

Abstract
The Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor involved in many physiological processes. Several studies indicate that AHR is also involved in energy homeostasis. Fibroblast growth factor 21 (FGF21) is an important regulator of the fasting and feeding responses. When administered to various genetic and diet-induced mouse models of obesity, FGF21 can attenuate obesity-associated morbidities. Here, we explore the role of AHR in hepatic Fgf21 expression through the use of a conditional, hepatocyte-targeted AHR knock-out mouse model (CreAlbAhrFx/Fx). Compared with the congenic parental strain (AhrFx/Fx), non-fasted CreAlbAhrFx/Fx mice exhibit a 4-fold increase in hepatic Fgf21 expression, as well as elevated expression of the FGF21-target gene Igfbp1. Furthermore, in vivo agonist activation of AHR reduces hepatic Fgf21 expression during a fast. The Fgf21 promoter contains several putative dioxin response elements (DREs). Using EMSA, we demonstrate that the AHR-ARNT heterodimer binds to a specific DRE that overlaps binding sequences for peroxisome proliferator-activated receptor α (PPARα), carbohydrate response element-binding protein (ChREBP), and cAMP response element-binding protein, hepatocyte specific (CREBH). In addition, we reveal that agonist-activated AHR impairs PPARα-, ChREBP-, and CREBH-mediated promoter activity in Hepa-1 cells. Accordingly, agonist treatment in Hepa-1 cells ablates potent ER stress-driven Fgf21 expression, and pre-treatment with AHR antagonist blocks this effect. Finally, we show that pre-treatment of primary human hepatocytes with AHR agonist diminishes PPARα-, glucose-, and ER stress-driven induction of FGF21 expression, indicating the effect is not mouse-specific. Together, our data show that AHR contributes to hepatic energy homeostasis, partly through the regulation of FGF21 expression and signaling.
Keywords: aryl hydrocarbon receptor (AhR) (AHR), endoplasmic reticulum stress (ER stress), energy metabolism, fibroblast growth factor (FGF), glucose metabolism, peroxisome proliferator-activated receptor (PPAR), cAMP-responsive element-binding protein hepatocyte-specific (CREBH), carbohydrate response element-binding protein (ChREBP)
Introduction
The Aryl hydrocarbon receptor (AHR)2 is a ligand-activated basic helix-loop-helix/Per-ARNT-Sim (bHLH-PAS) transcription factor, classically known for mediating 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced toxicity. AHR normally resides in the cytoplasm, bound to molecules of heat shock protein 90, X-associated protein 2, and p23. Upon agonist (i.e. TCDD) binding, AHR translocates to the nucleus and aryl hydrocarbon receptor nuclear translocator (ARNT) displaces the cytoplasmic complex to form an AHR-ARNT heterodimer. The AHR-ARNT complex is then able to bind dioxin response elements (DRE) in the promoter region of a wide array of genes, many of which are involved in endogenous and xenobiotic metabolism (e.g. CYP1A1, CYP1A2, and CYP1B1) (1).
Several lines of evidence indicate that AHR is involved in the regulation of metabolic homeostasis. For example, TCDD activation of AHR alters the expression of various genes involved in hepatic metabolism (2). Additionally, AHR is involved in the regulation of gluconeogenesis via the AHR-responsive gene TCDD-inducible poly(ADP-ribose) polymerase (Tiparp) (3). However, these studies primarily examine the role of AHR in metabolism when activated by TCDD, a xenobiotic compound. Therefore, the physiological role of AHR in metabolic homeostasis remains poorly understood.
Recently, there has been keen interest in the potential use of the metabolic hormone fibroblast growth factor 21 (FGF21) as a treatment for obesity. Administration of recombinant FGF21 in various animal models of obesity consistently results in weight loss, fat pad reduction, and improved insulin sensitivity (4, 5). Physiologically, FGF21 is induced by fasting, and acts as an endocrine hormone to induce gluconeogenesis, ketogenesis, and torpor (6). Further evidence shows that these FGF21-mediated responses depend upon direct binding of PPARα-RXRα to the Fgf21 promoter region to activate transcription (7). PPARα, in combination with cAMP-responsive element-binding protein, hepatocyte specific (CREBH), has also been implicated in the activation of Fgf21 expression (8). Alternatively, carbohydrate response element-binding protein (ChREBP) is known to activate Fgf21 expression under hyperglycemic conditions (9). Recent studies suggest that ChREBP-dependent transcription might also be directly involved in the FGF21-mediated control of sweet taste preference and sugar intake (10). Last, the unfolded protein response is also capable of regulating Fgf21 transcription via the transcription factor X-box binding protein 1 (XBP1) (11). In summary, Fgf21 is involved with several biological processes, and is therefore subject to complex regulatory control.
Recent studies have indicated that AHR can increase hepatic Fgf21 expression in the presence of TCDD (12). Similarly, a separate study revealed elevated Fgf21 expression in mice expressing a constitutively active form of AHR in the liver (13). However, data from the former study indicate that hepatic Fgf21 expression is also greater in Ahr−/− mice compared with Ahr+/+ mice. Furthermore, their data demonstrate that activation of AHR with relatively low doses of TCDD represses hepatic Fgf21 expression over time (12). Such contradicting results warrant further investigation into the role of AHR in regulating Fgf21 mRNA.
In this study, we examined the physiological role of AHR in hepatic Fgf21 expression using a mouse model that lacks functional AHR protein in hepatocytes (CreAlbAhrFx/Fx) (14). Compared with the parental strain (AhrFx/Fx), CreAlbAhrFx/Fx mice exhibit increased hepatic expression of Fgf21 during a non-fasting state, along with elevated serum FGF21 levels. Therefore, we hypothesize that AHR may constitutively, or through endogenous ligand binding, interfere with the activation of hepatic Fgf21 expression.
