overlap in Mot1 and Spt16 function arises from a combination of both their unique and shared functions in transcription complex assembly and chromatin structure regulation.
Keywords: ATPase, histone chaperone, nucleosome, SPT16, transcription, yeast, Mot1, TATA-binding protein
Introduction
Modifier of transcription 1 (Mot1)4 was originally identified genetically as a negative regulator of transcription from weak promoters (1–7). It is an ATPase that utilizes ATP hydrolysis to remove TBP from DNA and thereby repress transcription (8–11). Subsequent studies have shown that Mot1 can also positively regulate transcription, potentially by recycling inactive TBP and enabling TBP to bind its proper locations (12–16). Other studies have suggested that Mot1 may have a direct role in transcription activation, including facilitating transcription preinitiation complex (PIC) formation or remodeling chromatin (17–20). Mot1 regulates over one-third of the protein-coding genome in budding yeast (17, 21–23), affecting both mRNA levels and RNA synthesis precision. Using a novel method to determine transcription length defects, we showed that the most common length defect in the temperature-sensitive mot1-42 strain is premature termination, linking Mot1 to productive transcription elongation as well as initiation (23). Chromatin immunoprecipitation (ChIP) experiments showed that Mot1 is present mainly at promoters (15, 18, 21), raising the question of how it can promote elongation.
Spt16 is a histone chaperone with functions in transcription, replication, and DNA repair (24–28). It forms a heterodimer with Pob3, and together they loosely associate with the high mobility group protein Nhp6 to bind nucleosomes (29–32). This complex of Spt16, Pob3, and Nhp6 is known as facilitates chromatin transcription (FACT) or yFACT in budding yeast. Spt16 can bind H2A/H2B dimers in vivo and in vitro and H3/H4 tetramers in vitro but with higher specificity for H2A/H2B (30, 33–37). Recently it was demonstrated that the middle domain of human Spt16 interacts with the H3/H4 tetramer, and the AID domain interacts with H2B (38). The best studied transcriptional role for Spt16 involves the catalysis of nucleosomal reorganization during transcriptional elongation (25, 39–41). Spt16 has been suggested to travel with elongating RNA polymerase II (Pol II) (42, 43), enabling it to remove or reorganize nucleosomes blocking transcription and replace them after Pol II passage (24, 25, 33, 44). Consistent with this role, mutations in Spt16 led to exposure of adventitious promoters buried within the gene body, leading to a phenomenon known as cryptic transcription (45, 46). There is some evidence that Spt16 can function during normal initiation as well by promoting transcription preinitiation complex formation (37, 43, 47). The functions of Mot1 and Spt16 in initiation as well as elongation prompted us to test whether they overlap functionally.
Results
Mot1 and Spt16 Genetically and Physically Interact
Mot1 and Spt16 have been implicated in transcription initiation and elongation but through different mechanisms. However, a functional relationship between them was suggested by the identification of Spt16 as a Mot1-associated protein using mass spectrometry (48). To test for a functional relationship, we first compared growth of conditional single mutant strains with that of the mot1-42 spt16-197 double mutant. As shown in Fig. 1A, mot1-42 and spt16-197 strains were temperature-sensitive at 35 °C. The double mutant displayed a synthetic growth defect at 30 °C, indicating that MOT1 and SPT16 interact genetically. In reciprocal co-immunoprecipitation (co-IP) experiments, Spt16 and Mot1 co-associated in cell extracts (Fig. 1, B and C), substantiating the prior report of their physical association. Together, these results establish that the two factors interact both biochemically and genetically.
FIGURE 1.

Mot1 and Spt16 genetically and physically interact. A, 10-fold serial dilutions of the indicated strains were plated on rich medium and incubated at the indicated temperatures for 3 days. The mot1-42 spt16-197 strain exhibited synthetic sickness at 30 °C compared with the other strains. B, co-IP of Spt16 and Mot1 in whole cell extracts obtained from the strains with HA-tagged Spt16 and/or myc-tagged Mot1 as indicated. IPs were performed using the myc antibody, and blots were probed with the antibody indicated to the left of each panel. WCE lanes show results with whole cell extracts used as input in the IPs. C, a similar experiment as B using a polyclonal Spt16 antibody in the IP. IB, immunoblotting.
Genome-wide Analysis of RNA Expression Changes
To assess the global relationship between Mot1 and Spt16 in gene expression, we first compared mot1-42 genome-wide tiling array data from our previous study (23) with published spt16-197 genome-wide tiling array data (46). Initial analysis suggested overlap in genes regulated by Mot1 and Spt16 and significant correlation in expression changes between the two mutant strains (data not shown). The spt16-197 study used polyadenylated RNA and interrogated open reading frames (ORFs) with six probes per ORF for the main purpose of detecting cryptic initiation (46). In contrast, our mot1-42 study used total RNA and probes at 8-base pair (bp) intervals throughout the entire yeast genome to measure expression changes at higher resolution (23). For a more equivalent comparison, we repeated the spt16-197 genome-wide analysis using total RNA and probes at 8-bp intervals. log2 median differential expression correlated highly between previous and new data sets as expected (Fig. 2A; r = 0.72). Our initial observation of the correlation between the mot1-42 data set and the previous spt16-197 data set was upheld by the new spt16-197 data set, showing that the expression changes in mot1-42 and spt16-197 correlate (Fig. 2B; r = 0.34).
FIGURE 2.

Genome-wide expression changes in mot1-42 and spt16-197 cells are correlated. A, comparison of differential expression (spt16-197 cells versus WT) in this study with previously published data (46). The Pearson correlation coefficient r = 0.72 (p ≪ 1 × 10−277) indicates a high correlation. B, comparison of expression changes in mot1-42 cells (23) and spt16-197 cells (this study) versus WT. The expression changes are significantly correlated (r = 0.34; p < 1 × 10−277). C, the mot1-42 spt16-197 double mutant strain was obtained via a cross of the two single mutants, which are derived from different strain backgrounds. The plot shows that differential RNA levels in mot1-42 spt16-197 cells obtained by comparison with RNA from each of the parental WT strains are highly correlated (r = 0.74; p ≪ 1 × 10−277). D–F, differential gene expression thresholds in mot1-42, spt16-197, and double mutant strains. Distribution of log2 -fold median differential (diff.) expression in each mutant (blue) is compared with a normal distribution (red line) by plotting standard normal quantiles. Due to differences in magnitudes of expression changes in each mutant, different thresholds were determined for each strain at deviation from the normal distribution (green dots). D, differential expression in mot1-42 cells and thresholds for overexpression and underexpression of 0.1100 and −0.3245. E, differential expression in spt16-197 cells and thresholds of 0.3540 and −0.3910. F, differential expression in mot1-42 spt16-197 cells and thresholds of 0.5705 and −0.5010.
We next identified genes for which changes in expression were significant. For each data set, upper and lower thresholds of deviation from a normal distribution were defined to classify genes as repressed (up-regulated in a mutant), activated (down-regulated in a mutant), or unaffected (no substantial change) (Fig. 2, D, E, and F). Approximately 40% of genes classified as activated or repressed in one mutant were classified similarly in the other (Fig. 3A). The observed number of co-regulated genes (1,307 of 6,685 genes in the yeast genome) is much higher than chance expectation and indicates significant overlap in transcriptional regulation. Additionally, considering only genes significantly affected by Mot1 and Spt16, the initial correlation in Fig. 2B increases substantially (r = 0.52). To determine the effects in the double mutant, we analyzed RNA from the mot1-42 spt16-197 double mutant obtained from a cross of the mot1-42 and spt16-197 strains. Because the spt16-197 strain was derived from a different background than the mot1-42 strain, we first computed two sets of differential expression by comparing the double mutant with each parental wild type (WT) strain. The two sets correlated highly, indicating that differences in strain background contribute relatively little to the differential gene expression changes (Fig. 2C). For this reason, subsequent analyses for the double mutant are in comparison with the mot1-42 parental WT strain. We observed that differential expression was similar between single mutants, and in the double mutant these effects were exacerbated (Fig. 3B). The results show that at co-regulated genes Mot1 and Spt16 act in combination to control gene expression.
FIGURE 3.

Mot1 and Spt16 co-regulate gene expression. A, Mot1 and Spt16 co-regulate ∼1,300 genes. The Venn diagrams show the numbers of genes and their overlaps classified as activated (repressed in the mutants; p < 10−65) or repressed (overexpressed in the mutants; p < 10−40). p values were determined by a one-tailed Fisher's exact test. B, comparison of expression in the single and double mutants. The heat map shows log2 median differential expression for 6,685 genes with vertical lines as genes and colored as in the key. Genes regulated by both factors generally showed similar expression changes in the single mutants that were exacerbated in the double mutant. C, validation of gene expression effects at selected co-activated genes. The bar graph shows the relative levels of RNA for each gene relative to the level in the WT strain. The RNA levels for the Mot1 and Spt16 co-activated genes RPS23B, TRX1, LEU9, and ACT1 decreased compared with their levels in WT cells. PCF11 is an unaffected control gene. D, RNA levels for the Mot1 and Spt16 co-repressed genes INO1, GAD1, and GND2. In C and D, error bars show one standard deviation from at least three biological replicates, and asterisks denote p < 0.05 by Student's t test.
