

Abstract
Objective
NOX-1 and NOX-4 are key enzymes responsible for reactive oxygen species (ROS) generation in vascular smooth muscle cells (VSMC). The RNA-binding protein HuR is implicated in post-transcriptional regulation of gene expression; however, its role regulating NOX is unknown. We investigated transcriptional and post-transcriptional mechanisms underlying angiotensinII (AngII) and interleukin-1β (IL-1β) regulation of NOX-1 and NOX-4 in VSMC and their implications in cell migration.
Methods
Rat and human VSMC were stimulated with AngII (0.1 μM) and/or IL-1β (10 ng/mL). NOX-1 and NOX-4 mRNA and protein levels, NOX-1 and NOX-4 promoter and 3′UTR activities, NADPH oxidase activity, ROS production and cell migration were studied.
Results
IL-1β increased NOX-1 expression, NADPH oxidase activity and ROS production and decreased NOX-4 expression and H2O2 production in VSMC. AngII potentiated the IL-1β-mediated induction of NOX-1 expression, NADPH oxidase activity, ROS production and cell migration. However, AngII did not influence IL-1β-induced NOX-4 down-regulation. AngII+IL-1β interfered with the decay of NOX-1 mRNA and promoted HuR binding to NOX-1 mRNA. Moreover, HuR blockade reduced NOX-1 mRNA stability and AngII+IL-1β-induced NOX-1 mRNA levels. IL-1β decreased NOX-4 expression through a transcriptional mechanism that involved response elements situated in the proximal promoter. AngII and/or IL-1β-induced cell migration were prevented by NOX-1 and HuR blockade and were augmented by NOX-4 overexpression.
Conclusion
In VSMC HuR-mediated mRNA stabilization is partially responsible for AngII+IL-1β-dependent NOX-1 expression whereas transcriptional mechanisms are involved in decreased NOX-4 expression induced by IL-1β. NOX4 and HuR regulation of NOX-1 contributes to VSMC migration, important in vascular inflammation and remodeling.
Keywords: Vascular smooth muscle cells, angiotensin II, interleukin 1β, HuR, NADPH oxidase, cell migration
INTRODUCTION
Reactive oxygen species (ROS) are important modulators of vascular tone and structure. An imbalance between oxidants and antioxidant systems increases ROS steady-state levels and participates in multiple cardiovascular diseases such as hypertension by modulating vascular smooth muscle cell (VSMC) proliferation and migration [1]. It is well established that angiotensin II (AngII) and cytokines are key mediators of vascular damage in hypertension at least in part through ROS production [2,3]. NADPH oxidase is a major contributor of ROS production in VSMC [1]. Seven NADPH oxidase isoforms have been identified in mammals, each containing a catalytic subunit called NOX (NOX-1-5) or DUOX (DUOX-1/2) and various regulatory subunits [1]. Of the NOX isoforms, NOX-1 and NOX-4 appear to be particularly important in VSMC. Besides their different subcellular location, it has been suggested that each NOX isoform might produce different ROS, with NOX-1 generating O2•− and NOX-4 producing primarily H2O2 [4]. Functionally, NOX-1/NOX-4-derived ROS seem to promote VSMC proliferation and migration [5–7].
NOX-1 is an inducible gene and its expression is increased in vascular cells by several pro-inflammatory stimuli including AngII, interferon-γ (IFN-γ) and platelet derived growth factor (PDGF) [8–11]. Various transcription factors such as ATF-1 [12,13] or STAT [9] have binding sites in NOX-1 promoter and play an important role in its inducible expression. In addition, NOX-1 bears several AU-rich elements (AREs) in its 3′ untranslated region (3′UTR), which are involved in the control of mRNA stability [14]. Different RNA-binding proteins are responsible for stability of different mRNA. One of the best characterized is HuR (Hu antigen R; ELAVL1). HuR stabilizes different mRNAs by binding to AU-rich elements found in 3′UTR [14] and it is implicated in various physiological and pathological processes such as cell growth, differentiation and inflammation. However, no studies have evaluated whether post-transcriptional mechanisms mediated by HuR participate in the control of NOX-1 expression and activity in VSMC.