The Fgf21 promoter region contains several putative DREs, one of which overlaps a peroxisome proliferator-activated receptor response element (PPRE) and a carbohydrate response element (ChoRE). Furthermore, this DRE is found adjacent to a cAMP response element (CRE). Using EMSA, we demonstrate that AHR is able to bind to this specific DRE within the Fgf21 promoter region, while ligand-activated AHR in vitro impairs PPARα-, ChREBP-, and CREBH-mediated increases in promoter activity. In addition, AHR agonist treatment in Hepa-1 cells ablates potent, endoplasmic reticulum (ER) stress-driven activation of Fgf21 expression. Finally, we present evidence that ligand activation of AHR in human primary hepatocytes similarly attenuates PPARα-, glucose-, and ER stress-driven FGF21 expression.
Results
CreAlbAhrFx/Fx Mice Exhibit Increased Expression of the Fasting-induced Hormone Fgf21 during a Non-fasted State
FGF21 is a key regulator of the fasting response; therefore, hepatic Fgf21 expression occurs at a low basal level during a non-fasting state. However, non-fasting CreAlbAhrFx/Fx mice exhibit a significant 4-fold increase in hepatic Fgf21 expression compared with AhrFx/Fx mice (Fig. 1A). In addition, circulating FGF21 concentrations in non-fasted CreAlbAhrFx/Fx mice are 2-fold higher than the levels observed in AhrFx/Fx mice. Similarly, non-fasting Ahr−/− mice exhibit a significant 3.8-fold increase in hepatic Fgf21 expression compared with wild-type mice (Fig. 1B). Consistent with the known effects of elevated Fgf21 expression, we observe increased expression of the downstream target gene, insulin-like growth factor-binding protein 1 (Igfbp1), in conjunction with the down-regulation of genes involved in fatty acid synthesis (Fig. 1, C and D). Specifically, the expression of fatty acid synthase (Fasn) and sterol response element-binding protein 1c (Srebp1c) are reduced ∼2-fold in CreAlbAhrFx/Fx mice. In contrast, the Fgf21 expression levels in adipose tissue are comparable between AhrFx/Fx and CreAlbAhrFx/Fx mice (Fig. 1E). These results are consistent with the targeted deletion of AHR to hepatocytes, and not adipocytes. Interestingly, the repression of genes associated with fatty acid synthesis still occurs within white adipose tissue, despite no differences in Fgf21 expression (Fig. 1F). Whereas hepatic Fasn expression was reduced without a significant reduction in stearoyl-CoA desaturase 1 (Scd1) expression, we observe the opposite in white adipose tissue.
FIGURE 1.

A, hepatic Fgf21 expression and serum concentrations in AhrFx/Fx and CreAlbAhrFx/Fx mice. B, hepatic Fgf21 expression in C57BL6/J and Ahr-null mice. C, expression of Fgf21 target gene Igfbp1. D, hepatic expression of genes involved in de novo lipogenesis. E, Fgf21 expression and F, de novo lipogenesis gene expression in adipose tissue from AhrFx/Fx and CreAlbAhrFx/Fx mice. G, hepatic Fgf21 mRNA and serum FGF21 levels in fasted AhrFx/Fx mice, exposed to vehicle or 10 μg/kg of TCDD by gavage. Fasn, fatty acid synthase; Scd1, stearoyl-CoA desaturase 1; Srebp1c, sterol response element-binding protein 1c. All data are presented as mean ± S.E. from three or more mice. Statistical analyses were performed using either a two-tailed Student's t test or one-way ANOVA. The latter analysis was performed when there were more than two treatment groups; *, p < 0.05; **, p < 0.01. These experiments have been repeated twice.
Next, we investigated the effects of ligand-mediated AHR activation in fasting mice, given that FGF21 regulates the fasting response. For this experiment, 6-week-old male AhrFx/Fx mice were exposed for 24 h to 10 μg/kg of TCDD or vehicle (corn oil) by gavage, then fasted overnight. We observe that TCDD treatment significantly reduces hepatic Fgf21 expression by 50% compared with vehicle-treated mice (Fig. 1G). However, circulating levels of FGF21 are not altered with TCDD treatment.
The ChREBP-MLX and AHR-ARNT Heterodimer Complexes Bind to the Fgf21 Promoter at a Composite DRE-PPRE-CRE-ChoRE Regulatory Region
As depicted in Fig. 2A, the mouse Fgf21 promoter region contains four overlapping response elements (designated DRE, PPRE, CRE, and ChoRE). Previous studies have already shown direct binding of PPARα to the Fgf21 promoter at this site of DRE-PPRE-CRE-ChoRE overlap (7). However, direct binding of ChREBP and its heterodimeric partner, MLX, to this region has not been demonstrated. Utilizing EMSA, we show that the ChREBP-MLX complex does bind to the proximal Fgf21 ChoRE (Fig. 2B, lanes 5–8). Also shown is ChREBP-MLX complex formation with a positive control oligonucleotide containing a consensus ChoRE (Fig. 2B, lanes 1–4). Next, to demonstrate that the AHR-ARNT heterodimer binds to the putative DRE in this region, we incubated in vitro translated AHR and ARNT proteins in the presence of a 32P-labeled oligonucleotide that contains the region of response element overlap (−88 to −54), with or without the addition of AHR ligand. For a positive control, we utilized 32P-labeled Cyp1a1 oligonucleotide. As shown in Fig. 2C, TCDD results in AHR-ARNT heterodimerization and subsequent binding to the positive control oligonucleotide. We demonstrate that AHR agonists TCDD (Fig. 2D, lanes 1–5) and indolo[3,2b]carbazole (ICZ) (Fig. 2D, lanes 6–10) also result in AHR-ARNT heterodimerization and DRE binding to the 32P-labeled Fgf21 oligonucleotide. To confirm that this binding is specific, we incubated AHR and ARNT in an excess of unlabeled competitor Fgf21 oligonucleotide, with or without a mutation in the DRE, followed by incubation in the presence of labeled Fgf21 oligonucleotide. As shown in Fig. 2E, we observed that preincubating in an excess of unlabeled, non-mutated oligonucleotide prevents AHR-ARNT binding to the labeled Fgf21 oligonucleotide (lanes 1 and 2), whereas addition of an unlabeled Fgf21 mutant oligonucleotide does not (lanes 3 and 4).