To validate the differential effects inferred from the tiling array data and to select specific examples of co-regulated genes for further study, total RNA was isolated from each mutant, and gene-specific RNA levels were measured by RT-qPCR. Consistent with the tiling array data, RPS23B, TRX1, LEU9 and ACT1 all showed decreased expression in the three mutant strains, and most decreases were significant using a p value threshold of 0.05 (Fig. 3C). Three examples of genes identified by the global analysis as repressed by Mot1 and Spt16 were also confirmed by qPCR, and in most cases these were statistically significant (Fig. 3D).
Preferences for Promoter Elements in Mot1 and Spt16 Regulation
Mot1 preferentially represses genes containing the TATA consensus sequence (12, 15, 18, 23, 49) “TATAWAWR” (W = A/T and R = A/G) (50), so we tested whether Spt16 had a similar preference. A preference for regulation of TATA-containing genes by Spt16 would be consistent with the production of cryptic transcripts that initiate preferentially from sites containing a cryptic TATA element in spt16 cells (46). Most promoters in yeast historically classified as lacking a TATA element do contain a short degenerate TATA sequence that underlies the TBP binding location (51). Here, for simplicity, genes with TATA sequences in their promoters are termed “TATA-containing,” and genes without high affinity TBP binding sites are referred to as “TATA-less.” As expected for the mot1-42 data set, TATA-containing promoters were enriched in the repressed gene class, and TATA-less promoters were enriched in the activated gene class (Fig. 4A). Notably, spt16-197 and the double mutant strains had nearly identical enrichment patterns to the mot1-42 strain. In contrast, no such pattern was observed in a deletion strain for Set2, a histone methyltransferase that suppresses cryptic initiation (52), underscoring the significance of the preferential regulation for TATA or TATA-less promoters by both Mot1 and Spt16.
FIGURE 4.

Mot1 and Spt16 have similar preferences for promoter elements but regulate PIC components differently. A, genes are grouped by promoter attribute and proportioned by category of differential expression: co-repressed (yellow), co-activated (blue), or unaffected (gray). The set2Δ data (23) are a control that demonstrates a different relationship of TATA-containing and TATA-less genes to a regulatory factor. p values were calculated using a χ2 test. p values associated with enrichment or depletion of each group of genes are indicated by the symbols (* and #) as defined in the table below the figure. B, to examine effects of Mot1 on TBP levels at co-regulated promoters, relative TBP levels in WT and mutant strains were examined at three co-activated and three co-repressed genes. TBP in mot1-42 cells increased at promoters regardless of promoter attribute, was unaffected in spt16-197 cells, and in the double mutant cells resembled mot1-42 cells. C, TFIIB recruitment was affected in mot1-42 and spt16-197 cells at co-regulated promoters. Changes in TFIIB levels correlated with changes in RNA in each mutant. In B and C, error bars are one standard deviation from at least three biological replicates, and asterisks indicate p < 0.05.
Mot1 and Spt16 Affect Recruitment of PIC Components
Mot1 and Spt16 have been shown to be involved in TBP recruitment to promoters (15, 37, 53), but their functional relationship to PIC assembly at co-regulated promoters has not been reported. Consistent with published ChIP data (17, 20–22), in mot1-42 cells TBP levels increased at five of six Mot1-regulated promoters examined (Fig. 4B). TBP levels were unaffected in spt16-197 cells at all of these co-regulated promoters (Fig. 4B). In the mot1-42 spt16-197 double mutant, TBP levels increased at all co-regulated promoters and to a similar extent as in mot1-42 cells (Fig. 4B). These results suggest that Mot1 influences TBP recruitment at these promoters, whereas Spt16 does not. ChIP was also performed for TFIIB, which associates with TBP-DNA and forms a scaffold for association of Pol II (54). In contrast to TBP, TFIIB levels generally correlated with gene expression changes observed in each mutant (Fig. 4C) (23). These results suggest that the increased TBP occupancy observed at Mot1-activated promoters in mot1-42 cells is indicative of transcriptionally incompetent complexes. The results also suggest that Mot1 and Spt16 facilitate formation of a relatively early promoter-bound intermediate that promotes TFIIB binding but that they do so through distinct biochemical mechanisms with respect to TBP.
Roles for Mot1 and Spt16 in RNA Synthesis Precision
Given the roles for Mot1 and Spt16 in transcription initiation, we next compared their roles in RNA synthesis precision. Using our method described previously (23), tiling array data were mined for RNA length defects defined by four categories: upstream initiation (UI), downstream or cryptic initiation (DI), premature termination (PT), and downstream termination (DT). Fig. 5 shows the relationship between the change in level and precision of gene expression in each mutant strain. Previous studies have shown that characteristic defects in mot1-42 and spt16-197 cells are PT and DI, respectively (23, 46); however, we observed that spt16-197 RNA was highly enriched in all four defect categories relative to mot1-42 RNA, which indicates that Spt16 has a broader role in synthesis precision than reported previously. The mot1-42 spt16-197 double mutant RNA was also enriched in all four categories, and DI and PT were both prominent. The co-prominence of the main defect from each single mutant in the double mutant suggests distinct roles for Mot1 and Spt16 in RNA synthesis precision. Common to all three strains were significant enrichments in two of the four possible initiation/termination couplings (UI/DT and DI/PT), average differential expression near zero for DI and PT, and widely distributed and on average highly elevated differential expression for UI and DT. Despite non-equivalent roles for Mot1 and Spt16 in elongation, certain characteristics of each individual and coupled defect category are the same for both factors, suggesting a shared role that extends beyond the amount of RNA production to how RNA synthesis precision is determined.
FIGURE 5.

Mot1 and Spt16 have distinct and prominent roles in maintaining transcriptional precision. Distributions of differential expression for each length defect category, UI, DI, PT, and DT, are shown. Plot areas are proportional to the number of genes. Pairs of categories with similar distributions and mean differentials overlap significantly in affected genes. UI and DT co-occurred significantly in all mutants as did DI and PT. Asterisks denote p < 0.001 from a one-tailed Fisher's exact test.
Spt16 Affects Mot1 Localization Genome-wide
To better define the functional relationship between Mot1 and Spt16, we next determined the genome-wide localization profile of Mot1 by ChIP-Seq and compared the patterns in WT and spt16-197 cells, focusing on genes whose expression was regulated by both factors. Mot1 distributions at co-activated genes (Fig. 6A) and co-repressed genes (Fig. 6B) were plotted by computationally aligning genes to their transcription start site (TSS). On average, Mot1 ChIP signal was highest in the 5′ regulatory region as expected (9, 15, 18, 21). At both classes of genes, promoter-localized Mot1 signals decreased in spt16-197 cells compared with WT with the largest effect occurring at co-activated gene promoters. Mot1 ChIP-Seq signal was also higher at co-activated genes than co-repressed genes (Fig. 6, black lines), consistent with published results showing that Mot1 signal in general scales with gene expression level (21, 55). The global effect of Spt16 on the Mot1 profile supports a broad role for Spt16 in establishing the proper chromatin environment for Mot1 binding. Mutation of Spt16 also resulted in a decrease in Mot1 signal downstream of the TSS of co-activated genes (Fig. 6A, red line) and an increase at co-repressed genes (Fig. 6B, red line). Possible explanations for these differences are discussed below.
FIGURE 6.

Effect of Spt16 mutation on gene average localization of Mot1. A, Mot1 ChIP-Seq signal was determined as log2 -fold change from two replicates relative to an input control. Genes were computationally aligned by TSS (0 bp) to determine average localization in WT (black) and spt16-197 (red) strains. In spt16-197 cells, Mot1 ChIP signal decreased most dramatically in the promoter region (∼−300 to 0 bp), consistent with decreased expression of the Mot1 and Spt16 activated gene class. There were also changes in Mot1 signal upstream and downstream of the promoter region in the absence of Spt16, suggesting that Spt16 establishes a chromatin environment important for Mot1 binding. B, plot similar to that in A showing the Mot1 localization at Mot1 and Spt16 co-repressed genes. Mot1 ChIP signal increased in the ORF in the absence of Spt16, potentially due to exposed adventitious binding sites for TBP (and hence Mot1). In WT cells and for both classes of genes, Mot1 was notably enriched in the promoter region as expected. In A and B, the S.E. is shown by the transparent shading around the lines.