Similarly to NOX-1, inducible expression of NOX-4 has been reported in VSMC in response to different stimuli including hypoxia [15], transforming growth factor-β (TGF-β) [16], IFN-γ [9] or tumor necrosis factor-α (TNF-α) [17]. Transcriptional mechanisms involving the JAK/STAT signaling pathway [9] and NF-kB [17] are involved in these effects. However, other studies have demonstrated that pro-inflammatory stimuli such as AngII, interleukin-1β (IL-1β), thrombin and PDGF reduce NOX-4 expression [8,18], through poorly characterized processes.
In this study, we investigated transcriptional and post-transcriptional mechanisms involved in the regulation of NOX-1 and NOX-4 expression and activity by AngII and IL-1β in VSMC and their implications in cell migration. To ensure human relevance of our studies, we examined VSMC from both rats and humans.
MATERIALS AND METHODS
All experimental procedures were approved by the Reviewer Institutional Committee on Human Research of the Hospital de la Santa Creu i Sant Pau, that conforms to the Declaration of Helsinki and by the Institutional Animal Care and Use Committees of our institutions, according to the guidelines for ethical care of experimental animals of the International Principles for Biomedical Research Involving Animals, the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and the current Spanish and European laws (RD53/2013 and 2010/63/UE)
Cell culture
Primary cultures of VSMC were obtained from Sprague Dawley rat aortas and grown in DMEM-F12 medium supplemented with 10% (v/v) fetal bovine serum (FBS) containing 100 U/ml of penicillin and 100 μg/ml of streptomycin as previously reported [19]. Human VSMC were obtained from coronary arteries of hearts removed in transplant operations by using a modification of the explant technique [20]. Human VSMC were grown in M199 supplemented with 20% (v/v) FBS, 1% (v/v) human serum, 2 mmol/l of L-glutamine, 100 U/ml of penicillin and 100 μg/ml of streptomycin. Cells were starved in serum free media for 24 h prior to stimulation. All products were purchased to Sigma-Aldrich (St. Louis, MO, USA).
Cells were stimulated with vehicle (control), AngII (Sigma-Aldrich), IL-1β (Sigma-Aldrich) or AngII+IL-1β at the times and concentrations indicated. The effects of the following inhibitors were analyzed by their addition 30 min (or 4 h in the case of MS-444) before and throughout stimulation: actinomycin D, ML171 and cycloheximide (CHX) (Sigma-Aldrich); U0126, SP600125, SB203580 and LY294002 (Calbiochem, Darmstadt, Germany) and MS-444 (generously provided by Novartis, Basel, Switzerland). MS-444 was originally characterized as myosin light chain kinase inhibitor in the μM-range [21], however the concentrations used in these experiments did not affect myosin light chain phosphorylation induced by AngII+IL-1β or cell viability (Supplementary Figure S1A and B). AngII and IL-1β were dissolved in distilled water; the rest of compounds were dissolved in DMSO. Data of stimulated cells in the presence of inhibitors were normalized to the effect of the inhibitors alone.
