

Abstract
Phosphorylation of GluA1, a subunit of AMPA receptors (AMPARs), is critical for AMPAR synaptic trafficking and control of synaptic transmission. cGMP-dependent protein kinase II (cGKII) mediates this phosphorylation, and cGKII knockout (KO) affects GluA1 phosphorylation and alters animal behavior. Notably, GluA1 phosphorylation in the KO hippocampus is increased as a functional compensation for gene deletion, while such compensation is absent in the prefrontal cortex. Thus, there are brain region-specific effects of cGKII KO on AMPAR trafficking, which could affect animal behavior. Here, we show that GluA1 phosphorylation levels differ in various brain regions, and specific behaviors are altered according to region-specific changes in GluA1 phosphorylation. Moreover, we identified distinct regulations of phosphatases in different brain regions, leading to regional heterogeneity of GluA1 phosphorylation in the KO brain. Our work demonstrates region-specific changes in GluA1 phosphorylation in cGKII KO mice and corresponding effects on cognitive performance. We also reveal distinct regulation of phosphatases in different brain region in which region-specific effects of kinase gene KO arise and can selectively alter animal behavior.
AMPA receptors (AMPARs) are one type of glutamatergic postsynaptic receptor that mediates fast synaptic transmission in the central nervous system. They are tetrameric ligand-gated ion channels consisting of a combinatorial assembly of four subunits (GluA1–4) (Cull-Candy et al. 2006). GluA1 is the major subunit that is trafficked from recycling endosomes to the synaptic membrane in response to neuronal activity (Ehlers 2000). Altered GluA1 expression is important in various brain disorders, as aberrant GluA1 expression has been detected in brains of people who suffered from schizophrenia and bipolar disorder (Maksimovic et al. 2014). Several phosphorylation sites within the intracellular carboxyl-terminal domain (CTD) of GluA1 can regulate AMPAR membrane trafficking (Lee 2006; Derkach et al. 2007). Phosphorylation of serine 845 of GluA1 (GluA1-pS845) is important for activity-dependent trafficking of GluA1-containing AMPARs and increases the level of extrasynaptic receptors (Roche et al. 1996; Malinow and Malenka 2002; Derkach et al. 2007; Serulle et al. 2007; Shepherd and Huganir 2007). Behavioral studies of learning and memory show a contribution of GluA1-pS845 and experimental manipulations that prevent the phosphorylation of GluA1-pS845 have detrimental effects on learning and memory behaviors (Lee et al. 2003; Havekes et al. 2007; Clem and Huganir 2010). cAMP-dependent protein kinase A (PKA) and cGMP-dependent protein kinase II (cGKII) can mediate this phosphorylation (Derkach et al. 2007; Serulle et al. 2007). cGKII-mediated GluA1-pS845 is critical for hippocampal long-term potentiation (LTP) (Serulle et al. 2007). On the other hand, calcineurin dephosphorylates S845 of GluA1, which enables GluA1-containing AMPARs to be endocytosed from the plasma membrane during the induction of long-term depression (LTD) (Lee et al. 1998; Sanderson et al. 2012). Taken together, GluA1 phosphorylation is important for AMPAR-mediated synaptic plasticity.
Alterations in GluA1 trafficking and phosphorylation in various brain regions have different effects on different behaviors. In the lateral amygdala (LA) injecting a peptide that mimics the GluA1 carboxy terminus membrane proximal region has a detrimental effect on fear memory acquisition (Ganea et al. 2015). Phosphorylation of GluA1 S831 in the LA is important in fear renewal (Lee et al. 2013; Park et al. 2014), while GluA1-pS845 plays a role in the ventral striatum in spatial learning (Ferretti et al. 2015). Lithium, a common treatment used for bipolar disease, also has anti-depressant effects on behaviors assayed by the tail suspension test and forced swim test in an AMPAR dependent manner (Gould et al. 2008). These studies demonstrate the importance of AMPARs and the GluA1 subunit in various behaviors.
Although cGKII is important for GluA1-pS845 and LTP (Serulle et al. 2007), cGKII knockout (KO) shows functional compensation in the hippocampus via the calcineurin-mediated homeostatic mechanism (Kim et al. 2015a). In the KO hippocampus, synaptic GluA1-pS845 is increased, expressing GluA2-lacking Ca2+-permeable AMPARs, which provides a novel form of LTP as a compensatory response to the genetic deletion of cGKII (Kim et al. 2015a). However, we have shown that this compensation is restricted to the hippocampus since GluA1-pS845 is significantly reduced in the prefrontal cortex (PFC) (Wincott et al. 2013). Furthermore, impaired cGKII-mediated GluA1 S845 phosphorylation in the KO PFC, an indication of disrupted trafficking of AMPARs, results in defective working memory as determined using the PFC-dependent radial arm maze (RAM) (Wincott et al. 2014). Therefore, we propose that the functional compensation we described previously in the KO hippocampus is brain-region specific, thereby GluA1-pS845 differs in each brain area, which ultimately alters animal behavior. Here, we show that cGKII KO mice exhibit altered GluA1-pS845 in different brain regions, and such alteration affects animal behaviors. We also provide a mechanism that can account for these differences by identifying distinct regulation of phosphatases in the hippocampus and PFC, which leads to heterogeneity of GluA1 phosphorylation in the KO. Evaluation of tissue-specific effects of a particular gene KO on its functions is a challenging and important question in modern pharmacology, medical systems biology, and biomedicine. Therefore, this study provides evidence that the mechanism of control of GluA1 phosphorylation determines the outcome of gene KO for behavior. This is a novel situation in which neuronal biochemistry restricts the consequences of gene KO.