Mot1 Affects Spt16 Localization
For comparison with the Mot1 ChIP-Seq data just described, we performed ChIP-Seq for Spt16 in WT and mot1-42 cells. Spt16 profiles were plotted for Mot1-Spt16 co-activated genes (Fig. 7A) and co-repressed genes (Fig. 7B). Average Spt16 localization differed from Mot1 with enrichment throughout gene bodies and depletion from promoter regions. Like Mot1, Spt16 levels increased at co-activated genes and decreased at co-repressed genes in mot1-42 cells compared with WT (Fig. 7, A and B). The relative enrichment of Spt16 at co-activated compared with co-repressed genes in WT cells suggests that Spt16 directly activates its target genes, whereas Spt16-mediated repression may result from indirect effects. Spt16 enrichment in gene bodies is consistent with published work as expected for a factor with key roles in elongation (25, 41–46). Global trends in Spt16 enrichment inferred from ChIP-Seq data were validated for seven genes by locus-specific ChIP and qPCR (Fig. 7C). Notably, Mot1 was required for full Spt16 recruitment to co-activated promoters, and Mot1 affected recruitment of Spt16 to two of three co-repressed promoters. From these results, it is unclear whether Mot1 plays a direct role in Spt16 recruitment or whether Spt16 occupancy is linked to gene activity as dictated by Pol II density.
FIGURE 7.

Gene average chromatin localization of Spt16 and dependence on Mot1. A, average Spt16 ChIP-Seq signal in WT (black) and mot1-42 (blue) strains. At Mot1 and Spt16 co-activated genes, Spt16 was depleted from regions upstream of the TSS (0 bp) but was enriched in flanking transcribed regions. Spt16 levels decreased in the ORFs in mot1-42 cells, consistent with the decreased expression of these genes. B, in mot1-42 cells, Spt16 signal increased in the transcribed regions of co-repressed genes, consistent with their increased expression. In A and B, the S.E. is shown by the transparent shading around the lines. C, Spt16 ChIP at selected co-regulated genes. In mot1-42 cells, Spt16 ChIP signal decreased at co-activated genes and increased at co-repressed genes, consistent with changes observed in A and B and RNA levels (Fig. 3, C and D). Error bars are one standard deviation from the mean from at least three biological replicates, and asterisks denote p < 0.05.
Effects of Mot1 on RNA Polymerase II Levels
To better understand the relationship between Mot1 and Spt16 in transcription initiation and elongation, Pol II levels were measured by ChIP at a subset of co-activated and co-repressed genes. As shown in Fig. 8A, Pol II levels were affected in mot1-42 versus WT cells as expected based on the changes we observed in RNA levels at these same genes. Changes in Pol II levels were statistically significant and quantitatively in line with the relatively modest changes in gene expression (Fig. 3). A larger difference in Pol II was detectable at the GAL10 ORF during galactose induction (Fig. 8B), indicating that these modest changes are not due to insensitivity in the ChIP assay. Combined with the effects on RNA synthesis precision described above, although Mot1 and Spt16 are essential global transcriptional regulators, they appear to have primary roles in fine-tuning gene expression rather than being absolutely required for RNA synthesis. However, as cells have the capacity to buffer effects on key regulators of transcription by modifying the RNA decay rate (56–58), it may be that the analysis of steady-state RNA reported here underestimates the magnitude of the effects of Mot1 and Spt16 on RNA synthesis.
FIGURE 8.
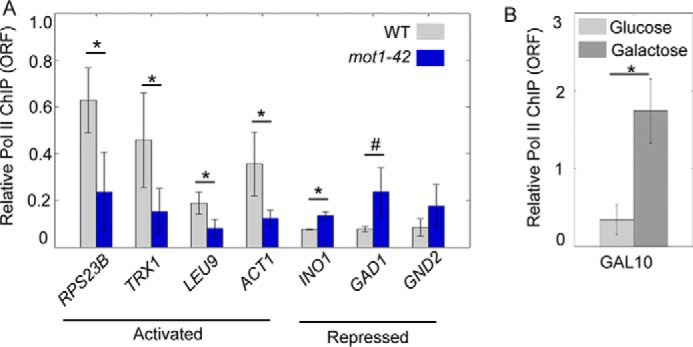
RNA polymerase II levels correlate with expression changes. A, ChIP of Pol II in the open reading frames of the indicated genes was performed using WT (gray) and mot1-42 cells (blue). Error bars indicate one standard deviation of at least three biological replicates. B, ChIP of Pol II in the ORF of GAL10 was performed in WT cells grown in glucose (light gray) and galactose (dark gray). Error bars are standard deviations from two biological replicates. In A and B, the asterisk indicates p < 0.05 from a Student's t test. The # indicates p = 0.055.
Roles for Mot1 and Spt16 in Nucleosome Organization
Next, we used micrococcal nuclease (MNase) ChIP-Seq to define the chromatin landscape in WT and mutant cells. Mononucleosomes were obtained by MNase digestion followed by histone H3 immunoprecipitation, DNA isolation, and high throughput sequencing as described previously (see “Experimental Procedures” and Ref. 59). As shown in Fig. 9A (green line), in an average plot of all genes in WT cells, we observed a nucleosome-depleted promoter region flanked by peaks of nucleosomal density. As expected, nucleosomal peaks were most precisely defined proximal to the TSS and became progressively less well defined within the gene body and moving away from the TSS (60–62). Nucleosome density and periodicity were diminished in spt16-197 cells, consistent with prior results implicating Spt16 in establishing chromatin structure on a global scale (32, 33, 39–41, 45, 46). In addition, nucleosome levels were higher in nucleosome-depleted promoter regions in spt16-197 cells than in WT cells. Chromatin structural changes in mot1-42 cells were more defined: loss of Mot1 activity resulted in decreased intensity of the +1 and +2 nucleosomes, but other nucleosomes in the transcribed region were largely unaffected. As discussed below, the positions of the +1 and +2 nucleosomes appear shifted in mot1-42 cells compared with their average positions in WT cells, and promoter regions show somewhat more pronounced nucleosomal depletion in cells lacking Mot1. Nucleosome organization in mot1-42 spt16-197 cells was more severely perturbed than in either single mutant with an overall pattern similar to spt16-197 cells but with a greater loss of nucleosome density, particularly at the 5′ ends of genes. Nucleosome density in promoter regions was restored to near WT levels in the double mutant compared with spt16-197 cells as well.
FIGURE 9.
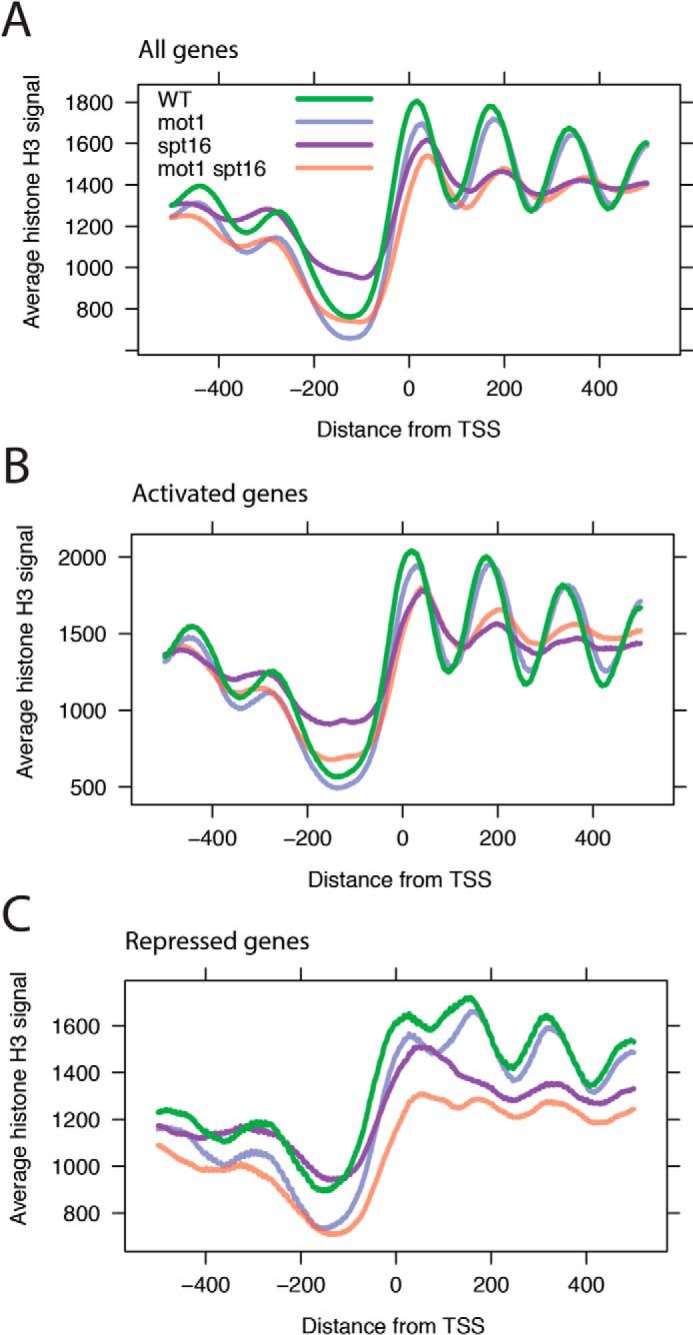
Effects of Mot1 and Spt16 on nucleosomal organization. A, gene average MNase ChIP-Seq read density for all genes aligned by TSS (0 bp). Data are plotted for each of the four strains as indicated in the legend. Of note are the depletion of reads in the promoter region immediately upstream (left) of the TSS and the periodicity in the signal for WT cells (green) emanating downstream (right) of the TSS indicative of positioned nucleosomes and the loss of this periodicity in spt16-197 and double mutant cells. In mot1-42 cells, the features of the genic nucleosomal array are still present, but the first two nucleosomal peaks are reduced and slightly shifted rightward into the gene body. The −1 and −2 nucleosomes immediately upstream of the promoter region were also reduced on average compared with WT. B, plot as in A but for all co-activated genes. C, plot as in A but for all co-repressed genes.