RNA analysis
Cells were harvested in TRI Reagent (Sigma-Aldrich) or TRIzol (Life Technologies Inc., Gaithersburg, Maryland, USA) according to the manufacturer’s recommendations to obtain total RNA and reverse transcribed using the High Capacity cDNA Archive Kit (Life Technologies) with random hexamers. Quantitative PCR (qPCR) was performed in a 7500 Fast ABI System (Life Technologies). qPCR for human NOX-1, NOX-4 and 18S rRNA and rat PPAR-γ were performed using Taqman Gene Expression Assays (rat PPAR-γ: Rn00440945_m1; human NOX-1 Hs00246589_m1; human NOX-4: Hs00418356_m1; human 18S rRNA: Hs99999901_s1, Applied Biosystems, Foster City, CA, USA). qPCR for rat NOX-1, NOX-4, Egr-1 and β2-microglobulin were performed using SYBR green PCR master mix (iTaq FAST SYBRGreen Supermix with ROX, Bio-Rad, USA). Rat primer sequences are: NOX-1 (FW: CGGCAGAAGGTCGTGATTA; RV: TGGAGCAGAGGTCAGAGT), rat NOX-4 (FW: GCCTCCATCAAGCCAAGA; RV: CCAGTCATCCAGTAGAGTGTT), Egr-1 (FW: CAGCGCTTTCAATCCTCAAG; RV: GCGATGTCAGAAAAGGACTCTGT) and β2-microglobulin (FW: ACCCTGGTCTTTCTGGTGCTT; RV: TAGCAGTTCAGTATGTTCGGCTT). PCR cycles proceeded as follows: initial denaturation for 30 s at 95°C, followed by 40 cycles at 95°C for 5 s and 60°C for 30 s. Melting curve analysis was performed in SYBR green reactions to show PCR product specificity. Data analyses used the 2−ΔΔCt method, where β2-microglobulin or 18S rRNA served as the internal control.
NOX-1 and NOX-4 mRNA stability was determined in 24 h-stimulated cells. Then (t=0) actinomycin D (5 μg/ml Sigma-Aldrich) was added to the growth medium. Total RNA was isolated at indicated times and NOX-1, NOX-4 and β2-microglobulin mRNA expression levels were measured by qPCR, as indicated above.
Western blot analysis
Whole-cell lysates were harvested in RIPA buffer containing: 50 mmol/l Tris pH 7.5, 150 mmol/l NaCl, 1 mmol/l MgCl2, 1 mmol/l EDTA, 1% (v/v) Nonidet-P40 (NP-40), 0.5% (w/v) deoxycholate Na, 1% (v/v) sodium dodecyl sulfate (SDS), a protease inhibitor cocktail (Roche Applied Science, Barcelona, Spain) and a mix of phosphatase inhibitors (1 mmol/l orthovanadate, 20 mmol/l β-glycerophosphate, 10 mmol/l NaF from Sigma-Aldrich). Protein content was determined with Lowry (Bio-Rad) or BCA protein assay reagent (Pierce, Rockford, IL, USA), using bovine serum albumin (BSA, Sigma-Aldrich or Pierce) as standard. Lysates (30 μg) were separated by 10% SDS-PAGE, transferred to PVDF or nitrocellulose membranes (Amersham, GE Healthcare, Buckinghamshire, UK) and probed with antibodies against NOX-1 and NOX-4 (1:200; SC25545 and SC30141 respectively; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), p-MLC and MLC (1:1,000; Cell Signaling, Boston, MA, USA). Detection was accomplished using horseradish peroxidase-coupled anti-rabbit (1:2,000; Bio-Rad) IgG antibody for 1 h at room temperature. Signal was detected using the Luminata Forte (Millipore Corporation, Billerica, MA, USA) detection system. Immunoblot signals were quantified by NIH ImageJ using β-actin expression as loading control.
Ribonucleoprotein complex immunoprecipitation
Ribonucleoprotein complex immunoprecipitation (RNP-IP) was performed as described [22]. Briefly, cells were lysed in polysome lysis buffer [20 mmol/l Tris-HCl pH 7.5, 150 mmol/l NaCl, 5 mmol/l MgCl2, 1 mmol/l DTT, 0.5% (v/v) NP-40, 1 mmol/l PMSF, protease inhibitors, 100 U RNAse inhibitor (Promega, Mannheim, Germany)]. 200 μg of cytoplasmic lysate was incubated with anti-HuR antibody or control IgG precoated to protein A/G PLUS agarose (Santa Cruz Biotechnology) overnight at 4°C in NT2 buffer (50 mmol/l Tris-HCl pH 7.5, 150 mmol/l NaCl, 1 mmol/l MgCl2, 0.05% (v/v) NP-40) adding 0.1 mmol/l DTT, 40 U RNAse inhibitor, 5 mmol/l EDTA, 100 μg/ml tRNA (Promega). Immunoprecipitates were collected by centrifugation and washed 4 times with NT2 buffer. Total RNA was isolated from immunoprecipitates using 1 ml TRIzol reagent and then used for cDNA synthesis and qPCR as described above.
