

Abstract
Most patients with myelodysplastic syndromes (MDS) have macrocytic or normocytic anemia as a result of ineffective erythropoiesis due to clonal hematopoiesis. Occasional patients with MDS have microcytic red blood cell (RBC) indices in the absence of iron deficiency, however, and a high proportion of such patients have decreased alpha globin expression associated with somatic mutations in the chromatin remodeling factor ATRX; an established alternative mechanism for acquired alpha thalassemia in myeloid neoplasia is clonal deletion of the alpha globin cluster on chromosome 16p. This clinicopathological phenomenon has been called “acquired alpha thalassemia – myelodysplastic syndrome” (ATMDS). Here we describe a patient with new-onset microcytic anemia associated with acquired deletion of the beta globin cluster on chromosome 11, resulting in a beta thalassemia-MDS (BTMDS) phenotype. Phenotype-genotype correlation studies may reveal associations of specific mutation patterns with other recurrent MDS-associated hematological findings.
Keywords: Myelodysplastic syndrome, beta thalassemia, Anemia, Cytogenetics
Introduction
The most common human disorders of hemoglobin synthesis are the inherited alpha and beta thalassemias endemic to tropical and subtropical global regions, and which result from germline mutations, rearrangements, or deletions of the globin gene clusters and their cis-acting regulatory elements. Acquired thalassemia has also been described, primarily alpha thalassemia due to somatic point mutations in a trans-acting regulatory factor, ATRX, or, less commonly, due to deletions of the alpha globin gene cluster on chromosome 16 as part of the clonal instability of myeloid neoplasia.1 Acquired mutations of ATRX, an X-linked chromatin associated factor that has distinct effects on alpha and beta globin gene expression, are observed most commonly in the context of myelodysplastic syndromes (MDS).1 In contrast to alpha thalassemia-MDS (ATMDS, Online Mendelian Inheritance in Man #300448), cases of MDS with acquired beta thalassemia are exceedingly rare and poorly chracterized.2–4
Here we describe a patient with previously normal erythrocyte indices who developed microcytic anemia and was subsequently found to have a myeloid neoplasm. Additional evaluation revealed acquired beta thalassemia in association with MDS (BTMDS), resulting from acquired loss of the beta globin gene on chromosome 11p in a neoplastic subclone.
Case history and results
A 65 year-old woman with previously normal blood count and erythrocyte indices developed microcytic anemia (hemoglobin 9.7 g/dL, hematocrit 30.7%, mean cell volume 65.7 fL, red cell distribution width (RDW) of 34%) with unremarkable white blood cell and platelet counts. Although the patient was iron deficient at the time of initial evaluation, no bleeding source was identified and anemia failed to improve with parenteral iron repletion (Figure 1).
Figure 1. Diagnostic findings leading up to and following the diagnosis of BTMDS in the patient described.