Results
Amygdala-dependent auditory cued fear conditioning is impaired in cGKII KO animals
Since cGKII is known to phosphorylate GluA1 (Serulle et al. 2007), we predicted that eliminating cGKII from the brains of mice should reduce the amount of GluA1-pS845 in various brain regions. Consistent with this prediction, we detected significantly reduced GluA1-pS845 in the amygdala of these mice, indicating disrupted AMPAR trafficking in synapses (Fig. 1A). In order to assess the behavioral contribution of reduced GluA1-pS845, we performed an auditory cued fear-conditioning task (Fig. 1B). Cued fear conditioning is dependent on the amygdala but not on the hippocampus (Phillips and LeDoux 1992). Therefore, we hypothesized that such reduction of GluA1-pS845 in the KO amygdala would lead to impaired cued fear conditioning. Mice were exposed to a neutral auditory cue for 30 sec where the final two seconds of the cue coincide with a mild shock. To test the fear memory, we measured freezing behavior 24 h later when these mice were exposed to the auditory cue alone while placed in an altered chamber (Fig. 1B). Consistent with our hypothesis, cGKII KO mice froze less than their WT littermates, consistent with reduced GluA1-pS845 in the amygdala disrupting AMPAR trafficking and impairing amygdala-dependent learning and memory.
Figure 1.
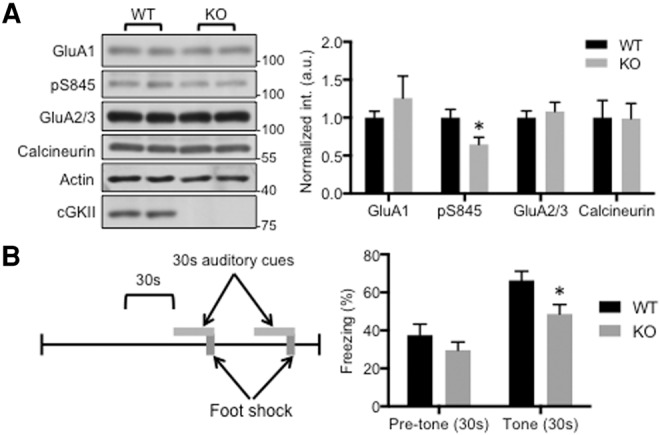
Reduction of GluA1 phosphorylation in the cGKII KO amygdala and impaired cued fear conditioning. (A) Representative immunoblots and quantitative analysis of whole amygdala lysates from the WT and KO animals showing significant reduction of GluA1-pS845 in the KO amygdala (n = 4 experiments, (*) P < 0.05, unpaired two-tailed student's t-test). (B) Diagram of auditory cued fear-conditioning protocol and summary of average percentage freezing during the 30 sec prior to the initial tone as well as the average freezing during each of the two tones for each genotype showing reduced freezing levels for KO mice during presentation of auditory cue (n = 20 WT and 21 KO mice, (*) P < 0.05, unpaired two-tailed Student's t-test).
Hippocampus-dependent contextual fear conditioning is enhanced in cGKII KO animals
We showed in a previous study that functional compensation occurs in the hippocampus, and synaptic GluA1-pS845 levels are increased in cGKII KO mice compared with WT littermates (Kim et al. 2015a). We wanted to examine whether this functional compensation via increased GluA1-pS845 in the hippocampus had a different behavioral effect than the reduced GluA1-pS845 levels we observed in the amygdala (Fig. 1A). To test this, we performed a contextual fear-conditioning task where freezing behavior is known to require hippocampal function along with amygdala activity (Phillips and LeDoux 1992). 24 h after the initial training session, KO mice were reexposed to the same environment where they received foot shocks previously. In contrast with performance in the auditory cued fear-conditioning task (Fig. 1B), the cGKII KO mice froze more than their WT littermates during the 5-min reexposure period (Fig. 2A), suggesting that they had enhanced contextual fear memory. In order to preclude the possibility that increased freezing behavior is due to reduction of memory extinction in KO mice, we measured fear memory 3 d after the shock and found that there was no significant difference in memory extinction between WT and KO animals (Fig. 2B). We next investigated short-term memory, which was determined by measuring freezing behavior 1 h after the initial training to identify whether enhanced fear conditioning in KO animals is dependent on memory formation or learning. There was no significant difference in short-term memory, suggesting that increased fear is due to enhanced memory rather than learning (Fig. 2C). This suggests that KO animals have enhanced contextual fear memory, consistent with an increase in synaptic GluA1-pS845 levels in the KO hippocampus.
Figure 2.