DNA and siRNA transfections
The luciferase reporter vector containing 2,700 bp of the rat NOX-1 promoter (−2547 +125) (pGL3-NOX-1rP) was kindly provided by Dr. J. Pfeilschifter (University of Frankfurt Medical School, Germany). 630 bp of NOX-1 3′UTR pGL3 control was purchased from Genewiz. NOX-4 fused to EGFP (NOX-4/EGFP) was kindly provided by Dr. L. Terada (University of Texas Southwestern Medical Center, Dallas, Texas, USA). Egr-1 overexpression was accomplished using Flag-tagged Egr-1 cDNA cloned in pcDNA3.1/Zeo+.
Cells were seeded in 12-well culture plates at a density of 6×104 cells per well and transiently transfected with 0.5 μg luciferase reporter gene construct per well using the Lipofectamine LTX Plus reagent (Life Technologies). After 6 h, medium was changed and cells were serum-starved overnight. After 24 h of stimulation, cells were harvested in reporter lysis buffer and luciferase activities were measured using a luminometer (Berthold Detection System, Sirius, Pforzheim, Germany) according to the manufacturer’s instructions. Relative luciferase activities were normalized to total protein.
Cells seeded in 6-well plates or transwells at a density of 1.2×105 or 3×104 cells per well, respectively, were transfected with a predesigned siRNA against HuR, NOX-1 (20 nmol/l; Life Technologies) or a negative control siRNA (Qiagen-Izasa, Barcelona, Spain) using the Lipofectamine LTX Plus reagent (Life Technologies) according to the manufacturer’s protocol. Silencing efficiency was determined by western blot or qPCR 48 h after transfection.
NADPH oxidase activity
The O2•− production generated by NADPH oxidase activity was determined by a chemiluminiscence assay using lucigenin (5 μmol/l; Sigma-Aldrich) and NADPH (100 μmol/l; Sigma-Aldrich). The reaction was started by the addition of a lucigenin/NADPH mixture to the cell lysate in a final volume of 250 μl. Chemiluminescence was determined every 2.4 seconds for 5 min in a plate luminometer (AutoLumat LB 953, Berthold, Germany). Buffer blank was subtracted from each reading. Luminescence was normalized by protein concentration measured by the Lowry assay and data were expressed as fold increase over the control.
Detection of ROS by fluorescence microscopy
The oxidative fluorescent dye dihydroethidium (DHE) was used to evaluate oxidative stress in situ in VSMC. DHE freely permeates cells and upon oxidation, becomes positively charged and accumulates in cells by intercalating into DNA. Briefly, VSMC were plated onto glass coverslips inserted into 6-well plates and cultured and stimulated as described above. Afterwards, cells were loaded with DHE (10 μmol/l; Sigma-Aldrich) in cell culture medium for 30 min at 37°C. Using the same imaging settings for all experimental conditions, images were then acquired with a confocal microscope (Ex561 and Em610 nm, Leica SP2, objective 40×) and fluorescence intensity was measured using Metamorph image analysis software. Total fluorescence of DHE is a sum of the composite spectra of ethidium possibly formed by non-specific redox reactions and 2-OH-ethidium which is a specific adduct of superoxide anion.