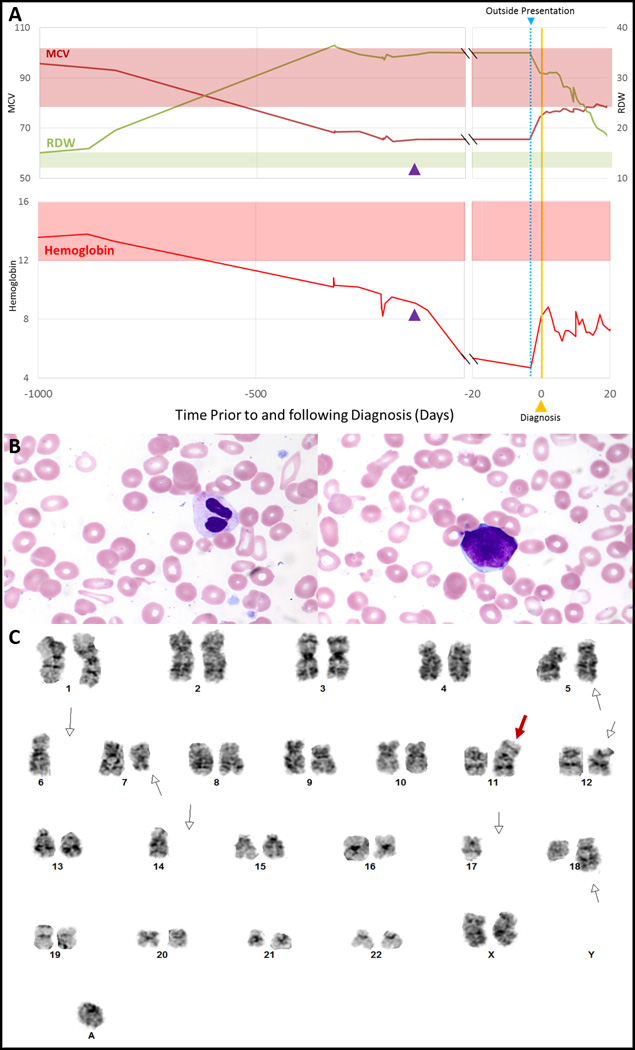
Approximately 2 years prior to diagnosis, this patient experienced a progressive decline in MCV and hemoglobin and an associated increase in RDW, suggesting acquired beta thalassemia was a result of an early clonal event in her MDS (A). The first date of transfusion is marked as the “outside presentation” (blue line, right graphs) and is associated with the nadir in hemoglobin. The date of intravenous iron administration (122 days prior to diagnosis) is indicated by a purple arrowhead. At diagnosis, her peripheral smear revealed anisopoikilocytosis and dysplastic neutrophils and rare circulating blasts (B; Wright-Giemsa, 400x). A supravital stain was performed (not shown) and did not reveal any HbH inclusions. Cytogenetic analysis revealed a complex karyotype (C); shown here is representative metaphase from the dominant clone. The white open arrows designate additional material on chromosome 5; loss of chromosome 6; a deletion at chromosome 7q21; a deletion at chromosome 12p11.2, loss of chromosome 14, an addition at chromosome 18q21, and an extra unidentified ring chromosome. Of these 15 metaphases, 2 had trisomy 8 (not shown). The red arrow identifies a derivative chromosome comprised of the long arm of chromosomes 11 and 17 [der(11;17)(q10;q10)], resulting in deletion of 11p and deletion of 17p. An additional 5 metaphases (not shown) were 43~45,XX,add(5)(q31),-6,add(7)(q21),-8,-9,+r. Abbreviations: MCV = mean cell volume; RDW = red cell distribution width; MDS = myelodysplastic syndromes.
She subsequently developed worse anemia (hemoglobin of 4.7 g/dL, again with microcytic indices) and new splenomegaly and thrombocytopenia (platelet count of 65 × 109/L); on transfer to our hospital she was noted to have 22% peripheral blasts, leukocyte dysmorphology and anisopoikilocytosis. Hemoglobin electrophoresis (after transfusion) showed hemoglobin A 94.5%, hemoglobin A2 2.4%, and hemoglobin F 3.1%; no hemoglobin variants were detected on isoelectric focusing, and supravital staining with new methylene blue and did not reveal any HbH inclusions.
The marrow was hypercellular and left-shifted, with increased reticulin and scattered blast cells with a myeloid phenotype. Next-generation sequencing including 39 genes recurrently mutated in cancer identified single nucleotide variants in TP53 (R175H), APC (E1317Q), and CDH1 (A592T), as well as an insertion/deletion in TP53 at codon Val97 (290delT) resulting in a frame shift mutation.
The karyotype was 43~45,XX,add(5)(q31),-6,add(7)(q21),-8,-9,+r[5]/43~45,XX,add(5),-6,del(7)(q21),+8[2],der(11;17)(q10;q10),del(12)(p11.2),-14,add(18)(q21),+r,+mar[15]. The derivative chromosome der(11;17)(q10;q10) resulted in loss of chromosomes 11p and 17p, including the locus of beta globin cluster (11p15.5) and the TP53 gene (17p13). Sequencing of the alpha and beta globin genes showed no sequence alterations, but Multiplex Ligation-dependent Probe Amplification (MLPA) demonstrated abnormal copy number of chromosome 11p.