FIGURE 2.

A, diagram of overlapping response elements within the Fgf21 promoter region. The boxed (dashed line) region of the promoter was utilized for EMSA. B, formation of the ChREBP-MLX complex with a control labeled oligonucleotide (lanes 1–4) and at the Fgf21 ChoRE (lanes 5–8). C, TCDD induces AHR-ARNT heterodimerization and binding to a control 32P-labeled oligonucleotide. D, TCDD and ICZ treatment results in formation of the AHR-ARNT complex at the Fgf21 DRE. E, addition of unlabeled oligonucleotide interrupts AHR-ARNT binding to the Fgf21 DRE (lanes 1 and 2). Introducing a mutation into the DRE of the unlabeled oligonucleotide eliminates this competitive binding (lanes 3 and 4). All data are representative of at least two independent experiments.
Ligand-activated AHR Attenuates PPARα-, ChREBP-, and ER Stress-dependent Fgf21 Expression in Hepa-1 Cells
Having demonstrated that the AHR-ARNT heterodimer can bind to the composite DRE-PPRE-CRE-ChoRE, we next explored the possibility of cross-talk between AHR and promoter-driven PPARα, ChREBP, or CREBH signaling. To do so, we transfected a luciferase reporter construct containing the −1906 to +52 Fgf21 promoter region into Hepa-1 cells, along with expression vectors for the different transcription factors of interest. In the absence of AHR ligands, ectopic PPARα, ChREBP, and CREBH expression results in significant Fgf21 promoter-dependent expression. However, treatment with AHR agonist ICZ ablates PPARα-, ChREBP-, and CREBH-dependent induction (Fig. 3A), suggesting that ligand-activated AHR can interfere with the promoter-driven activation of Fgf21 expression by these transcription factors. We chose to utilize ICZ in our cell culture experiments as it represents a common dietary AHR ligand. ICZ is formed from the acid condensation of its parent compound, indole-3-carbinol, a breakdown product of glucobrassicin, which naturally occurs at high concentrations within vegetables of the Brassica genus (15).
FIGURE 3.

A, ICZ treatment of Hepa-1 cells that transiently express PPARα, ChREBP, or CREBH suppresses promoter-driven expression. B, treatment of Hepa-1 cells with 1 mm DTT or 1 μm thapsigargin leads to the activation of ER stress. C, ligand activation of AHR in Hepa-1 cells diminishes thapsigargin and DTT-mediated induction of Fgf21. D, 1 h preincubation with only 100 nm ICZ ablates thapsigargin- and DTT-mediated induction of Fgf21. E, ICZ activates Cyp1a1 expression in Hepa-1 cells. F, GNF351 fails to antagonize ICZ-mediated activation of AHR. G, GNF351 successfully antagonizes TCDD-dependent Cyp1a1 expression. H, activation of AHR with 2 nm TCDD inhibits thapsigargin-driven Fgf21 expression. However, a 1-h preincubation with AHR antagonist GNF351 blocks this effect. All data are representative of two or more experiments. Statistical analyses were performed using one-way ANOVA; significance *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To further investigate AHR-mediated repression of Fgf21 expression in the presence of ligand, we examined the ability of AHR to attenuate ER stress-mediated activation of Fg21 in Hepa-1 cells. To stimulate ER stress, we incubated cells overnight in serum-free medium supplemented with 0.2% BSA, then refreshed the medium and added 1 mm dithiothreitol (DTT) or 1 μm thapsigargin for 4 h. Confirming activation of ER stress, we observe significantly elevated expression of Ddit3 and Atf4 in DTT- and thapsigargin-treated cells (Fig. 3B). In the absence of AHR ligand, DTT and thapsigargin both markedly increase Fgf21 expression >150-fold. However, 1 h pre-treatment with 500 nm ICZ reduces thapsigargin- and DTT-driven expression by 74 and 75%, respectively (Fig. 3C). Notably, significant ICZ-mediated repression is evident at a 5-fold lower ICZ concentration (Fig. 3D). Consistent with AHR activation, we also observe a statistically significant increase in Cyp1a1 transcriptional levels in ICZ-treated cells (Fig. 3E). To confirm that the repressive action of AHR agonists occurs specifically through AHR, we attempted to suppress agonist-mediated activity by pretreating cells with 1 μm N-(2-(1H-indol-3-yl)ethyl)-9-isopropyl-2-(5-methylpyridin-3-yl)-9H-purin-6-amine (GNF351), a known AHR antagonist (16). Surprisingly, GNF351 was unable to antagonize 100 nm ICZ-mediated AHR activity (Fig. 3F). In contrast, GNF351 does successfully antagonize 2 nm TCDD-mediated AHR activity, as evidenced by the significant suppression of TCDD-driven Cyp1a1 expression (Fig. 3G). Shown in Fig. 3H, treatment with 1 μm thapsigargin results in a 32.6-fold induction of Fgf21 mRNA. Similar to pretreatment with ICZ, 1 h pretreatment with 2 nm TCDD reduces this effect by ∼70%. However, the addition of 1 μm GNF351 for 1 h prior to TCDD treatment reverses TCDD suppression of Fgf21 expression (Fig. 3H).