Similar trends for each mutant were observed at co-activated genes (Fig. 9B) compared with the all-gene average. Mot1 exerted effects on the nucleosomes proximal to the TSS, and there were more widespread effects on nucleosome structure at co-activated genes in spt16-197 and double mutant strains. At co-repressed genes (Fig. 9C), mutation of Mot1 led to diminished nucleosome occupancies at the 5′ ends of genes, there was global disruption of nucleosome structure in spt16-197 cells, and the most severe effects were observed in mot1-42 spt16-197 cells. These effects are consistent with nucleosome disruption accompanying increased expression of these genes in the mutants. However, there were trends at co-repressed genes that were markedly different from patterns at other classes of genes as well. Comparison of the mot1-42 pattern in Fig. 9C with A and B shows that nucleosome depletion at the promoter is attributable primarily to repressed genes. For spt16-197, elevated histone H3 levels at the promoter specifically were not observed at repressed genes. For all strains, there is a less pronounced trough between the +1 and +2 peaks, which may be related to chromatin organization that occurs with up-regulation. Additionally, although mutation of Mot1 by itself mainly affected the +1 and +2 nucleosomes regardless of gene expression outcome, comparison of spt16-197 with the double mutant reveals a more expansive co-regulatory role for Mot1 in gene repression with effects on nucleosome occupancy that extend at least several hundred base pairs in both directions from the TSS.
To better understand the nature of nucleosome changes that gave rise to the different patterns in Fig. 9, A–C, a computational approach (60, 63) was applied to identify nucleosomal peaks that show statistically significant changes in occupancy versus positioning (also known as fuzziness; see Refs. 60, 64, and 65). Occupancy was defined by the height of the MNase ChIP-Seq peak, whereas fuzziness refers to an estimate of the width of the peak. The statistically significant (false discovery rate-corrected p values <0.05) differential nucleosome effects observed in each mutant strain compared with WT are summarized in Table 1. In general, those nucleosomes with significant changes in occupancy showed decreased occupancy in a given mutant compared with the WT strain. In addition, there were more changes in fuzziness than occupancy with most affected nucleosomes having increased fuzziness in the mutant compared with WT. As shown in Fig. 10A, the overlaps in nucleosomes showing differential occupancy and fuzziness were statistically significant with the greatest overlaps occurring between differentially fuzzy nucleosomes in spt16-197 and the double mutant and in agreement with their similar nucleosomal patterns shown in Fig. 9.
TABLE 1.
Effects on nucleosomes in mutants compared with wild type
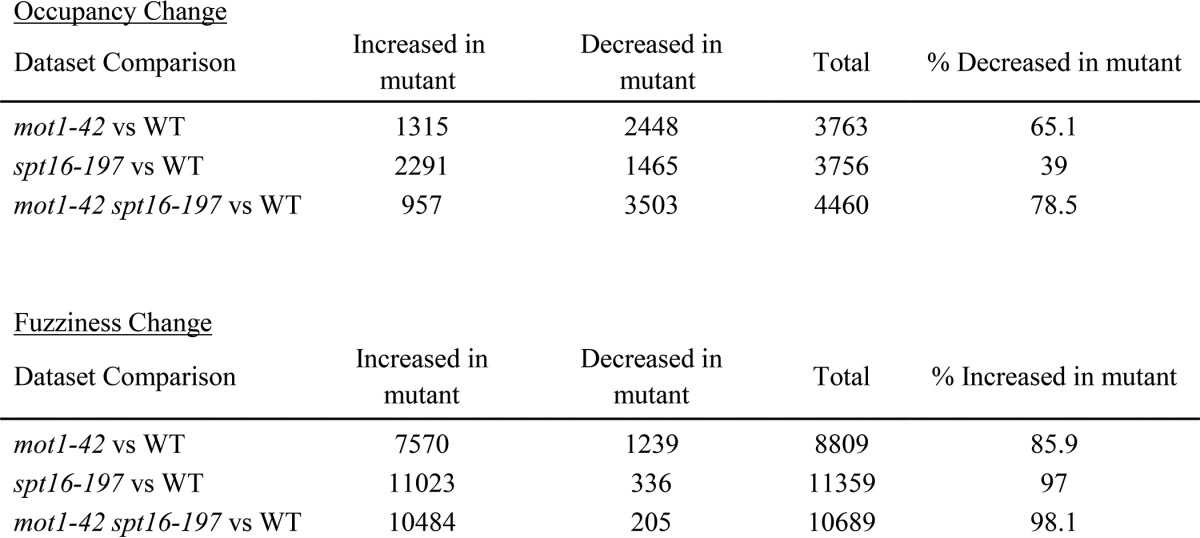
FIGURE 10.

Overlapping sets of nucleosomes are affected in mutant strains. A, the table shows the overlap in sets of nucleosomes affected in mot1-42, spt16-197, and double mutant cells. False discovery rate-corrected p values are <0.007 (*) and <2.6 × 10−196 (**). B, differentially fuzzy nucleosomes in mutant strains were analyzed for position with respect to the TSS. Analysis of the results in Fig. 9 established that the +1 nucleosome was located from approximately −50 to +100 bp with respect to the TSS. Nucleosomes downstream of the +1 nucleosome were assigned to non-overlapping 150-bp bins based on the +1 nucleosome position. The distance from the nucleosome dyad to the TSS was then determined for each nucleosome and in each strain. The plot shows the distribution of median differences in position with respect to the TSS for each nucleosome in the indicated mutant compared with WT. The +1, +2, and +3 nucleosomes in mot1-42 (blue), spt16-197 (red), and mot1-42 spt16-197 (green) cells had significant shifts away from the TSS. The −1, +4, and +5 nucleosomes all shifted away from the TSS in spt16-197 and double mutant cells. Asterisks indicate p < 0.05 as determined by a Kolmogorov-Smirnov test. NDR, nucleosome-depleted region.
Mot1 and Spt16 Affect the Position of the +1 Nucleosome
In addition to affecting occupancy and fuzziness, it appeared from the gene average plots in Fig. 9 that loss of one or both of these factors shifted the median position of promoter-proximal nucleosomes with respect to the TSS. Such an effect has been reported previously for Spt16 (65). Nucleosomal peaks were canonically designated −1, +1, +2, etc. based on the distance of the nucleosome to the TSS. Nucleosomal dyad positions were tabulated for each differentially fuzzy nucleosome, and position shifts were calculated from the difference in dyad position for each nucleosome in the mutant compared with WT. Position shift patterns for spt16-197 cells were similar to published data (65) with the largest (+10-bp) shift at the +1 nucleosome. A +10-bp median shift at the +1 nucleosome was also observed in mot1-42 cells and the double mutant, which demonstrates that the two factors play non-redundant roles in positioning the +1 nucleosome properly (Fig. 10B). Nucleosomes at +2 and +3 positions also showed significant position shifts in all three mutants as did +4 and +5 nucleosomes in spt16-197 cells, consistent with the loss of nucleosome organization in this mutant and the double mutant.
Discussion
Mot1 and Spt16 Co-Regulate Transcription
Here we report a functional overlap in transcriptional control between Mot1 and Spt16 that operates on a global scale with ∼1,300 genes controlled by both factors. Co-regulated genes have similar differential expression patterns in the single mutant strains, and in the double mutant these effects tend to be exacerbated. The physical and genetic interactions between Mot1 and Spt16 further suggest that these two factors function together in vivo. This relationship is particularly interesting because they have distinct genome-wide localizations (Figs. 6 and 7). Spt16 is broadly distributed on the transcribed regions of genes but notably depleted in promoter regions where Mot1 is enriched. The localization of Spt16 is distinct from the localization of other elongation factors such as Spt6 and Bur1 (66). Mutation of Mot1 led to redistribution of Spt16 in accordance with changes in expression, most likely due to changes in Pol II levels and the requirement for Spt16 during elongation. It is interesting that mutation of Spt16 decreased Mot1 localization at co-regulated promoters regardless of whether expression was positively or negatively affected. This suggests that although Spt16 is not localized to promoters it is nonetheless required to establish the chromatin environment responsible for the genome-wide distribution of Mot1.