Discussion
Inherited alterations in expression of the beta globin genes are associated with a broad spectrum of clinical presentations, from an asymptomatic beta thalassemia trait that is only of reproductive consequences, to the severe transfusion-dependent phenotype of beta thalassemia major. Acquired clonally restricted deletion of the alpha globin gene cluster or mutation of ATRX is well described in MDS; however, this report is one of only a very small number of descriptions of acquired beta thalassemia. In one such report, two patients with beta thalassemia trait (β39C→T) were found to have a somatic mutation that led to a beta thalassemia intermedia phenotype due to a somatic deletions of the wildtype β39C globin gene locus.4 Other reports of acquired beta thalassemia in MDS identified beta thalassemia using hemoglobin electrophoresis,2,5 including one case of acquired delta beta thalassemia, without genetic analysis.3 In the present case, cytogenetic and molecular testing revealed beta thalassemia due to a subclone with chromosome 11p loss resulting in HBB haploinsufficiency. Hemoglobin electrophoresis can help distinguish underlying abnormalities in globin synthesis; normal results include ~98% HbA, 1–2% HbA2, and <1% HbF. Patients with beta thalassemia have different electrophoresis phenotypes depending on the number of and type of alterations. Beta thalassemia trait has 92–95% HbA, >3,8% HbA2, and 1–4% HbF. Patients with beta thalassemia intermedia have residual variable beta globin synthesis with HbA of 10–30%, HbA2 of 2–5%, and HbF 70–90%. Beta thalassemia major results in no beta globin synthesis and shows 0% HbA, >95% HbF, and 2–5% HbA2. Our patient had 94.5% HbA, 2.4% HbA2, and 3.1% HbF, most consistent with acquired beta thalassemia trait.
MDS is commonly associated with ineffective erythropoiesis and normocytic or macrocytic anemia; microcytic red cell indices should prompt assessment for another contributing cause such as iron deficiency, acquired thalassemia, copper deficiency or severe inflammation.
Gains and losses in genetic material are characteristic of MDS, including recurrent chromosome 11 abnormalities. Trisomy 11 is observed in 1.5% of cases and deletions in 11q in another 1%, while, in one study of 1084 MDS patients, haploinsufficiency of chromosome 11 or deletions of 11p were present in 2%, including del(11p) in 6 patients, add(11p) in 2, and der(11) in 5; all of these patients had complex karyotypes.6 Therefore, beta thalassemia may be more common than described. These findings underscore the spectrum of phenotypic alterations that may occur in MDS and other hematologic malignancies and highlight another mechanism of ineffective hematopoiesis that may contribute to anemia associated with MDS.
Acknowledgments
we thank Drs. Karen Ballen, Elizabeth Van Cott, Aliyah Sohani, and Paola Dal Cin for their assistance in the evaluation of this patient. AMB is funded in part by the NIH, grant number T32 CA 071345.
Footnotes
Disclosures: The authors report no relevant financial conflicts.
References
- 1.Steensma DP, Gibbons RJ, Higgs DR. Acquired α-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies. Blood. 2005;105(2):443–452. doi: 10.1182/blood-2004-07-2792. [DOI] [PubMed] [Google Scholar]
- 2.Hoyle C, Kaeda J, Leslie J, Luzzatto L. ACQUIRED β THALASSAEMIA TRAIT IN MDS. Br. J. Haematol. 1991;79(1):116–117. doi: 10.1111/j.1365-2141.1991.tb08017.x. [DOI] [PubMed] [Google Scholar]
- 3.Markham RE, Butler F, Goh K, Rowley PT. Erythroleukemia manifesting delta beta-thalassemia. Hemoglobin. 1983;7(1):71–78. doi: 10.3109/03630268309038402. [DOI] [PubMed] [Google Scholar]
- 4.Galanello R, Perseu L, Perra C, et al. Somatic deletion of the normal β-globin gene leading to thalassaemia intermedia in heterozygous β-thalassaemic patients. Br. J. Haematol. 2004;127(5):604–606. doi: 10.1111/j.1365-2141.2004.05237.x. [DOI] [PubMed] [Google Scholar]
- 5.Peters RE, May A, Jacobs A. Increased alpha:non-alpha globin chain synthesis ratios in myelodysplastic syndromes and myeloid leukaemia. J. Clin. Pathol. 1986;39(11):1233–1235. doi: 10.1136/jcp.39.11.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385–4395. doi: 10.1182/blood-2007-03-082404. [DOI] [PubMed] [Google Scholar]