Comparable with treatment using DTT or thapsigargin, incubating Hepa-1 cells for 24 h in glucose-free medium also activates ER stress and Fgf21 expression (Fig. 4A). Exposing glucose-starved cells to various doses of ICZ, ranging from 250 to 1000 nm, results in a dose-dependent increase in the suppression of Fgf21 expression (Fig. 4B). Furthermore, we observe the same dose-dependent suppression of Fgf21 expression upon exposure to various doses of TCDD, ranging from 0.1 to 5 nm (Fig. 4C).
FIGURE 4.
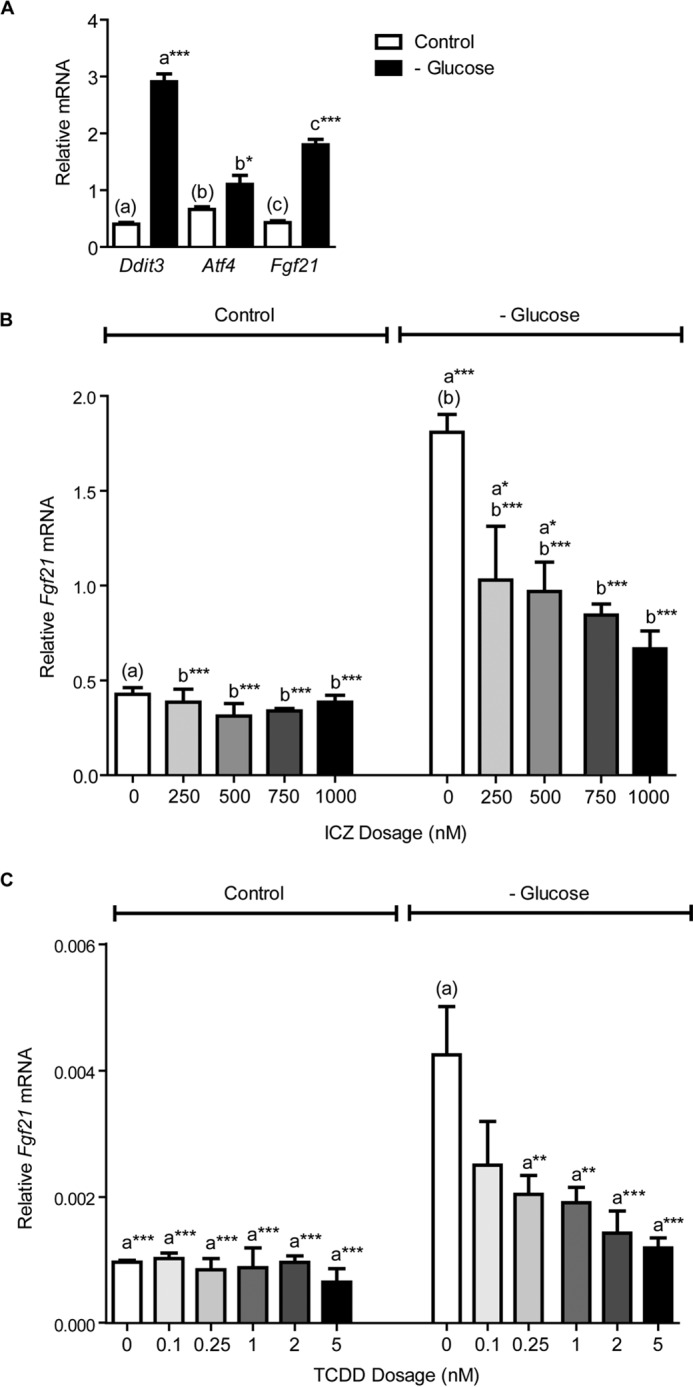
A, 24 h glucose starvation activates the expression of ER stress-response genes Ddit3 and Atf4, and increases Fgf21 mRNA in Hepa-1 cells. B, ICZ exposure results in a dose-dependent suppression of Fgf21 expression in glucose-starved Hepa-1 cells. For this experiment, cells were re-treated with ICZ after 12 h. C, in glucose-starved Hepa-1 cells, 24 h TCDD exposure also suppresses Fgf21 expression in a dose-dependent manner. Treatment groups were performed in triplicate, and the data presented are representative of data from at least two independent experiments. Statistical analyses were performed using one-way ANOVA; significance *, p < 0.05; **, p < 0.01; ***, p < 0.001.
AHR Activation Suppresses PPARα-, ChREBP-, and ER Stress-mediated Induction of FGF21 Expression in Primary Human Hepatocytes
To further demonstrate that AHR activity attenuates PPARα-agonist, glucose-mediated, and ER stress-driven FGF21 expression, we examined the effects of AHR activation in primary human hepatocytes. As shown in Fig. 5A, treatment with PPARα ligand GW7647 activates CPT1A expression and significantly increases FGF21 expression >3-fold, whereas pretreatment of cells with AHR agonist significantly inhibits this response. Similarly, incubation of primary human hepatocytes in medium supplemented with 30 mm glucose activates FGF21 expression after 6 and 24 h (Fig. 5, B and C). However, 10 nm TCDD or 500 nm ICZ treatment significantly suppresses FGF21 expression by 58 and 46%, respectively. Confirming ChREBP activation, we observe that incubating primary cells in 30 mm glucose increases the expression of liver and RBC pyruvate kinase (PKLR) (Fig. 5D). Finally, to activate ER stress, we incubated primary human hepatocytes in the presence of 1 mm DTT for 4 h, with or without a 500 nm ICZ pretreatment for 1 h. In the presence of DTT, we observe an increase in ATF4 and DDIT3 gene expression, thereby confirming the onset of ER stress (Fig. 5E). Consistent with previous experiments, DTT increases FGF21 expression 69-fold, whereas a 1-h pretreatment with 500 nm ICZ significantly impairs DTT-induced FGF21 expression by 46% (Fig. 5F).
FIGURE 5.