Although they are found at distinct locations in the genome, the physical interaction between Mot1 and Spt16 in extracts suggests how their activities may be coordinated. The physical interaction with Mot1 may increase the local concentration of Spt16 near gene start sites, allowing Spt16 to more efficiently act on promoter-proximal nucleosomes and/or to be handed off to Pol II during the early stages of elongation (25, 42, 43). Although Mot1 and Spt16 associate with one another in extracts, this may not be a direct interaction, and indeed, an interaction mediated by other regulatory factors could help explain how the activities of these factors are coordinated when they are largely found at different chromosomal locations. Regardless of the nature of their physical association, such a mechanism would be consistent with previous observations of Spt16 localization within or near promoters and affecting nucleosome occupancy to properly initiate transcription (37, 66–68). The synthetic genetic interaction observed between Mot1 and Spt16 is consistent with the observation that mutations in Mot1 are synthetically lethal with deletions of the FACT subunit genes NHP6A and NHP6B (69). Overall, these synthetic effects strongly suggest that Mot1 and the entire FACT complex function together to coordinate PIC assembly and nucleosome organization.
Although the results presented here demonstrate broad overlap in the set of genes controlled by Mot1 and the set controlled by Spt16, there are many genes apparently controlled by only one or the other as well (Fig. 3A). The transcriptome-wide overlap that we observe is limited by the achievable statistical power typical of such studies; allele specificity, growth conditions, and other technical issues may also have led to an underestimate of the functional overlap of Mot1 and Spt16. Conversely, we would expect that, given the complexity in the chromatin landscape and in gene regulatory mechanisms, not all affected genes require both factors for WT expression.
Regulation of TBP
A previous study (37) found that Spt16 was important for establishing the levels of TBP at the SER3 and ELP3 promoters, so it is possible that Spt16 activity impacts TBP directly at some promoters but not all. In our analysis, we did not detect any effect on TBP promoter localization in spt16-197 cells, but increased TBP levels were detected at co-regulated promoters in mot1-42 and mot1-42 spt16-197 cells. This is consistent with the global role of Mot1 in limiting TBP binding to chromatin that we reported previously (23). In contrast, both Mot1 and Spt16 apparently control TFIIB levels at promoters, which are in general better correlated with gene expression levels than TBP (23). Because mutation of Spt16 promotes cryptic initiation (45, 46), the genome-wide redistribution of Mot1 in spt16-197 cells reported here is consistent with Mot1 tracking the presumed redistribution of TBP to DNA sites exposed as a result of the global perturbation of chromatin integrity that occurs when Spt16 activity is compromised.
Mot1 and Spt16 Have Roles in Elongation
Mutations in Mot1 and Spt16 gave rise to prominent but distinct RNA length defects, and the double mutant showed the combination of defects present in the single mutants. These results suggest that Mot1 and Spt16 have distinct roles in elongation. However, they do show similar associations between length precision and differential expression. In particular, both factors overlap in suppressing upstream initiation and downstream termination at co-repressed genes (Fig. 5). One conundrum is why Mot1 and Spt16 are essential when their effects on steady-state RNA levels, although affecting many genes, are relatively small in magnitude. As mentioned above, changes in transcription may be underestimated as a result of the cellular capacity to buffer RNA turnover rates in the mutants. However, the large numbers of RNA length defects observed in the mot1-42 and spt16-197 strains suggest that the essential roles of these factors may be more intimately related to ensuring proper start site and termination site selection rather than driving abundance of proper RNA species per se.
Mot1 and Spt16 Globally Affect Nucleosome Organization
Spt16 has been widely studied as a histone chaperone, but the role of Mot1 in nucleosomal organization had not been established. Mot1 was required for nucleosome remodeling at the induced GAL1 promoter (19), but the chromatin organization at the Mot1-activated URA1 promoter was unaffected by a mutation in Mot1 (20). A direct role for Mot1 in chromatin remodeling is attractive as most other members of the Swi2/Snf2 enzyme family are ATP-dependent chromatin remodelers (70–72). The results presented here implicate Mot1 in determining the occupancy and position of the +1 and +2 nucleosomes. It remains to be determined precisely how these nucleosomal changes arise, but as they were observed at genes regardless of the effect of Mot1 on expression, it is unlikely that these changes are entirely an indirect result of Mot1-mediated effects on transcription via interaction of Mot1 with TBP. The main roles for Mot1 in chromatin structure involve positioning and fuzziness of the +1 and +2 nucleosomes. These nucleosomes tended to be less precisely positioned (more fuzzy) in mot1-42 cells than WT, and the +1 and +2 nucleosomes had a median position shift of 10 bp away from the TSS. A shift was also apparent in spt16-197 cells and the double mutant, indicating that both factors contribute to establishing and/or maintaining this important aspect of the native chromatin state.
Experimental Procedures
Yeast Strains and Growth Conditions
Saccharomyces cerevisiae strains used in this study are listed in Table 2. The WT strain AY51 is referred to as MOT1-WT because it was used to generate the mot1-42 (AY87) strain; these strains were described previously (73, 74). FY56 is referred to as SPT16-WT because it was used to generate the spt16-197 strain (L577); both of these strains were provided by Fred Winston (39). The mot1-42 spt16-197 double mutant was constructed by mating the spt16-197 strain with a MOT1 shuffling strain (AY138). Diploids were sporulated, and tetrads were dissected and screened for temperature sensitivity and resistance to kanamycin. Candidates from the screen were then transformed with plasmid pMOT221 (74) containing the mot1-42 allele, and the MOT1-WT plasmid was shuffled out by plating on synthetic complete medium without leucine and containing 5-fluoroorotic acid. MOT1-WT, mot1-42, SPT16-WT, and spt16-197 cells were all grown at 30 °C in YPD (yeast extract, peptone, dextrose), whereas mot1-42 spt16-197 cells were grown at 25 °C in YPD. For the RNA isolation, ChIP, ChIP-Seq, and MNase ChIP-Seq experiments, all strains were grown at their permissive temperatures to an optical density (OD) of ∼1.0; heat-shocked with addition of an equal volume of 42 °C prewarmed YPD; and incubated at 35 °C for 45 min. For spot assays, strains were grown to an OD of ∼1.0 at their permissive temperatures, and then 10-fold serial dilutions were plated on YPD and incubated at 25, 30, or 35 °C for 3 days.
TABLE 2.
Yeast strains used in this study
| Strain name | Genotype | Ref. |
|---|---|---|
| YPH499 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1 | 73 |
| AY51 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1. mot1Δ::TRP1 pAV20(EE-MOT1, LEU2, CEN ARS) | 20 |
| AY87 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1. mot1Δ::TRP1 pMot221(mot1-42, LEU2, CEN ARS) | 20 |
| AY138 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1. mot1Δ::kanMX pMR13(MOT1, URA3, CEN ARS) | 82 |
| FY56 | MATα, his4-912δ, lys2-128δ, ura3-52 | 39 |
| L577 | MATα, his4-912δ, lys2-128δ, ura3-52, spt16-197 | 39 |
| YMW054 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1, mot1Δ::kanMX, SPT16-13xMyc(HIS3), pAV20(EE-MOT1, LEU2, CEN ARS) | This study |
| YMW055 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1, mot1Δ::kanMX, SPT16-13xMyc(HIS3), pMot221(mot1-42, LEU2, CEN ARS) | This study |
| YMW066 | MATa, ura3-52, trp1-Δ63, his3-Δ200, spt16-197, mot1Δ::kanMX, pMOT221(mot1–42, LEU2, CEN ARS) | This study |
| YMW070 | MATa, ura3-52, lys2-801a, ade2-101o, trp1-Δ63, his3-Δ200, leu2-Δ1, MOT1-13xMyc(HIS3), SPT16-HA(TRP1) | This study |
Co-immunoprecipitation
Yeast strains were grown as above to an OD of ∼1.0, and cells were then collected and lysed in BA/150 lysis buffer (20 mm HEPES-KOH, pH 7.6, 2 mm EDTA, 2 mm EGTA, 0.25% Nonidet P-40, 150 mm potassium acetate, 5 mm DTT, and a protease inhibitor mixture tablet (one tablet/25 ml of buffer) (Roche Applied Science)). A total of 1 mg of protein was immunoprecipitated overnight at 4 °C with either 15 μl of α-myc (9E10, 1 mg/ml) or 2.5 μl α-Spt16 (rabbit polyclonal antibody from Tim Formosa). Immunoprecipitated samples were incubated with Protein A-Sepharose beads (GE Healthcare) and washed with BA/250 lysis buffer (same as BA/150 lysis buffer but with 250 mm potassium acetate). Samples were separated on 4–15% precast Bio-Rad SDS-polyacrylamide gels. After transfer to nitrocellulose membranes, samples were blocked in Tris-buffered saline with Tween (50 mm Tris HCl, pH 7.6, 150 mm NaCl, 0.05% Tween 20) plus 5% nonfat milk, then incubated with either α-myc (9E10; 1 mg/ml), α-HA (12CA5, Abcam), or α-Spt16 (rabbit polyclonal antibody) followed by incubation with secondary antibody, and developed using the ECL Prime kit (GE Healthcare).