Enhanced contextual fear conditioning in cGKII KO animals. (A) Average percentage freezing 24 h after the initial training for WT and KO mice during each minute of the 4-min exposure to the training chamber showing increased freezing behavior in the KO mice during the final 2 min of the fear-conditioning test (n = 28 WT and 28 KO mice, (*) P < 0.05 and (**) P < 0.01, unpaired two-tailed student's t-test). (B) Average percentage freezing for WT and KO mice during reexposure to the training context 1 and 3 d after training showing no change in rate of extinction after day 3 (n = 28 WT and 28 KO mice). (C) Average percentage freezing 1 h after the initial training for WT and KO mice during each minute of the 4-min exposure to the training chamber showing no significant difference (n = 9 WT and 8 KO mice).
cGKII KO animals show less depression-like behaviors
Nitric oxide (NO) has been implicated as a secondary messenger in various cellular functions, including synaptic plasticity (Huang 1997). Ca2+ influx through NMDA receptors (NMDARs) activates neuronal nitric oxide synthase (nNOS), which generates NO (Garthwaite et al. 1989). NO activates guanylate cyclase that produces cGMP, and cGKII is a downstream signaling target of cGMP (Rameau et al. 2007; Serulle et al. 2007; Ota et al. 2008; Kim et al. 2015a). Several studies suggest that the inhibition of both nNOS and guanylate cyclase shows antidepressant-like effects (Eroglu and Caglayan 1997; Heiberg et al. 2002; Kaster et al. 2005; Joca and Guimaraes 2006; Jesse et al. 2008; Zomkowski et al. 2010). Therefore, we hypothesized that cGKII KO would have the same antidepressant effects as those inhibitors, via alteration of GluA1-mediated synaptic transmission. We focused on GluA1-pS845 levels in the nucleus accumbens (NAc) because it is a part of the mesolimbic dopamine system that is strongly associated with depression (Nestler and Carlezon 2006). We examined tissue from the NAc in cGKII KO mice. Similar to the amygdala tissue (Fig. 1A), the NAc had reduced synaptic GluA1-pS845 compared with WT littermates (Fig. 3A). Moreover, synaptic GluA1 and GluA2/3 levels were significantly decreased in the KO NAc (Fig. 3A), suggesting that there is impaired AMPAR trafficking in synapses of the KO NAc. We next performed two different depression-associated behavioral tasks that require NAc activity, the sucrose preference test (SPT) and the tail suspension test (TST). During the SPT, mice were exposed to 2% sucrose overnight. KO mice demonstrated an increased preference for sucrose compared with WT littermates (Fig. 3B) suggesting that KO mice exhibited less anhedonia. In addition, we performed the TST, in which mice were suspended by their tails for 6 min and the total amount of immobile hanging time was recorded as a measure of depressive behavior. KO mice displayed less immobile time compared with WT littermates (Fig. 3C). Taken together, the decrease in GluA1 phosphorylation in the NAc is thought to be responsible for the decreased expression of depressive behaviors in rodents where reduced GluA1 phosphorylation can have an antidepressive effect.
Figure 3.

Reduction of AMPAR levels in the NAc of KO animals and KO-mediated antidepressant effects. (A) Representative immunoblots and quantitative analysis of PSD from the NAc of the WT and KO animals showing significant reduction of GluA1, GluA1-pS845, and GluA2/3 in the KO NAc (n = 7 experiments, (****) P < 0.0001, unpaired two-tailed Student's t-test). (B) Percentage of sucrose water consumed out of total liquid consumption overnight for both WT and KO mice during the sucrose preference test showing significant increase of sucrose consumption for the KO mice (n = 15 WT and 15 KO mice, (*) P < 0.05, unpaired two-tailed Student's t-test). (C) Average of the total time WT and KO mice spent immobile during the 6-min tail suspension test showing significant reduction in immobility for KO mice (n = 16 WT and 16 KO mice, (*) P < 0.05 and (**) P < 0.01, unpaired two-tailed Student's t-test).
No alteration of GluA1 phosphorylation in the olfactory bulb and olfaction
It has been shown that cGKII is not present in parts of olfactory bulb, including the olfactory tubercle, external plexiform layer, and inner part of granular layer, which are critical for olfaction-related behavior (de Vente et al. 2001; Werner et al. 2004). Thus, we hypothesized that GluA1 phosphorylation would not be altered in the KO olfactory bulb. As expected, we detected no change in synaptic GluA1-pS845 in the olfactory bulb of cGKII KO mice (Fig. 4A). We predicted that the absence of altered GluA1-pS845 would result in no difference in olfactory dependent behavior tasks in the cGKII KO mice. In order to test this we performed a buried food test where an odorous snack was buried under the bedding and mice were allowed 20 min to find the hidden snack (Yang and Crawley 2009). Consistent with our hypothesis, there was no significant difference in the amount of time it took KO mice to find the food compared with WT littermates (Fig. 4B).
Figure 4.

No alteration in GluA1 phosphorylation in the KO olfactory bulb and normal olfactory function in KO animals. (A) Representative immunoblots and quantitative analysis of PSD from the olfactory bulb of the WT and KO animals showing no alteration of AMPAR trafficking in the KO olfactory bulb. (B) Average latency in seconds of the amount of time it took WT and KO mice to find and commence consumption of the hidden food showing no difference between WT and KO mice. (n = 13 WT and 15 KO mice).
Taken together these studies in various brain regions, amygdala, hippocampus, nucleus accumbens and olfactory bulb, plus our previous study of prefrontal cortex (Wincott et al. 2013; Kim et al. 2015a), show a strong correlation between the regional expression of GluA1-pS845 and behaviors requiring those brain areas, suggesting a predictive role for GluA1-pS845 in a behavioral readout.