A, ICZ-mediated AHR activation attenuates GW7647-driven FGF21 expression after 24 h co-incubation, without any effect on the expression of PPARα target gene CPT1. B, liganded AHR diminishes glucose activation of FGF21 expression in primary human hepatocytes at 6 h and (C) 24 h. D, incubation of primary human hepatocytes in medium containing 30 mm glucose activates ChREBP-target gene PKLR. E, treating primary human hepatocytes with 1 mm DTT for 4 h activates gene pathways involved in ER stress. F, DTT treatment induces FGF21 expression in primary human hepatocytes, whereas, 1 h preincubation with 500 nm ICZ attenuates this effect. Treatment groups were performed in triplicate and the data presented are representative of experiments from at least two individual donors. Statistical analyses were performed using one-way ANOVA; significance *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
To date, pharmacological administration of FGF21 is well characterized and known to exert beneficial effects in various animal models of obesity (4, 5). Additionally, FGF21 overexpression is linked to increased longevity in mice (17). However, the regulation of this metabolic hormone remains poorly understood. Our data reveal that AHR is an important regulator of Fgf21 expression during the non-fasting state. When fed a semi-purified diet, CreAlbAhrFx/Fx mice display elevated hepatic Fgf21 expression and serum FGF21 concentrations. Importantly, the AhrFx/Fx background used to generate CreAlbAhrFx/Fx mice carries the Ahrd allele, which encodes a form of AHR exhibiting reduced ligand affinity relative to the Ahrb allele (18). Therefore, a study that utilized Ahrb conditional knock-out mice (which are not currently available) would likely yield a greater increase of Fgf21 expression relative to hepatocyte-targeted AHR knock-out mice. Consistent with elevated hepatic Fgf21 expression, we observed a repression of fatty acid synthesis genes in the liver. Interestingly, we observed a similar repression of fatty acid synthesis gene expression in adipose tissue collected from CreAlbAhrFx/Fx mice, without any increase in adipose Fgf21 expression. Such results are consistent with the ongoing hypothesis in the literature that liver-excreted FGF21 acts in an endocrine fashion (6).
In agreement with recent data (12), our results suggest that AHR can bind directly to the Fgf21 promoter at the same location to which the PPARα-RXRα and PPARα-CREBH heterodimers bind (7, 8). Evidence implicates the PPARα-RXRα heterodimer in regulating lipid metabolism (19), whereas CREBH, independent of PPARα, induces a systemic inflammatory response upon ER stress (20). CREBH also modulates lipid metabolism in response to metabolic stress (21). However, the exact function of the PPARα-CREBH complex is not known. To date, formation of this complex has only been observed at the −62 to −93 region of the Fgf21 promoter (8). Furthermore, whether CREBH can participate with other transcription factors (e.g. activating transcription factor 6) to activate Fgf21 expression upon ER stress remains unknown. Also unclear is whether AHR can compete with the PPARα-CREBH complex for DNA binding at these sites.
Our study presents novel data indicating the ChREBP-MLX heterodimer, which mediates the insulin-independent response to glucose (22), is able to bind a ChoRE that overlaps the binding sites for the AHR-ARNT, PPARα-RXRα, and PPARα-CREBH heterodimers. Recently, investigators determined that ChREBP plays a crucial role in the FGF21-dependent control of simple sugar intake and sweet taste preference (10). By extension, the ability of AHR to attenuate ChREBP-dependent Fgf21 expression therefore presents the possibility that dietary AHR ligands, or the direct administration of AHR ligands can influence simple sugar intake and/or sweet taste preference. We are currently investigating the validity of this hypothesis through the modulation of AHR activity with different classes of ligand.
Importantly, 31 bp upstream from the site of overlapping response elements lies a characterized ER stress-response element. XBP1, which facilitates the mammalian unfolded protein response (23), binds to this site to activate Fgf21 expression in response to ER stress (11). Given the close proximity of this site to the DRE, this element likely competes with AHR for binding to the Fgf21 promoter. In fact, we hypothesize that mutually antagonistic interactions must inherently exist between all the transcription factors that bind to the proximate binding elements (i.e. DRE, ChoRE, PPRE, CRE, ER stress-response element) in this region of the Fgf21 promoter.
Our data indicate that ligand-activated AHR successfully ablates Fgf21 induction within Hepa-1 cells. Specifically, our reporter transfection experiments demonstrate that ICZ-stimulated AHR can compete with transcriptional activators of Fgf21 to repress promoter-driven activity. Using a Hepa-1 cell line, we demonstrate that ICZ-mediated AHR activation ablates potent, ER stress-mediated Fgf21 induction. Importantly, this effect is specific to AHR because AHR antagonists successfully block agonist-driven repression. Unexpectedly, our data indicate that thapsigargin, in combination with AHR agonist, synergistically increases Cyp1a1 expression. We hypothesize that this increase likely represents a cellular mechanism for preventing further toxicity, after the onset of ER stress. Last, we demonstrate that ligand activation of AHR in primary human hepatocytes suppresses PPARα-ligand, glucose-, or ER stress-mediated induction of Fgf21 expression, indicating that the ability of the AHR to attenuate Fgf21 expression is conserved between mice and humans.
Throughout all of our cell culture experiments, AHR activation solely modulated Fgf21 expression, and not the expression of known PPARα, ChREBP, or XBP1 target genes. This indicates that the AHR does not affect the underlying pathways that each transcription factor regulates, and instead impacts Fgf21 expression directly. However, whether AHR-mediated repression of Fgf21 expression occurs directly through the interruption of transcription factor binding to the proximal Fgf21 promoter in vivo will require additional studies.