RNA Isolation for Tiling Array Analysis and RT-PCR
Strains were grown as above in duplicate for tiling array analysis and in triplicate for RT-PCR. Total RNA was isolated, and samples were prepared for tiling array analysis as described previously (23). The University of Virginia Microarray Core Facility hybridized the samples to S. cerevisiae 1.0 Tiling Arrays (Affymetrix) and generated the raw data. After the total RNA was isolated for RT-qPCR analysis, the RNA was reverse transcribed with the iScript Select cDNA Synthesis kit (Bio-Rad) according to the manufacturer's instructions with the exception that twice the amount of starting RNA and reagents were used. Real time PCR was carried out as described previously (23).
Tiling Array Data Analysis
Integrated Genome Browser-compatible files were produced from the raw tiling array data using Tiling Analysis Software. Signal profiles were smoothed over a 50-bp bandwidth (101-bp sliding window), and differential profiles were smoothed over a 250-bp bandwidth (501-bp sliding window). Additional Tiling Analysis Software parameters were as follows: log2 signal scale, scale to target intensity of 100, two-sided test type, plus or minus 0.3 threshold, 100-bp maximum gap, and 50-bp minimum run. The Mot1 differential signal was generated by subtracting the mot1-42 signal (log2 scale) by the MOT1-WT signal. The Spt16 differential signal was generated by subtracting the spt16-197 signal by the SPT16-WT signal. Two double mutant differentials were generated by subtracting the mot1-42 spt16-197 signal by each of the two WT signals. Two types of analysis were conducted with the differential profiles: unbiased and biased. The unbiased analysis considered genome-wide differential expression, whereas the biased analysis considered only the 6,685 annotated genes. For the biased analysis, median differential expression was calculated for each gene, and for the 309 intron-containing genes only the longest exon was considered. Because each mutant produces different magnitudes of changes in transcript levels, different thresholds were applied to each differential profile for classifying genes as underexpressed or overexpressed. These thresholds were determined by comparing the distributions of median differential expression with a normal distribution and identifying the levels above or below which median gene expression deviated from the normal distribution using quantile-quantile plots. Because both WT strains had relatively high correlations between the data sets (r = 0.74), further analyses for the single and double mutants were conducted using comparisons with MOT1-WT.
Chromatin Immunoprecipitation
Yeast strains were grown as described above. After cultures reached an OD of ∼1.0 and were heat-shocked, cells were collected, lysed, and sonicated as described previously (20). Immunoprecipitations were conducted using 1 mg of protein as described previously with either 2.5 μl of α-TFIIB (Sua7 rabbit polyclonal), 5 μl of α-TBP (58C9, Abcam), 5 μl of α-myc (9E10; 1 mg/ml), or 5 μl of α-Pol II (8WG16, Covance). After immunoprecipitation and cross-link reversal, DNA was purified using a Qiagen PCR purification kit according to the manufacturer's instructions. Purified DNA was then quantified by qPCR using the oligonucleotides (Life Technologies) listed in Table 3 and SYBR Green Master Mix (Bio-Rad). The mock values were subtracted from the IP values and then normalized to the input.
TABLE 3.
Primer sequences used in this study
| Primer name | Sequence |
|---|---|
| RPS23B Promoter F | 5′-TGC TAA GCA CTA CCG CAT TG-3′ |
| RPS23B Promoter R | 5′-GAA AGC GTG GAG ACA AGG AG-3′ |
| TRX1 Promoter F | 5′-CCA AAA CCC TGA AAC TGC AT-3′ |
| TRX1 Promoter R | 5′-ATT CGC TGG CAG TTT TGA AT-3′ |
| ACT1 Promoter F | 5′-CAG CTT TTA GAT TTT TCA CGC TTA-3′ |
| ACT1 Promoter R | 5′-TTT TCG ATC TTG GGA AGA AAA A-3′ |
| INO1 Promoter F | 5′-GTT GGC GGC AAT GTT AAT TT-3′ |
| INO1 Promoter R | 5′-CGA CAA CAG AAC AAG CCA AA-3′ |
| GAD1 Promoter F | 5′-CAC TGA ACT GCA ACG CAC TC-3′ |
| GAD1 Promoter R | 5′-TTT TAG CAT CGC CAA AAG GT-3′ |
| GND2 Promoter F | 5′-CGT CAG AAA TTG AAC GTT TCC-3′ |
| GND2 Promoter R | 5′-GGC ACT CGT GGT TAA AGA GC-3′ |
| RPS23B ORF F | 5′-GAA CCA CCG AAT GGA GAA GA-3′ |
| RPS23B ORF R | 5′-GTT GGG CCG AAA ACA ACT AT-3′ |
| TRX1 ORF F | 5′-GAA GTT GCA AAG GTT GTT GG-3′ |
| TRX1 ORF R | 5′-TTA GCA GCA ATG GCT TGC TT-3′ |
| LEU9 ORF F | 5′-AGA AAT TGA ACC CAG AGC GT-3′ |
| LEU9 ORF R | 5′-TTC AGT AAA TTG GAT AGC GCA-3′ |
| ACT1 ORF F | 5′-GCA AAA GGA AAT CAC CGC TT-3′ |
| ACT1 ORF R | 5′-AAG CCA AGA TAG AAC CAC CAA-3′ |
| PCF11 ORF F | 5′-GGT TGA ATC CTA ATG ACA CCG-3′ |
| PCF11 ORF R | 5′-TTG CTT GCA GGT TTT TCT GG-3′ |
| INO1 ORF F | 5′-GGC TGA GCA TGA GGG TAC AT-3′ |
| INO1 ORF R | 5′-CAA CTT GGT TTG TCC CGA CT-3′ |
| GAD1 ORF F | 5′-AAC GGA TGG ATC CGA TGA GAA-3′ |
| GAD1 ORF R | 5′-CAT CTC CGA TCT GAA AAC CA-3′ |
| GND2 ORF F | 5′-AAT GTG GAG AGG TGG CTG TAT-3′ |
| GND2 ORF R | 5′-GAA GCG AAG AAC TCG TTG AA-3′ |
| GAL10 ORF F | 5′-GCA GCC CTG CAA TAC CTA GA-3′ |
| GAL10 ORF R | 5′-TTC CAC TCA CGA CAC AAA CC-3′ |
Analysis of RNA Length Precision
Categories of RNA length defects were assigned to genes using our previously published method (23). Intron-containing genes were excluded from the analysis. For the spt16-197 and mot1-42 spt16-197 differential RNA signals compared with WT, the signal cutoff was raised from 0.30 to 0.36 to account for overall greater magnitudes of differential expression and to reduce the likelihood of calling false positives. The other parameters were held constant.
ChIP-Seq
Immunoprecipitated DNA was prepared as above for ChIP. The resulting purified DNA was then processed using the TruSeq ChIP Library Prep kit from Illumina (IP-202-1012) according to the manufacturer's instructions. Briefly, DNA fragments were end-repaired and adenylated, adapter 6 was ligated onto the adenylated ends and gel-purified, and purified products were enriched and quantified via Qubit. The DNA samples were then analyzed using a Bioanalyzer to ensure that the samples mostly contained a single peak of mononucleosomal DNA. A total of 0.2 pmol of DNA was sequenced by the University of Virginia Sequencing Core using an Illumina MiSeq instrument, yielding 25.6 ± 1.6 million 150-bp raw reads per sample. Data were obtained from two independent replicate samples for each strain.
MNase ChIP-Seq
MNase ChIP-Seq was performed as described previously with minor modifications (59). Strains were grown as described above. After collection, cells were lysed as described above for ChIP. MNase titrations were performed, and samples with the highest proportion of mononucleosomes and without subnucleosomal fragments were selected for each sample. Cross-links were reversed with heat, and protein was digested with proteinase K (Thermo Fisher). Samples were then immunoprecipitated with 10 μg of α-H3 (ab1791, Abcam) overnight at 4 °C, then incubated with Protein A-Sepharose, washed, and eluted as described previously. The immunoprecipitated material was prepared for sequencing as described above, and sequencing data were obtained using the Illumina MiSeq instrument in the University of Virginia Sequencing Core, yielding 27.9 ± 5.7 million 150-bp raw reads per sample. Data were obtained from two independent replicate samples for each strain.