Distinct regulation of phosphatases in the PFC and hippocampus leads to heterogeneity of GluA1-pS845 in the KO
In order to investigate the mechanism that underlies the different levels of GluA1-pS845, we focused on the region-specific control of phosphatases in the hippocampus and PFC although it has been proposed that there are regional differences in kinase regulation and expression (Ortiz et al. 1995; de Vente et al. 2001; Mazzucchelli et al. 2002; Havekes et al. 2007). Two different phosphatases are known to dephosphorylate GluA1-pS845, calcineurin and protein phosphatase 1 (PP1) (Lee et al. 1998, 2000). We have previously shown that a decrease in calcineurin activity is sufficient to elevate GluA1-pS845 in the KO hippocampus (Kim et al. 2015a). PP1 activity is regulated by endogenous PP1 inhibitors such as DARPP-32 (dopamine-and cAMP-regulated neuronal phosphoprotein-32) (Walaas and Greengard 1991). Phosphorylation of DARPP-32 at threonine 34 (DARPP-32-pT34) is mediated by dopamine signaling and inhibits PP1 activity (Cohen et al. 1989). As expected, DARPP-32 was highly enriched in the striatum (Fig. 5A). Interestingly, its expression in the PFC was much higher than the hippocampus (Fig. 5A). This suggests that PP1 activity in the PFC and hippocampus is differentially regulated, which could contribute to region-specific effects of cGKII KO on GluA1 phosphorylation. To further address this region specific effect, we investigated the role of PP1 in GluA1-pS845 in the PFC and hippocampus. We first treated cultured hippocampal neurons with specific phosphatase inhibitors and measured GluA1-pS845 (Fig. 5B). As shown previously (Kim and Ziff 2014; Kim et al. 2015a), 5 µM FK506, a calcineurin inhibitor, was sufficient to increase GluA1-pS845 in cultured hippocampal neurons (Fig. 5B). Of note, 500 nM okadaic acid (OA) treatment, sufficient for inhibiting both PP1 and PP2A, was able to increase GluA1-pS845 in hippocampal neurons (Fig. 5B). Moreover, treating hippocampal neurons with both FK506 and OA increased GluA1-pS845 but no more than each inhibitor alone (Fig. 5B). This suggests that calcineurin and PP1 work on the same pathway to regulate GluA1-pS845 in hippocampal neurons (Fig. 5E). We next measured alteration of GluA1-pS845 levels in cultured PFC neurons. Unlike hippocampal neurons, neither FK506 or OA alone, nor the two together had an effect on GluA1 phosphorylation (Fig. 5C). Notably, when we induced synaptic scaling by treating neurons with 2 µM tetrodotoxin (TTX) in PFC neurons, GluA1-pS845 levels were significantly increased in response to homeostatic synaptic plasticity (data not shown). This suggests that there are unknown pathways induced by TTX but not by FK506+OA, which are able to increase GluA1 phosphorylation in PFC neurons. This supports the idea that there are distinct pathways of phosphatase regulation in the hippocampus and PFC.
Figure 5.

Distinct regulation of phosphatases in the hippocampus and PFC affects GluA1 phosphorylation differently in the region-specific manner. (A) Representative immunoblots showing that DARPP-32 expression is higher in the PFC than the hippocampus. (B) Representative immunoblots and quantitative analysis of hippocampal neurons treated with FK506, OA, or FK506 and OA showing that inhibition of phosphatases is sufficient to increase GluA1-pS845 in cultured hippocampal neurons (n = 10 experiments, (**) P < 0.01 and (***) P < 0.001, one-way analysis of variance (ANOVA) followed by Fisher's Least Significant Difference (LSD) test). (C) Representative immunoblots and quantitative analysis of PFC neurons treated with FK506, OA, or FK506 and OA showing no alteration of GluA1 phosphorylation (n = 10 experiments). (D) Representative immunoblots showing that DARPP-32 phosphorylation is not altered in the KO PFC. (E) In the hippocampus, calcineurin directly regulates I-1 phosphorylation, so that calcineurin and PP1 work in the same Ca2+ pathway to control GluA1 phosphorylation and trafficking. (F) In the PFC, unlike the hippocampus, DARPP-32 would play a central role in control of PP1 activity. Because DARPP-32 is mainly regulated by the dopamine signaling pathway, Ca2+-dependent calcineurin activity and dopamine-mediated PP1 activity regulate GluA1 phosphorylation independently in the PFC.
Discussion
It is not surprising to see that loss of a kinase affects the functions of its targets in a region-specific manner (Mazzucchelli et al. 2002). In addition, there are regional differences in kinase regulation, including regulation by PKA and ERK (Ortiz et al. 1995; Havekes et al. 2007). However, the molecular mechanism of such specificity is not clear. Furthermore, tissue or region-specificity of inhibition of target functions in the brain is particularly important for drug design (Petrovskiy et al. 2015). Our previous studies of the hippocampus and PFC in cGKII KO mice have suggested that effects of cGKII KO on GluA1 phosphorylation are region-specific (Wincott et al. 2013; Kim et al. 2015a). Studies of various mental illnesses have demonstrated an important role for AMPARs in maintaining behavior (Kessels and Malinow 2009; Chang et al. 2012). This raises two questions; (1) How does the region specificity of changes in GluA1 phosphorylation affect behavior? and (2) What governs the region specificity of the effects of the KO of a kinase? In an effort to examine whether bidirectional changes in GluA1-pS845 levels correlate with changes in behavior in KO animals, we examined GluA1-pS845 in several different brain regions and tested behaviors dependent on those brain regions. Here, we demonstrate region-specific effects of the KO on GluA1-pS845 and cognitive performance associated with these brain regions. We also describe distinct pathways regulating phosphatases in the hippocampus and PFC that could underlie the region specific differences of GluA1-pS845 in the KO.