H2O2 production by amplex red
Cells were seeded in 12-well plate, transfected with NOX-4/EGFP, EGFP alone or without transfection and then stimulated 24 h with AngII and/or IL-1β. In order to prevent interference with the resorufin measurement, we used phenol red-free medium. Supernatants were used to determine H2O2 release and cell lysates to measure total protein content. Amplex Red (100 μmol/l; Sigma-Aldrich) and horseradish peroxidase type II (0.2 U/ml; Sigma-Aldrich) were added to 50 μl of supernatants. Fluorescence readings were made in duplicate in a 96-well plate at Ex/Em = 530/580 nm. H2O2 concentration was estimated using a standard curve between 0–4.8 μmol/l of H2O2. Total protein of cell lysates as well as the volume of the supernatants was measured in order to normalize H2O2 values.
Cell viability and cell migration assays
Cell viability was assessed using the CellTiter 96 Non-Radioactive Cell Proliferation Assay MTT (Sigma-Aldrich). 8×103 cells were seeded on 96-well plates in DMEM-F12 medium. After stimulation, cell survival was quantified by adding MTT tetrazolium solution according to the manufacturer’s protocol. Absorbance was measured at 540 nm in an ELx800TM Absorbance Microplate Reader (BIOTek).
VSMC migration was examined using a 6.5 mm Transwell chamber with an 8 μm pore size (Corning Costar Inc., New York, NY, USA). 3×104 cells were serum-starved in the upper compartment of each chamber for 16 h; inhibitors were added to the upper chamber and the stimuli (AngII and/or IL-1β) were added to the bottom chamber. Cells were allowed to migrate 24 h and cells of the upper membrane surface were removed with a cotton swab. Then, the membrane was washed with PBS and migrating cells were fixed in 4% (v/v) paraformaldehyde. Migration values were determined by counting three fields per chamber after staining the migrated cells with Hoechst 33342 or DAPI (Life Technologies).
Cell migration and proliferation in response to physical damage was determined using a wound healing assay. VSMC monolayers were wounded using a sterile 10 μl pipette tip. Phase contrast images were taken immediately after wounding and at 24 h post-stimulation using a Nikon microscope (Tokyo, Japan) connected to a video camera (Sony Corporation, Tokyo, Japan). To measure migration, wound area was quantified using Adobe Photoshop and expressed as fold increase of wound closure in control cells.
Data analysis and statistics
All values are expressed as mean±SEM of independent cell culture-based experiments. Statistical analysis was done by Student’s t test or by one-way ANOVA followed by a Bonferroni test. Values were considered to be significant when P<0.05.
RESULTS
AngII and IL-1β synergistically induce NOX-1 expression and decrease NOX-4 expression in VSMC
Stimulation of cells with AngII (0.1 μmol/l) or IL-1β (10 ng/ml) led to an increase in NOX-1 mRNA and protein expression (Figure 1A, B; Supplementary Figure S2A). Co-stimulation of cells with AngII+IL-1β resulted in a synergistic increase in NOX-1 mRNA and protein levels that become evident after 8 h stimulation and persisted out to 24 h (Figure 1A, B; Supplementary Figure S2A). In contrast, AngII reduced NOX-4 mRNA levels without modifying the protein expression and IL-1β substantially decreased NOX-4 mRNA and protein levels (Figure 1C, D; Supplementary Figure S2B). The addition of AngII to IL-1β-treated cells had no further effect on NOX-4 levels at any time analyzed (Figure 1C, D; Supplementary Figure S2B). Studies were also performed in human VSMC and results are shown in Supplementary Figure S3; in human VSMC stimulated for 24 h, the effects of AngII, IL-1β or AngII+IL-1β on NOX-1 and NOX-4 mRNA levels were similar indicating an interspecies conserved effect.
Figure 1.