ChIP-Seq and MNase ChIP-Seq Data Analysis
Reads were aligned to the SacCer3 reference genome using Bowtie 2 2.2.6 with default mapping settings (75). More than 95% of the raw reads from each data set were mapped under these conditions. Unmapped reads and reads with mapping quality scores <30 were removed from the Bowtie 2-generated bam file using SAMtools 0.1.19 (76). This mapping quality threshold resulted in removal of 10% or fewer of the mapped reads. For viewing the data in Integrative Genomics Viewer (IGV) (77, 78), bam files were converted to BigWig by first sorting and indexing using SAMtools, then creating .bed and .cov files using BEDTools 2.18.2 (79), and finally converting the .cov files to BigWig using the bedGraphToBigWig converter (80). Gene average plots were produced using ngs.plot 2.4.7 (81). Nucleosome mapping and differential nucleosome analyses were performed using the dpos tool in the DANPOS 2.2.2 package with default settings that include -fold change normalization (multiplying each sample by a scale factor to account for differences in mapped read coverage) rather than quantile normalization as originally described (63). dpos yielded normalized wig files of the processed data. We observed that the total signal nonetheless varied among these normalized data sets by a small (∼5%) but not insignificant amount. For this reason, the dpos-generated wig data files were further normalized globally based on the total signal. The gene average plots shown in Fig. 9 were generated in R from the renormalized wig data by computing the mean signal at base pair resolution for all genes, activated genes, or repressed genes (as indicated) with respect to the TSS. Other statistical analyses and data visualization were performed using R v3.0.2. All of the genome-wide data sets from this study have been submitted to the NCBI Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) under SuperSeries accession number GSE80235.
Author Contributions
J. D. T., J. J. M., M. N. C., and S. J. S. conducted the experiments. J. D. T., J. J. M., and D. T. A. performed the data analysis. J. D. T., J. J. M., and D. T. A. wrote the manuscript. K. P. and S. B. assisted with the data analysis.
Acknowledgments
We thank Mike Guertin for discussions, advice, and sharing code for MNase ChIP-Seq data analysis. We are thankful to Fred Winston for providing yeast strains; Tim Formosa for the Spt16 antiserum; Patrick Grant for input and advice about MNase ChIP-Seq; and Jeff Smith, Patrick Grant, and Elizabeth Hoffman for critical reading of the manuscript.
This work was supported in part by National Institutes of Health Grant GM55763 (to D. T. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ChIP-Seq, MNase ChIP-Seq, and RNA tiling array data are accessible via Gene Expression Omnibus (GEO) SuperSeries accession number GSE80235.
- Mot1
- modifier of transcription 1
- TBP
- TATA-binding protein
- PIC
- preinitiation complex
- Pol II
- RNA polymerase II
- UI
- upstream initiation
- DI
- downstream initiation
- PT
- premature termination
- DT
- downstream termination
- MNase
- micrococcal nuclease
- TSS
- transcription start site
- FACT
- facilitates chromatin transcription
- qPCR
- quantitative PCR
- TF
- transcription factor
- Seq
- sequencing
- IP
- immunoprecipitation
- YPD
- yeast extract, peptone, dextrose.
References
- 1. Davis J. L., Kunisawa R., and Thorner J. (1992) A presumptive helicase (MOT1 gene product) affects gene expression and is required for viability in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 12, 1879–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piatti S., Tazzi R., Pizzagalli A., Plevani P., and Lucchini G. (1992) Control of DNA synthesis genes in budding yeast: involvement of the transcriptional modulator MOT1 in the expression of the DNA polymerase α gene. Chromosoma 102, S107–S113 [DOI] [PubMed] [Google Scholar]
- 3. Karnitz L., Morrison M., and Young E. T. (1992) Identification and characterization of three genes that affect expression of ADH2 in Saccharomyces cerevisiae. Genetics 132, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munn A. L., and Riezman H. (1994) Endocytosis is required for the growth of vacuolar H+-ATPase-defective yeast: identification of six new END genes. J. Cell Biol. 127, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldman-Levi R., Miller C., Bogoch J., and Zak N. B. (1996) Expanding the Mot1 subfamily: 89B helicase encodes a new Drosophila melanogaster SNF2-related protein which binds to multiple sites on polytene chromosomes. Nucleic Acids Res. 24, 3121–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang Y. W., and Stillman D. J. (1996) Epigenetic effects on yeast transcription caused by mutations in an actin-related protein present in the nucleus. Genes Dev. 10, 604–619 [DOI] [PubMed] [Google Scholar]
- 7. Prelich G. (1997) Saccharomyces cerevisiae BUR6 encodes a DRAP1/NC2α homolog that has both positive and negative roles in transcription in vivo. Mol. Cell. Biol. 17, 2057–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Timmers H. T., Meyers R. E., Sharp P. A. (1992) Composition of transcription factor B-TFIID. Proc. Natl. Acad. Sci. U.S.A. 89, 8140–8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Auble D. T., and Hahn S. (1993) An ATP-dependent inhibitor of TBP binding to DNA. Genes Dev. 7, 844–856 [DOI] [PubMed] [Google Scholar]
- 10. Auble D. T., Hansen K. E., Mueller C. G., Lane W. S., Thorner J., and Hahn S. (1994) Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev. 8, 1920–1934 [DOI] [PubMed] [Google Scholar]
- 11. Adamkewicz J. I., Mueller C. G., Hansen K. E., Prud'homme W. A., and Thorner J. (2000) Purification and enzymic properties of Mot1 ATPase, a regulator of basal transcription in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 275, 21158–21168 [DOI] [PubMed] [Google Scholar]
- 12. Collart M. (1996) The NOT, SPT3, and MOT1 genes functionally interact to regulate transcription at core promoters. Mol. Cell. Biol. 16, 6668–6676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gumbs O. H., Campbell A. M., and Weil P. A. (2003) High-affinity DNA binding by a Mot1p-TBP complex: implications for TAF-independent transcription. EMBO J. 22, 3131–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muldrow T. A., Campbell A. M., Weil P. A., and Auble D. T. (1999) MOT1 can activate basal transcription in vitro by regulating the distribution of TATA binding protein between promoter and nonpromoter sites. Mol. Cell. Biol. 19, 2835–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zentner G. E., and Henikoff S. (2013) Mot1 redistributes TBP from TATA-containing to TATA-less promoters. Mol. Cell. Biol. 33, 4996–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Madison J. M., and Winston F. (1997) Evidence that Spt3 functionally interacts with Mot1, TFIIA, and TATA-binding protein to confer promoter-specific transcriptional control in Saccharomyces cerevisiae. Mol. Cell. Biol. 17, 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrau J.-C., Van Oevelen C. J., Van Teeffelen H. A., Weil P. A., Holstege F. C., and Timmers H. T. (2002) Mot1p is essential for TBP recruitment to selected promoters during in vivo gene activation. EMBO J. 21, 5173–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dasgupta A., Darst R. P., Martin K. J., Afshari C. A., and Auble D. T. (2002) Mot1 activates and represses transcription by direct, ATPase-dependent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 99, 2666–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Topalidou I., Papamichos-Chronakis M., Thireos G., and Tzamarias D. (2004) Spt3 and Mot1 cooperate in nucleosome remodeling independently of TBP recruitment. EMBO J. 23, 1943–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dasgupta A., Juedes S. A., Sprouse R. O., and Auble D. T. (2005) Mot1-mediated control of transcription complex assembly and activity. EMBO J. 24, 1717–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geisberg J. V., Moqtaderi Z., Kuras L., and Struhl K. (2002) Mot1 associates with transcriptionally active promoters and inhibits association of NC2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 22, 8122–8134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sprouse R. O., Wells M. N., and Auble D. T. (2009) TATA-binding protein variants that bypass the requirement for Mot1 in vivo. J. Biol. Chem. 284, 4525–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poorey K., Sprouse R. O., Wells M. N., Viswanathan R., Bekiranov S., and Auble D. T. (2010) RNA synthesis precision is regulated by preinitiation complex turnover. Genome Res. 20, 1679–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Formosa T., Ruone S., Adams M. D., Olsen A. E., Eriksson P., Yu Y., Rhoades A. R., Kaufman P. D., and Stillman D. J. (2002) Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics 162, 1557–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jamai A., Puglisi A., and Strubin M. (2009) Histone chaperone spt16 promotes redeposition of the original H3-H4 histones evicted by elongating RNA polymerase. Mol. Cell 35, 377–383 [DOI] [PubMed] [Google Scholar]
- 26. Wittmeyer J., Formosa T. (1997) The Saccharomyces cerevisiae DNA polymerase α catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol. Cell. Biol. 17, 4178–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han J., Li Q., McCullough L., Kettelkamp C., Formosa T., and Zhang Z. (2010) Ubiquitylation of FACT by the cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev. 24, 1485–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foltman M., Evrin C., De Piccoli G., Jones R. C., Edmondson R. D., Katou Y., Nakato R., Shirahige K., and Labib K. (2013) Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep. 3, 892–904 [DOI] [PubMed] [Google Scholar]
- 29. Brewster N. K., Johnston G. C., and Singer R. A. (1998) Characterization of the CP Complex, an abundant dimer of Cdc68 and Pob3 proteins that regulates yeast transcriptional activation and chromatin repression. J. Biol. Chem. 273, 21972–21979 [DOI] [PubMed] [Google Scholar]