The cGKII KO reduces Ca2+ levels and calcineurin activity in the hippocampus as shown previously (Kim et al. 2015a). This reduced calcineurin activity in the KO hippocampus can have an effect on PP1 as well. PP1 inhibitor-1 (I-1) is an endogenous PP1 inhibitor, and the phosphorylated form of I-1 blocks PP1 activity (Nimmo and Cohen 1978). Notably, calcineurin dephosphorylates and inactivates I-1 in the hippocampus, which in turn increases PP1 activity to regulate hippocampal synaptic transmission (Fig. 5E; Mulkey et al. 1994). Because hippocampal expression of a second PP1 inhibitor, DARPP-32, is significantly lower than in the PFC (Fig. 5A), I-1 would serve as the major PP1 inhibitor in the hippocampus (Fig. 5E). This suggests that the concomitant decreases of calcineurin and PP1 activities stabilize GluA1 phosphorylation in hippocampal neurons (Fig. 5E). Indeed, we found that blocking both calcineurin and PP1 increased GluA1-pS845 in cultured hippocampal neurons but not in PFC neurons (Fig. 5B,C). Conversely, we found that DARPP-32 was abundantly expressed in the PFC but not in the hippocampus (Fig. 5A). We also found that DARPP-32-pT34 levels were not altered in the KO PFC compared with the WT PFC (Fig. 5D), suggesting that DARPP-32-mediated regulation of PP1 activity is independent of cGKII in the PFC, so that PP1 activity could remain active in the KO PFC (Fig. 5F). This suggests that in the PFC, unlike in the hippocampus, a dopamine-dependent and Ca2+-independent pathway regulates PP1 activity (Fig. 5F). Thus, normal PP1 activity in the KO PFC would lead to reduction of GluA1 phosphorylation and failure of synaptic trafficking of AMPARs, as observed previously (Wincott et al. 2013), contributing to impairment of PFC-dependent learning behavior (Wincott et al. 2014). Hence, the Ca2+-calcineurin dependence of GluA1 dephosphorylation in the hippocampus and the Ca2+-independence in the PFC could account for the observed region specific effects of cGKII KO on GluA1 phosphorylation, synaptic trafficking, and animal behaviors.
Notably, cGKII KO animals express Ca2+-permeable AMPARs (CP-AMPARs) at hippocampal synapses, which provide a basis for a novel form of LTP (Kim et al. 2015a). Expression of this form of AMPARs in the hippocampus enhances LTP (Jia et al. 1996; Wiltgen et al. 2010; Sanderson et al. 2012; Jin et al. 2013), which utilizes a distinct pathway compared with a traditional NMDAR-dependent LTP (Jia et al. 1996; Wiltgen et al. 2010; Jin et al. 2013; Kim et al. 2015a). In this study, we reveal that KO animals show enhanced hippocampus-dependent contextual fear learning and memory (Fig. 2). In contrast to our study, Wiltgen et al. (2010) report that GluA2 KO mice expressing CP-AMPARs in the hippocampus show impaired contextual fear conditioning. This discrepancy of the fear learning and memory could be due to the presence of GluA2-containing AMPARs. All AMPARs in GluA2 KO are GluA1 homomeric channels, which allows hippocampal LTP to be independent of NMDARs (Jia et al. 1996; Wiltgen et al. 2010), and impairs NMDAR-mediated learning (Wiltgen et al. 2010). Conversely, the hippocampus of cGKII KO that expresses CP-AMPARs alongside GluA2-containing AMPARs exhibit NMDAR-dependent LTP (Sanderson et al. 2012; Jin et al. 2013; Kim et al. 2015a). It is possible that expression of CP-AMPARs in the presence or absence of GluA2 would activate distinct pathways that result in different behavior outcomes. CP-AMPAR expression in the KO hippocampus could enhance contextual learning and memory if the CP-AMPARs can enhance memory or recognition of the context. A previous study shows that CP-AMPAR expression in the hippocampus is required for contextual discrimination (Jin et al. 2013), thus CP-AMPAR expression in the KO hippocampus could increase contextual discrimination, leading to enhanced fear learning. Since GluA1-pS845 levels are lower in the KO amygdala (Fig. 1A) and cued fear conditioning is impaired in the KO (Fig. 1B), it precludes amygdala activity as the source of this enhanced fear learning. Moreover, CP-AMPAR expression in the amygdala is required for proper amygdala-dependent fear learning and memory (Clem and Huganir 2010), which further confirms that the amygdala has no effect on this enhanced contextual learning in KO animals. Although our work demonstrates high region specificity for the compensatory increases in GluA1 phosphorylation, we cannot exclude that compensation in unidentified regions apart from hippocampus also contribute to the memory enhancement.