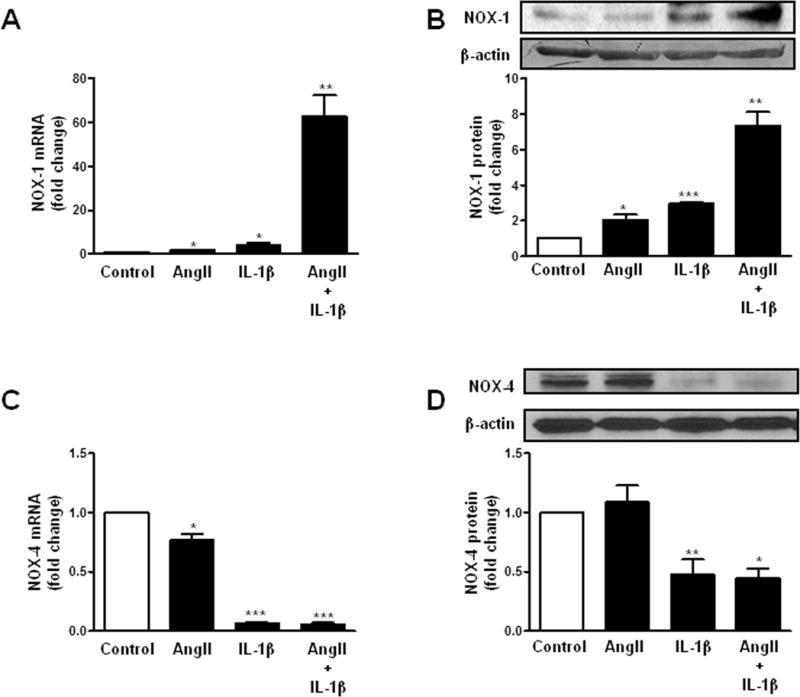
AngII potentiates IL-1β-induced NOX-1 expression and does not modify IL-1β-dependent NOX-4 decrease expression in VSMC. Effect of AngII, IL-1β and AngII+IL-1β on NOX-1 mRNA (A) and protein (B) levels, and in NOX-4 mRNA (C) and protein (D) levels in rat VSMC. Representative blots are also shown. Data are expressed as mean±SEM. *P <0.05, **P<0.01, ***P<0.001 vs Control. n=4–10.
We therefore analyzed the signaling pathways involved in the synergistic effect of AngII+IL-1β on NOX-1 expression and in IL-1β-induced NOX-4 down-regulation. Supplementary Figure S4 shows that AngII and/or IL-1β promoted a rapid phosphorylation of ERK1/2, JNK, p38 MAPK and Akt as soon as 5 min after treatment. At 24 h, only p38 and Akt were activated by AngII+IL-1β (Supplementary Figure S4). Interestingly, pre-treatment with specific inhibitors of ERK1/2 (U0126, 10 μmol/l), JNK (SP600125, 20 μmol/l) and p38 MAPK (SB203580, 10 μmol/l) reduced the AngII+IL-1β-induced NOX-1 mRNA levels at 24 h (Figure 2A). On the other hand, U0126, SP600125 and a PI3K inhibitor (LY294002, 10 μmol/l) partially blocked the IL-1β-dependent NOX-4 down-regulation (Figure 2B). These findings suggest that early activation of MAPK and PI3K/Akt are likely responsible for the observed effects on NOX-1 and NOX-4 mRNA levels.
Figure 2.
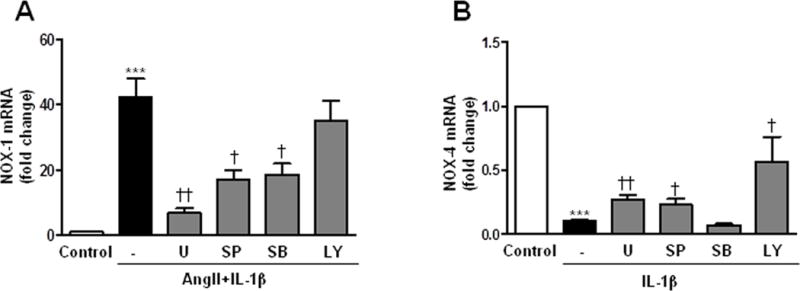
Signaling pathways involved in the expression of NOX-1 and NOX-4 induced by AngII and/or IL-1β. Effect of inhibitors of ERK1/2 (U, U0126), JNK (SP, SP600125) and p38 (SB, SB203580) MAPKs and PI3K (LY, LY294002) on NOX-1 (A) and NOX-4 (B) mRNA levels in VSMC stimulated with AngII and/or IL-1β (24 h). ***P<0.01 vs Control; †P<0.05, ††P<0.05 vs AngII+IL-1β or IL-1β. n=7–11.