We provide in vitro and in vivo data that suggest that the AHR plays a role in the constitutive repression of hepatic Fgf21 expression. Data presented in a recent publication (12) implicating Fgf21 as an AHR target gene supports our conclusions. The authors observed long-term, time-dependent repression of hepatic Fgf21 expression at 24 h and beyond with low dose (0.1, 1, and 10 μg/kg) TCDD treatments in C57BL6/J mice. Also consistent with our results, their data demonstrate elevated hepatic Fgf21 expression in Ahr-null mice. However, the authors demonstrated a marked increase in hepatic Fgf21 expression in C57BL6/J mice given daily i.p. injections of TCDD (40 μg/kg) or β-naphthoflavone (100 mg/kg) for 4 days. In addition, they reported >100-fold increase of Fgf21 expression in human and mouse hepatoma cell lines treated with 10 nm TCDD. Although we repeated their mouse hepatoma cell line experiment, we failed to observe this TCDD-mediated induction of Fgf21.3 In a related, separate study involving mice that express a constitutively active form of AHR in the liver, investigators also observed increased hepatic Fgf21 expression (13). However, this constitutively active form of AHR was previously determined to exhibit AHR activity similar to high-dose TCDD exposure (24). Data obtained using high-dose treatments are often hard to interpret due to acute toxicity and AHR-independent effects on metabolism (1, 25). In addition, ER stress resulting from high-dose TCDD treatments might also contribute to the activation of Fgf21, further complicating data interpretation (26). Despite these complications, we maintain that high-dose TCDD-mediated induction of Fgf21 represents a new marker for TCDD exposure.
We believe our data illustrates a homeostatic role of AHR in the liver, when activated by dietary ligands. We speculate that ligand-activated AHR may play a previously unrecognized role in maintaining low basal Fgf21 expression, given its ability to attenuate Fgf21 activation by PPARα, a key regulator of the fasting response, and ChREBP, a glucose-sensing transcription factor. We further speculate that AHR may modulate the cellular response to ER stress, given the ability of agonist-stimulated AHR to attenuate potent ER stress-driven Fgf21 expression in cell culture experiments. Ultimately, the physiological role of AHR in attenuating Fgf21 expression presented here contrasts with the ability of high-dose TCDD exposure to increase Fgf21, as others have observed (12, 13). Therefore, additional research is required to fully understand the intricate role of AHR in Fgf21 expression. Nonetheless, our data firmly supports a physiological role for AHR in metabolic homeostasis.
To conclude, we show that AHR is involved in the homeostatic regulation of Fgf21, possibly through attenuation of ChREBP-, PPARα-, and/or ER stress-dependent activation of FGF21 expression. Although we demonstrate that AHR binds to the Fgf21 promoter at a DRE that overlaps other response elements, the exact mechanism of AHR repression remains unclear. Current work in our laboratory aims to identify whether or not AHR impairs Fgf21 activation via direct binding to its core promoter region, or through an alternative DRE-dependent or -independent (e.g. sequestration of transcription factors in the cytoplasm) mechanism. Most importantly, the AHR may represent a useful target to modulate FGF21 expression levels. The fact that many non-nutritive dietary components, such as the flavonoids present in a variety of plants, can repress or activate AHR highlights the potential for regulating FGF21 expression through dietary modulation of AHR activity.
Experimental Procedures
Animal Experiments
6-Week-old, age-matched male C57BL6/J, congenic C57BL6/J-Ahr−/−, C57BL6/J-AhrFx/Fx, and C57BL6/J-CreAlbAhrFx/Fx mice (n = 3–6 per group) were maintained on semi-purified diet AIN-93G (Dyets, NJ) for 3 weeks. AhrFx/Fx and CreAlbAhrFx/Fx mice were kindly provided by Chris Bradfield (University of Wisconsin). Mice were housed on corncob bedding in a pathogen-free, temperature- and light-controlled facility, and were given access to food ad libitum. Upon sacrifice, serum samples were collected and FGF21 concentration measured using an ELISA kit, according to manufacturer's instructions (R&D Systems). All mouse experiments were carried out humanely with approval and in accordance to the Animal Care and Use Committee of the Pennsylvania State University guidelines.
Cell Culture
Hepa-1 cells were obtained from the American Type Culture Collection and maintained as previously described (27). Enriched normal primary human hepatocytes were obtained through the Liver Tissue Cell Distribution System (University of Pittsburgh, PA). The isolated hepatocytes were seeded at >90% confluence in 24-well collagen-coated dishes and cultured as previously described, with minor modifications (28). Briefly, the cells were overlaid with 225 μg/ml of BD MatrigelTM Basement Membrane Matrix (Corning, NY) and cultured in serum-free Leibovitz's L-15 media (Life Technologies), supplemented with 10 mm HEPES, 100 units/ml of penicillin, 100 μg/ml of streptomycin, 25 nm dexamethasone, 10 nm insulin, 5 ng/ml of selenium, 5 μg/ml of transferrin, 1% linoleic acid, 1% albumin, and 1% sodium bicarbonate. For ER stress experiments, we replaced the above stated medium with Williams E Medium containing GlutaMAXTM (Thermo Fisher Scientific) and 5 mg/ml of BSA, immediately prior to addition of compounds. Due to possible human variability, primary hepatocyte experiments were repeated using two or more donors.
RNA Extraction and Quantitative RT-PCR
RNA was isolated as previously described (29). Gene expression was measured using quantitative RT-PCR as previously described (29), with the primers described in Table 1. The relative level of expression was normalized to ribosomal protein L13a mRNA (Rpl13a).
TABLE 1.