Although inhibition of the NO-cGMP signaling is involved in anti-depressive behaviors (Eroglu and Caglayan 1997; Heiberg et al. 2002; Kaster et al. 2005; Joca and Guimaraes 2006; Jesse et al. 2008; Zomkowski et al. 2010), the exact mechanism of action of the antidepressant-like effect in this model is not fully understood. cGKs are direct downstream effectors of NO-cGMP signaling (Francis and Corbin 1999), thus antidepressant action induced by inhibition of this pathway could be due to reduction of cGK activity. Indeed, we reveal that cGKII KO-mediated reduction of GluA1-pS845 in the NAc show antidepressant effects on behavior assays (Fig. 3). Therefore, the NO-cGMP-cGKII signaling-mediated GluA1 phosphorylation in the NAc would be required for expression of depressive behavior. In contrast to this study, it has been shown that chronic treatment with fluoxetine increases GluA1 phosphorylation in the striatum, and could potentiate AMPAR function as antidepressant actions (Svenningsson et al. 2002). This difference could be due to dual roles of GluA1 phosphorylation in the NAc. It has been suggested that various brain regions operate in a series of highly interacting parallel circuits, which formulate a neural circuitry involved in depression (Nestler et al. 2002). It is possible that GluA1 phosphorylation in the NAc during development would be required for formation of depression-like behavior by proper wiring of the circuitry. Conversely, after development is completed, GluA1 phosphorylation would potentiate AMPARs for antidepressant action. Thus, cGKII KO would fail to form adequate wiring in the brain during the development, which could contribute to antidepressive-like behavior.
Following on from the previous study (Kim et al. 2015a), we have raised two main questions—what are the effects of the brain region-specific GluA1 phosphorylation on behavior of cGKII KO animals and what are the mechanisms governing the region specificity of the effects of the cGKII KO? Alterations in GluA1 pS845 levels are known to affect behaviors, and transgenic mice expression a nonphosphorylatable mutant of GluA1, S845A, have altered learning and memory (Lee et al. 2003; Havekes et al. 2007; Clem and Huganir 2010). In this study, we demonstrate that GluA1 phosphorylation levels differed in various regions in the cGKII KO brain, and behaviors dependent on those brain regions were correspondingly altered in KO animals. Moreover, we identified distinct regulation of phosphatases in different brain regions, accounting for the heterogeneity of GluA1 phosphorylation in the KO brain. These observations suggest that the mechanisms of control of GluA1 phosphorylation determine the outcome for behavior. Thus this work provides a novel example in which neuronal biochemistry restricts the effects of gene KO on its functions, an important issue for understanding genetic control of behavior and drug design.
Materials and Methods
Animal
cGKII KO animals were backcrossed to C57BL/6 and bred at Taconic Farms in Germantown, NY. Animals were housed under 12:12 h light–dark cycle. All behavioral assays were conducted on adult mice between the ages of 10–12 wk. Animal experiments were conducted in compliance with the Institutional Animal Care and Use Committee at the New York University School of Medicine.
Rat hippocampal neuron cultures and drug treatment
Hippocampal primary neurons were carried out by the previously described method (Restituito et al. 2011). Neurons were isolated from embryonic day 19 Sprague Dawley (Charles River). To inhibit calcineurin or PP1 activity, 5 µM FK506 (Tocris Biosciences) or 500 nM okadaic acid (Tocris Biosciences) was treated to DIV14 neurons for 12 h. After treatment, whole-cell lysates were collected from cultured hippocampal neurons as shown previously (Restituito et al. 2011) and applied to immunoblots.
Tissue sample preparation and immunoblots
Whole-brain tissue and PSD fraction from mouse brain were prepared as described previously (Restituito et al. 2011; Kim et al. 2015b). Equal amounts of protein were loaded on 10% SDS-PAGE gel and transferred to the nitrocellulose or PVDF membranes. Membranes were blotted with GluA1 (Millipore, 1:5000), GluA2/3 (Millipore, 1:500), GluA1-pS845 (Millipore, 1:1000), actin (Sigma, 1:5000), DARPP-32-pT32 (Millipore, 1:1000), DARPP-32 (BD biosciences, 1:250), and cGKII (Serulle et al. 2007) (1:1000) antibodies and developed with ECL (PerkinElmer).
Fear conditioning
Mice were trained and tested using the FreezeFrame system (Coulbourn Instruments) as described previously with modifications (Lai et al. 2012). Briefly, Mice were habituated for 150 sec on a shocking grid (shocking floor grids, ethanol scent). Fear conditioning was conducted with two tone–foot shock pairings (30-sec, 400-Hz, 80-dB auditory cue coterminating with a 2-sec, 0.6-mA scrambled foot shock) separated by 60 sec. Contextual memory tests were carried out 24 h later for long-term memory or 1 h later for short-term memory in the same environment where mice were allowed to explore for 4 min. Cued memory tests were carried out 24 h after training in an altered environment (test floor grids, 1% Pinesol) where mice were allowed to explore for 150 sec before reexposure to the same auditory cues. During the extinction tests, mice were reexposed to the same environment for 4 min 2 d after the contextual memory test. Behavior scoring was automated by the FreezeFrame System (Coulbourn Instruments).
Tail suspension test (TST)
TST was performed as described previously (Cryan et al. 2005). In brief, male mice were suspended by their tails from a rod suspended 30 cm above the tabletop surface for 6 min. Immobile time was recorded for each mouse and averaged for each genotype.
Sucrose preference test (SPT)
The sucrose preference test was performed by presenting each male mouse with two identical bottles containing either 2% sucrose solution or water. Bottles were placed in the housing chamber overnight. At the end of each test, sucrose preference was calculated as volume of sucrose consumed divided by total liquid consumption for each mouse.