- 30. Formosa T., Eriksson P., Wittmeyer J., Ginn J., Yu Y., and Stillman D. J. (2001) Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J. 20, 3506–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruone S., Rhoades A. R., Formosa T. (2003) Multiple Nhp6 molecules are required to recruit Spt16-Pob3 to form yFACT complexes and to reorganize nucleosomes. J. Biol. Chem. 278, 45288–45295 [DOI] [PubMed] [Google Scholar]
- 32. Formosa T. (2012) The role of FACT in making and breaking nucleosomes. Biochim. Biophys. Acta 1819, 247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Belotserkovskaya R., Oh S., Bondarenko V. A., Orphanides G., Studitsky V. M., and Reinberg D. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093 [DOI] [PubMed] [Google Scholar]
- 34. Orphanides G., Wu W. H., Lane W. S., Hampsey M., and Reinberg D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288 [DOI] [PubMed] [Google Scholar]
- 35. Stuwe T., Hothorn M., Lejeune E., Rybin V., Bortfeld M., Scheffzek K., and Ladurner A. G. (2008) The FACT Spt16 “peptidase” domain is a histone H3-H4 binding module. Proc. Natl. Acad. Sci. U.S.A. 105, 8884–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winkler D. D., Muthurajan U. M., Hieb A. R., Luger K. (2011) Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J. Biol. Chem. 286, 41883–41892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Biswas D., Yu Y., Prall M., Formosa T., and Stillman D. J. (2005) The yeast FACT complex has a role in transcriptional initiation. Mol. Cell. Biol. 25, 5812–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsunaka Y., Fujiwara Y., Oyama T., Hirose S., and Morikawa K. (2016) Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev. 30, 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Malone E. A., Clark C. D., Chiang A., and Winston F. (1991) Mutations in SPT16/CDC68 suppress cis- and trans-acting mutations that affect promoter function in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 5710–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rowley A., Singer R. A., Johnston G. C. (1991) CDC68, a yeast gene that affects regulation of cell proliferation and transcription, encodes a protein with a highly acidic carboxyl terminus. Mol. Cell. Biol. 11, 5718–5726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Orphanides G., LeRoy G., Chang C. H., Luse D. S., Reinberg D. (1998) FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92, 105–116 [DOI] [PubMed] [Google Scholar]
- 42. Saunders A., Werner J., Andrulis E. D., Nakayama T., Hirose S., Reinberg D., Lis J. T. (2003) Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301, 1094–1096 [DOI] [PubMed] [Google Scholar]
- 43. Mason P. B., and Struhl K. (2003) The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol. Cell. Biol. 23, 8323–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xin H., Takahata S., Blanksma M., McCullough L., Stillman D. J., and Formosa T. (2009) yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol. Cell 35, 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaplan C. D., Laprade L., and Winston F. (2003) Transcription elongation factors repress transcription initiation from cryptic sites. Science 301, 1096–1099 [DOI] [PubMed] [Google Scholar]
- 46. Cheung V., Chua G., Batada N. N., Landry C. R., Michnick S. W., Hughes T. R., and Winston F. (2008) Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 6, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shimojima T., Okada M., Nakayama T., Ueda H., Okawa K., Iwamatsu A., Handa H., and Hirose S. (2003) Drosophila FACT contributes to Hox gene expression through physical and functional interactions with GAGA factor. Genes Dev. 17, 1605–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arnett D. R., Jennings J. L., Tabb D. L., Link A. J., and Weil P. A. (2008) A proteomics analysis of yeast Mot1p protein-protein associations: insights into mechanism. Mol. Cell. Proteomics 7, 2090–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Venters B. J., Irvin J. D., Gramlich P., and Pugh B. F. (2011) Genome-wide transcriptional dependence on conserved regions of Mot1. Mol. Cell. Biol. 31, 2253–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Basehoar A. D., Zanton S. J., and Pugh B. F. (2004) Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709 [DOI] [PubMed] [Google Scholar]
- 51. Rhee H. S., and Pugh B. F. (2012) Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483, 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Venkatesh S., Smolle M., Li H., Gogol M. M., Saint M., Kumar S., Natarajan K., and Workman J. L. (2012) Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489, 452–455 [DOI] [PubMed] [Google Scholar]
- 53. Zanton S. J., and Pugh B. F. (2004) Changes in genomewide occupancy of core transcriptional regulators during heat shock. Proc. Natl. Acad. Sci. U.S.A. 101, 16843–16848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hahn S. (2004) Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 11, 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van Werven F. J., van Bakel H., van Teeffelen H. A., Altelaar A. F., Koerkamp M. G., Heck A. J., Holstege F. C., and Timmers H. T. (2008) Cooperative action of NC2 and Mot1p to regulate TATA-binding protein function across the genome. Genes Dev. 22, 2359–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun M., Schwalb B., Schulz D., Pirkl N., Etzold S., Larivière L., Maier K. C., Seizl M., Tresch A., and Cramer P. (2012) Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 22, 1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun M., Schwalb B., Pirkl N., Maier K. C., Schenk A., Failmezger H., Tresch A., and Cramer P. (2013) Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 52, 52–62 [DOI] [PubMed] [Google Scholar]
- 58. Helenius K., Yang Y., Tselykh T. V., Pessa H. K., Frilander M. J., and Mäkelä T. P. (2011) Requirement of TFIIH kinase subunit Mat1 for RNA Pol II C-terminal domain Ser5 phosphorylation, transcription and mRNA turnover. Nucleic Acids Res. 39, 5025–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wal M., and Pugh B. F. (2012) Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq. Methods Enzymol. 513, 233–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Z., and Pugh B. F. (2011) High-resolution genome-wide mapping of the primary structure of chromatin. Cell 144, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kaplan N., Moore I. K., Fondufe-Mittendorf Y., Gossett A. J., Tillo D., Field Y., LeProust E. M., Hughes T. R., Lieb J. D., Widom J., and Segal E. (2009) The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Albert I., Mavrich T. N., Tomsho L. P., Qi J., Zanton S. J., Schuster S. C., and Pugh B. F. (2007) Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446, 572–576 [DOI] [PubMed] [Google Scholar]
- 63. Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., and Li W. (2013) DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 23, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu C. L., Kaplan T., Kim M., Buratowski S., Schreiber S. L., Friedman N., and Rando O. J. (2005) Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 3, e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Bakel H., Tsui K., Gebbia M., Mnaimneh S., Hughes T. R., and Nislow C. (2013) A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet. 9, e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mayer A., Lidschreiber M., Siebert M., Leike K., Söding J., and Cramer P. (2010) Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 17, 1272–1278 [DOI] [PubMed] [Google Scholar]
- 67. Kang S. W., Kuzuhara T., and Horikoshi M. (2000) Functional interaction of general transcription initiation factor TFIIE with general chromatin factor SPT16/CDC68. Genes Cells 5, 251–263 [DOI] [PubMed] [Google Scholar]
- 68. Ransom M., Williams S. K., Dechassa M. L., Das C., Linger J., Adkins M., Liu C., Bartholomew B., Tyler J. K. (2009) FACT and the proteasome promote promoter chromatin disassembly and transcriptional initiation. J. Biol. Chem. 284, 23461–23471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Biswas D., Yu Y., Mitra D., and Stillman D. J. (2006) Genetic interactions between Nhp6 and Gcn5 with Mot1 and the Ccr4-Not complex that regulate binding of TATA-binding protein in Saccharomyces cerevisiae. Genetics 172, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dürr H., Flaus A., Owen-Hughes T., and Hopfner K. P. (2006) Snf2 family ATPases and DExx box helicases: differences and unifying concepts from high-resolution crystal structures. Nucleic Acids Res. 34, 4160–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ryan D. P., and Owen-Hughes T. (2011) Snf2-family proteins: chromatin remodellers for any occasion. Curr. Opin. Chem. Biol. 15, 649–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Singleton M. R., Dillingham M. S., and Wigley D. B. (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 [DOI] [PubMed] [Google Scholar]
- 73. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Darst R. P., Dasgupta A., Zhu C., Hsu J.-Y., Vroom A., Muldrow T., and Auble D. T. (2003) Mot1 regulates the DNA binding activity of free TATA-binding protein in an ATP-dependent manner. J. Biol. Chem. 278, 13216–13226 [DOI] [PubMed] [Google Scholar]
- 75. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., and Durbin R.; 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., and Mesirov J. P. (2011) Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Thorvaldsdóttir H., Robinson J. T., and Mesirov J. P. (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Quinlan A. R., and Hall I. M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kent W. J., Zweig A. S., Barber G., Hinrichs A. S., and Karolchik D. (2010) BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shen L., Shao N., Liu X., and Nestler E. (2014) ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Auble D. T., Wang D., Post K. W., and Hahn S. (1997) Molecular analysis of the SNF2/SWI2 protein family member MOT1, an ATP-driven enzyme that dissociates TATA-binding protein from DNA. Mol. Cell. Biol. 17, 4842–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]