Primers used for RT-qPCR
| Gene name | Sequence (5′ → 3′) | |
|---|---|---|
| Acaca | Fa | TAACAGAATCGACACTGGCTGGCT |
| R | ATGCTGTTCCTCAGGCTCACATCT | |
| Fasn | F | GGTGTGGTGGGTTTGGTGAATTGT |
| R | TCACGAGGTCATGCTTTAGCACCT | |
| Fgf21 | F | GACTGCTGCTGGCTGTCTTC |
| R | AGGAGACTTTCTGGACTGCG | |
| FGF21 | F | CAGAGCCCCGAAAGTCTCC |
| R | GTGGGCTTCGGACTGGTAAA | |
| Igfbp1 | F | AGATCGCCGACCTCAAGAAATGGA |
| R | TGTTGGGCTGCAGCTAATCTCTCT | |
| Rpl13a | F | TTCGGCTGAAGCCTACCAGAAAGT |
| R | GCATCTTGGCCTTTTTCCGTT | |
| RPL13A | F | CCTGGAGGAGAAGAGGAAAGAGA |
| R | GAGGACCTCTGTGTATTTGTCAA | |
| Scd1 | F | TCCCTCCGGAAATGAACGAGAGAA |
| R | AGTGCAGCAGGACCATGAGAATGA | |
| Srebp1c | F | TATGGAGGGCATGAAACCCGAAGT |
| R | TTGACCTGGCTATCCTCAAAGGCT |
a F, forward; R, reverse.
Plasmid Constructs
For the luciferase reporter assay, the −1906 to +52 upstream regulatory region of Fgf21 was cloned into pGL3-basic vector (Promega). The resulting plasmid is referred to as pGL3-FGF21.
Transfections
Hepa-1 cells were seeded onto 6-well plates and grown overnight, then transfected using LipofectAMINE3000 reagent (Promega) with pGL3-FGF21, pSV-β-galactosidase, and pcDNA3, along with pCMV-TNT-PPARα, pChREBP, or pcDNA3-hCREBH(N), according to the manufacturer's instructions. pChREBP was a gift from Isabelle Leclerc (Addgene plasmid number 39235) (30). pcDNA3-hCREBH(N) was a gift from Dr. Laurie Glimcher at Weill Cornell Medical College. After a 24-h recovery, cells were treated overnight with dimethyl sulfoxide vehicle or 500 nm ICZ (Sigma).
Electromobility Shift Assay
EMSA was performed as previously described (31), using in vitro translated (Promega) mouse AHR, ARNT, ChREBP, and MLX. As a positive control for AHR, we used a 32P-labeled Cyp1a1 oligonucleotide as previously described (32). In addition to the oligonucleotide described in Fig. 2A, the following 32P-labeled oligonucleotides were used: ChoRE, 5′-GCGACATGTGATCAAGCCATGAACCC; competitor Fgf21, 5′-ACTCCTGACGCGTGATATTTGACACACTTG; mutated competitor Fgf21, 5′-ACTCCTGACGCGCAATATTTGACACACTTG.
Statistical Analysis
All experiments were performed at least twice. All data are presented as mean ± S.E. Statistical analyses were performed using a two-tailed Student's t test or one-way ANOVA; *, p < 0.05; **, p < 0.01; or ***, p < 0.001.
Author Contributions
N. G. G. wrote the manuscript and conducted the experiments, with technical assistance from I. A. M. C. J. O. provided the primary human hepatocytes used in this study. G. H. P. is responsible for conceiving and coordinating the project, in addition to assisting with data analysis and manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Denise Coslo for technical assistance with maintaining primary human hepatocyte cultures and Kelly Wagner for assistance with animal husbandry. The Liver Tissue Cell Distribution System at the University of Pittsburgh, Pittsburg, PA, is funded by National Institutes of Health Contract HHSN276201200017C.
This work was supported by the National Institute of Food and Agriculture, U.S. Dept. of Agriculture award 2014-06624, and National Institutes of Health Grants ES004869 and ES019964 (to G. P.) and GM066411 (to C. O.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
N. G. Girer, I. A. Murray, C. J. Omiecinski, and G. H. Perdew, unpublished results.
- AHR
- aryl hydrocarbon receptor
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- ARNT
- aryl hydrocarbon receptor nuclear translocater
- DRE
- dioxin response element
- PPAR
- peroxisome proliferator-activated receptor
- ChREBP
- carbohydrate response element-binding protein
- MLX
- MAX-like protein X
- CREBH
- cAMP-responsive element-binding protein hepatocyte specific
- ER
- endoplasmic reticulum
- ICZ
- indolo[3,2-b]carbazole
- GW7647
- 2-methyl-2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-propanoic acid
- GNF351
- N-(2-(1H-indol-3-yl)ethyl)-9-isopropyl-2-(5-methylpyridin-3-yl)-9H-purin-6-amine
- ANOVA
- analysis of variance
- ChoRE
- carbohydrate response element
- PPRE
- peroxisome proliferator-activated receptor response element.