Buried food assay
The buried food assay was performed as described previously (Yang and Crawley 2009). Several days before the test, 2–3 Froot Loops (Kellogg's) were placed in each cage overnight to confirm that the food is palatable to the mice. Mice that did not consume the Froot Loops were omitted from the test. Mice were food-deprived for 24 h prior to the test. Three Froot Loops were placed in a clean cage and buried under fresh bedding prior to placing the mice in the cage. Mice were allowed 20 min to explore the cage searching for the hidden food and the latency to find and begin to nibble on the food was recorded. Mice that did not find the food after 20 min were excluded from the results.
Statistics
All statistical comparisons were analyzed with the GraphPad Prism6 software. Unpaired two-tailed Student's t-tests were used in single comparisons. For multiple comparisons, we used one-way analysis of variance (ANOVA) followed by Fisher's least significant difference (LSD) test to determine statistical significance. Results were represented as a mean ± sem, and P value <0.05 was considered statistically significant.
Acknowledgments
We appreciate discussions and comments on the manuscript by all members of the Ziff laboratory. We thank Dr. Wenbiao Gan (NYU) for sharing the fear-conditioning apparatus. This work was supported by a NIH grants, 5R01MH067229 (E.B.Z.) and T32DA007254 (J.E.P.).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.042960.116.
References
- Chang PK, Verbich D, McKinney RA. 2012. AMPA receptors as drug targets in neurological disease—advantages, caveats, and future outlook. Eur J Neurosci 35: 1908–1916. [DOI] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. 2010. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330: 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PT, Schelling DL, da Cruz e Silva OB, Barker HM, Cohen P. 1989. The major type-1 protein phosphatase catalytic subunits are the same gene products in rabbit skeletal muscle and rabbit liver. Biochim Biophys Acta 1008: 125–128. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Mombereau C, Vassout A. 2005. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29: 571–625. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. 2006. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol 16: 288–297. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. 2007. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8: 101–113. [DOI] [PubMed] [Google Scholar]
- de Vente J, Asan E, Gambaryan S, Markerink-van Ittersum M, Axer H, Gallatz K, Lohmann SM, Palkovits M. 2001. Localization of cGMP-dependent protein kinase type II in rat brain. Neuroscience 108: 27–49. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. 2000. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511–525. [DOI] [PubMed] [Google Scholar]
- Eroglu L, Caglayan B. 1997. Anxiolytic and antidepressant properties of methylene blue in animal models. Pharmacol Res 36: 381–385. [DOI] [PubMed] [Google Scholar]
- Ferretti V, Perri V, Cristofoli A, Vetere G, Fragapane P, Oliverio A, Teule MA, Mele A. 2015. Phosphorylation of S845 GluA1 AMPA receptors modulates spatial memory and structural plasticity in the ventral striatum. Brain Struct Funct 220: 2653–2661. [DOI] [PubMed] [Google Scholar]
- Francis SH, Corbin JD. 1999. Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit Rev Clin Lab Sci 36: 275–328. [DOI] [PubMed] [Google Scholar]
- Ganea DA, Dines M, Basu S, Lamprecht R. 2015. The membrane proximal region of AMPA receptors in lateral amygdala is essential for fear memory formation. Neuropsychopharmacology 40: 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Garthwaite G, Palmer RM, Moncada S. 1989. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol 172: 413–416. [DOI] [PubMed] [Google Scholar]
- Gould TD, O'Donnell KC, Dow ER, Du J, Chen G, Manji HK. 2008. Involvement of AMPA receptors in the antidepressant-like effects of lithium in the mouse tail suspension test and forced swim test. Neuropharmacology 54: 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Timmer M, Van der Zee EA. 2007. Regional differences in hippocampal PKA immunoreactivity after training and reversal training in a spatial Y-maze task. Hippocampus 17: 338–348. [DOI] [PubMed] [Google Scholar]
- Heiberg IL, Wegener G, Rosenberg R. 2002. Reduction of cGMP and nitric oxide has antidepressant-like effects in the forced swimming test in rats. Behav Brain Res 134: 479–484. [DOI] [PubMed] [Google Scholar]
- Huang EP. 1997. Synaptic plasticity: a role for nitric oxide in LTP. Curr Biol 7: R141–143. [DOI] [PubMed] [Google Scholar]
- Jesse CR, Bortolatto CF, Savegnago L, Rocha JB, Nogueira CW. 2008. Involvement of L-arginine-nitric oxide-cyclic guanosine monophosphate pathway in the antidepressant-like effect of tramadol in the rat forced swimming test. Prog Neuropsychopharmacol Biol Psychiatry 32: 1838–1843. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, et al. 1996. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945–956. [DOI] [PubMed] [Google Scholar]
- Jin SX, Arai J, Tian X, Kumar-Singh R, Feig LA. 2013. Acquisition of contextual discrimination involves the appearance of a RAS-GRF1/p38 mitogen-activated protein (MAP) kinase-mediated signaling pathway that promotes long term potentiation (LTP). J Biol Chem 288: 21703–21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joca SR, Guimaraes FS. 2006. Inhibition of neuronal nitric oxide synthase in the rat hippocampus induces antidepressant-like effects. Psychopharmacology 185: 298–305. [DOI] [PubMed] [Google Scholar]
- Kaster MP, Rosa AO, Santos AR, Rodrigues AL. 2005. Involvement of nitric oxide-cGMP pathway in the antidepressant-like effects of adenosine in the forced swimming test. Int J Neuropsychopharmacol 8: 601–606. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. 2009. Synaptic AMPA receptor plasticity and behavior. Neuron 61: 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ziff EB. 2014. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol 12: e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Titcombe RF, Zhang H, Khatri L, Girma HK, Hofmann F, Arancio O, Ziff EB. 2015a. Network compensation of cyclic GMP-dependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc Natl Acad Sci 112: 3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Violette CJ, Ziff EB. 2015b. Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol Aging 36: 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan WB. 2012. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 483: 87–91. [DOI] [PubMed] [Google Scholar]
- Lee HK. 2006. Synaptic plasticity and phosphorylation. Pharmacol Ther 112: 810–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. 1998. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151–1162. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. 2000. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955–959. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, et al. 2003. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112: 631–643. [DOI] [PubMed] [Google Scholar]
- Lee S, Song B, Kim J, Park K, Hong I, An B, Song S, Lee J, Park S, Kim J, et al. 2013. GluA1 phosphorylation at serine 831 in the lateral amygdala is required for fear renewal. Nat Neurosci 16: 1436–1444. [DOI] [PubMed] [Google Scholar]
- Maksimovic M, Vekovischeva OY, Aitta-aho T, Korpi ER. 2014. Chronic treatment with mood-stabilizers attenuates abnormal hyperlocomotion of GluA1-subunit deficient mice. PLoS One 9: e100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. 2002. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, et al. 2002. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34: 807–820. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. 1994. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369: 486–488. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA Jr. 2006. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59: 1151–1159. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. 2002. Neurobiology of depression. Neuron 34: 13–25. [DOI] [PubMed] [Google Scholar]
- Nimmo GA, Cohen P. 1978. The regulation of glycogen metabolism. Phosphorylation of inhibitor-1 from rabbit skeletal muscle, and its interaction with protein phosphatases-III and -II. Eur J Biochem 87: 353–365. [DOI] [PubMed] [Google Scholar]
- Ortiz J, Harris HW, Guitart X, Terwilliger RZ, Haycock JW, Nestler EJ. 1995. Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J Neurosci 15: 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KT, Pierre VJ, Ploski JE, Queen K, Schafe GE. 2008. The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn Mem 15: 792–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Song B, Kim J, Hong I, Song S, Lee J, Park S, Kim J, An B, Lee HW, et al. 2014. ABA renewal involves enhancements in both GluA2-lacking AMPA receptor activity and GluA1 phosphorylation in the lateral amygdala. PLoS One 9: e100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovskiy ED, Saik OV, Tiys ES, Lavrik IN, Kolchanov NA, Ivanisenko VA. 2015. Prediction of tissue-specific effects of gene knockout on apoptosis in different anatomical structures of human brain. BMC Genomics 16Suppl 13: S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. 1992. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106: 274–285. [DOI] [PubMed] [Google Scholar]
- Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L, Getzoff ED, Ziff EB. 2007. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci 27: 3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restituito S, Khatri L, Ninan I, Mathews PM, Liu X, Weinberg RJ, Ziff EB. 2011. Synaptic autoregulation by metalloproteases and γ-secretase. J Neurosci 31: 12083–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. 1996. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16: 1179–1188. [DOI] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML. 2012. AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J Neurosci 32: 15036–15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. 2007. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56: 670–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. 2007. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 23: 613–643. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Witkin JM, Fienberg AA, Nomikos GG, Greengard P. 2002. Involvement of striatal and extrastriatal DARPP-32 in biochemical and behavioral effects of fluoxetine (Prozac). Proc Natl Acad Sci 99: 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walaas SI, Greengard P. 1991. Protein phosphorylation and neuronal function. Pharmacol Rev 43: 299–349. [PubMed] [Google Scholar]
- Werner C, Raivich G, Cowen M, Strekalova T, Sillaber I, Buters JT, Spanagel R, Hofmann F. 2004. Importance of NO/cGMP signalling via cGMP-dependent protein kinase II for controlling emotionality and neurobehavioural effects of alcohol. Eur J Neurosci 20: 3498–3506. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Royle GA, Gray EE, Abdipranoto A, Thangthaeng N, Jacobs N, Saab F, Tonegawa S, Heinemann SF, O'Dell TJ, et al. 2010. A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincott CM, Kim S, Titcombe RF, Tukey DS, Girma HK, Pick JE, Devito LM, Hofmann F, Hoeffer C, Ziff EB. 2013. Spatial memory deficits and motor coordination facilitation in cGMP-dependent protein kinase type II-deficient mice. Neurobiol Learn Mem 99: 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincott CM, Abera S, Vunck SA, Tirko N, Choi Y, Titcombe RF, Antoine SO, Tukey DS, DeVito LM, Hofmann F, et al. 2014. cGMP-dependent protein kinase type II knockout mice exhibit working memory impairments, decreased repetitive behavior, and increased anxiety-like traits. Neurobiol Learn Mem 114C: 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Crawley JN. 2009. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci Chapter 8: Unit 8 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomkowski AD, Engel D, Gabilan NH, Rodrigues AL. 2010. Involvement of NMDA receptors and L-arginine-nitric oxide-cyclic guanosine monophosphate pathway in the antidepressant-like effects of escitalopram in the forced swimming test. Eur Neuropsychopharmacol 20: 793–801. [DOI] [PubMed] [Google Scholar]