References
- 1. Beischlag T. V., Luis Morales J., Hollingshead B. D., and Perdew G. H. (2008) The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 18, 207–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Angrish M. M., Mets B. D., Jones A. D., and Zacharewski T. R. (2012) Dietary fat is a lipid source in 2,3,7,8-tetrachlorodibenzo-ρ-dioxin (TCDD)-elicited hepatic steatosis in C57BL/6 mice. Toxicol. Sci. 128, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diani-Moore S., Ram P., Li X., Mondal P., Youn D. Y., Sauve A. A., and Rifkind A. B. (2010) Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 285, 38801–38810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coskun T., Bina H. A., Schneider M. A., Dunbar J. D., Hu C. C., Chen Y., Moller D. E., and Kharitonenkov A. (2008) Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 149, 6018–6027 [DOI] [PubMed] [Google Scholar]
- 5. Berglund E. D., Li C. Y., Bina H. A., Lynes S. E., Michael M. D., Shanafelt A. B., Kharitonenkov A., and Wasserman D. H. (2009) Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 150, 4084–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Potthoff M. J., Kliewer S. A., and Mangelsdorf D. J. (2012) Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 26, 312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J. K., Gerard R. D., Burgess S. C., Hammer R. E., Mangelsdorf D. J., and Kliewer S. A. (2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 8. Kim H., Mendez R., Zheng Z., Chang L., Cai J., Zhang R., and Zhang K. (2014) Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology 155, 769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iizuka K., Takeda J., and Horikawa Y. (2009) Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 583, 2882–2886 [DOI] [PubMed] [Google Scholar]
- 10. von Holstein-Rathlou S., BonDurant L. D., Peltekian L., Naber M. C., Yin T. C., Claflin K. E., Urizar A. I., Madsen A. N., Ratner C., Holst B., Karstoft K., Vandenbeuch A., Anderson C. B., Cassell M. D., Thompson A. P., et al. (2016) FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab. 23, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang S., Yan C., Fang Q. C., Shao M. L., Zhang Y. L., Liu Y., Deng Y. P., Shan B., Liu J. Q., Li H. T, Yang L., Zhou J., Dai Z., Liu Y., and Jia W. P. (2014) Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J. Biol. Chem. 289, 29751–29765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng X., Vispute S. G., Liu J., Cheng C., Kharitonenkov A., and Klaassen C. D. (2014) Fibroblast growth factor (Fgf) 21 is a novel target gene of the aryl hydrocarbon receptor (AhR). Toxicol. Appl. Pharmacol. 278, 65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu P., Yan J., Liu K., Garbacz W. G., Wang P., Xu M., Ma X., and Xie W. (2015) Activation of aryl hydrocarbon receptor dissociates fatty liver from insulin resistance by inducing FGF21. Hepatology 61, 1908–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walisser J. A., Glover E., Pande K., Liss A. L., and Bradfield C. A. (2005) Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 102, 17858–17863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bjeldanes L. F., Kim J. Y., Grose K. R., Bartholomew J. C., and Bradfield C. A. (1991) Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc. Natl. Acad. Sci. U.S.A. 88, 9543–9547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith K. J., Murray I. A., Tanos R., Tellew J., Boitano A. E., Bisson W. H., Kolluri S. K., Cooke M. P., and Perdew G. H. (2011) Identification of a high-affinity ligand that exhibits complete aryl hydrocarbon receptor antagonism. J. Pharmacol. Exp. Ther. 338, 318–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y., Xie Y., Berglund E. D., Coate K. C., He T. T., Katafuchi T., Xiao G., Potthoff M. J., Wei W., Wan Y., Yu R. T., Evans R. M., Kliewer S. A., and Mangelsdorf D. J. (2012) The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. eLife 1, e00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Poland A., and Glover E. (1990) Characterization and strain distribution pattern of the murine Ah receptor specified by the Ahd and Ahb-3 alleles. Mol. Pharmacol. 38, 306–312 [PubMed] [Google Scholar]
- 19. Mandard S., Müller M., and Kersten S. (2004) Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci. 61, 393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D. T., Back S. H., and Kaufman R. J. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599 [DOI] [PubMed] [Google Scholar]
- 21. Zhang C., Wang G., Zheng Z., Maddipati K. R., Zhang X., Dyson G., Williams P., Duncan S. A., Kaufman R. J., and Zhang K. (2012) Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 55, 1070–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma L., Tsatsos N. G., and Towle H. C. (2005) Direct role of ChREBP·Mlx in regulating hepatic glucose-responsive genes. J. Biol. Chem. 280, 12019–12027 [DOI] [PubMed] [Google Scholar]
- 23. Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 24. McGuire J., Okamoto K., Whitelaw M. L., Tanaka H., and Poellinger L. (2001) Definition of a dioxin receptor mutant that is a constitutive activator of transcription. J. Biol. Chem. 276, 41841–41849 [DOI] [PubMed] [Google Scholar]
- 25. Hsu H.-F., Tsou T.-C., Chao H.-R., Kuo Y.-T., Tsai F.-Y., and Yeh S.-C. (2010) Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on adipogenic differentiation and insulin-induced glucose uptake in 3T3-L1 cells. J. Hazard. Mater. 182, 649–655 [DOI] [PubMed] [Google Scholar]
- 26. Duan Z., Zhao J., Fan X., Tang C., Liang L., Nie X., Liu J., Wu Q., and Xu G. (2014) The PERK-eIF2α signaling pathway is involved in TCDD-induced ER stress in PC12 cells. NeuroToxicology 44, 149–159 [DOI] [PubMed] [Google Scholar]
- 27. Meyer B. K., Petrulis J. R., and Perdew G. H. (2000) Aryl hydrocarbon (Ah) receptor levels are selectively modulated by hsp90-associated immunophilin homolog XAP2. Cell Stress Chaperones 5, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Page J. L., Johnson M. C., Olsavsky K. M., Strom S. C., Zarbl H., and Omiecinski C. J. (2007) Gene expression profiling of extracellular matrix as an effector of human hepatocyte phenotype in primary cell culture. Toxicol. Sci. 97, 384–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanos R., Patel R. D., Murray I. A., Smith P. B., Patterson A. D., and Perdew G. H. (2012) Ah receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology 55, 1994–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. da Silva Xavier G., Rutter G. A., Diraison F., Andreolas C., and Leclerc I. (2006) ChREBP binding to fatty acid synthase and L-type pyruvate kinase genes is stimulated by glucose in pancreatic beta-cells. J. Lipid Res. 47, 2482–2491 [DOI] [PubMed] [Google Scholar]
- 31. Flaveny C. A., Murray I. A., Chiaro C. R., and Perdew G. H. (2009) Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol. Pharmacol. 75, 1412–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiaro C. R., Patel R. D., Marcus C. B., and Perdew G. H. (2007) Evidence for an aryl hydrocarbon receptor-mediated cytochrome P450 autoregulatory pathway. Mol. Pharmacol. 72, 1369–1379 [DOI] [PubMed] [Google Scholar]