AngII induces HuR-dependent NOX-1 mRNA stabilization
Next, we examined mechanisms by which AngII+IL1β potentiated NOX-1 expression. NOX-1 promoter activity increased in the presence of AngII and/or IL-1β, with responses being higher in the presence of both stimuli (Figure 3A). Furthermore, a time-course mRNA decay analysis showed that NOX-1 mRNA was stabilized in the presence of AngII+IL-1β (Figure 3B). Moreover, NOX-1 mRNA expression was increased in AngII+IL-1β-stimulated cells pretreated with actinomycin D, a transcriptional inhibitor (Supplementary Figure S5A). Together these results suggest that transcriptional and post-transcriptional regulatory mechanisms are involved in the effect of AngII+IL-1β. NOX-1 3′UTR luciferase activity was also increased in response to AngII+IL-1β, effects that were partially reversed by the inhibitors that partially restored the AngII+IL-1β-mediated NOX-1 mRNA increase (U0126, SP600125 and SB203580) (Supplementary Figure S5B).
Figure 3.
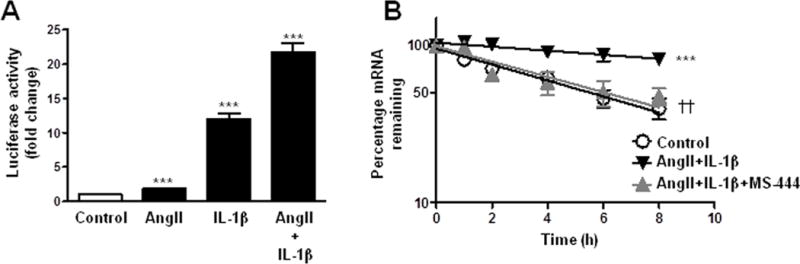
AngII potentiates IL-1β-induced NOX-1 expression in VSMC through transcriptional activity and NOX-1 mRNA stability. (A) Luciferase activity of NOX-1 promoter in cells stimulated with AngII, IL-1β or AngII+IL-1β for 24 h. (B) Effect of MS-444 on NOX-1 mRNA stability after 24 h of stimulation with AngII+IL-1β. NOX-1 mRNA levels were measured by qPCR at indicated times after actinomycin D addition (t=0). Data are expressed as mean±SEM. ***P<0.001 vs Control; ††P<0.01 vs AngII+IL-1β. n=5–7.
HuR is an RNA-binding protein that stabilizes different mRNAs by binding to AU-rich elements found in 3′UTR [14]. To test whether HuR was responsible for NOX-1 mRNA stabilization, we performed the mRNA stability assay in the presence of a HuR inhibitor (MS-444; [23]). As shown in Figure 3B, MS-444 (8 μmol/l) abolished the AngII+IL-1β-dependent NOX-1 mRNA stabilization. Furthermore, HuR blockade with MS-444 or with HuR siRNA (Supplementary Figure S6A) reduced AngII+IL-1β-induced NOX-1 mRNA levels (Figure 4A). To determine whether AngII+IL-1β signaling promotes HuR binding to NOX-1 mRNA, ribonucleoprotein immunoprecipitation assays were done. VSMC were stimulated for 24 h with AngII+IL-1β and immunoprecipitation of cytoplasmic lysates was performed using an antibody against HuR or a control IgG. We observed an enrichment in NOX-1 mRNA in anti-HuR precipitates from cells treated with AngII+IL-1β, but not in control cells or in anti-IgG precipitates (Figure 4B). These data suggest that AngII+IL-1β induce HuR binding to NOX-1 mRNA, which is responsible for increased NOX-1 mRNA stability.
Figure 4.
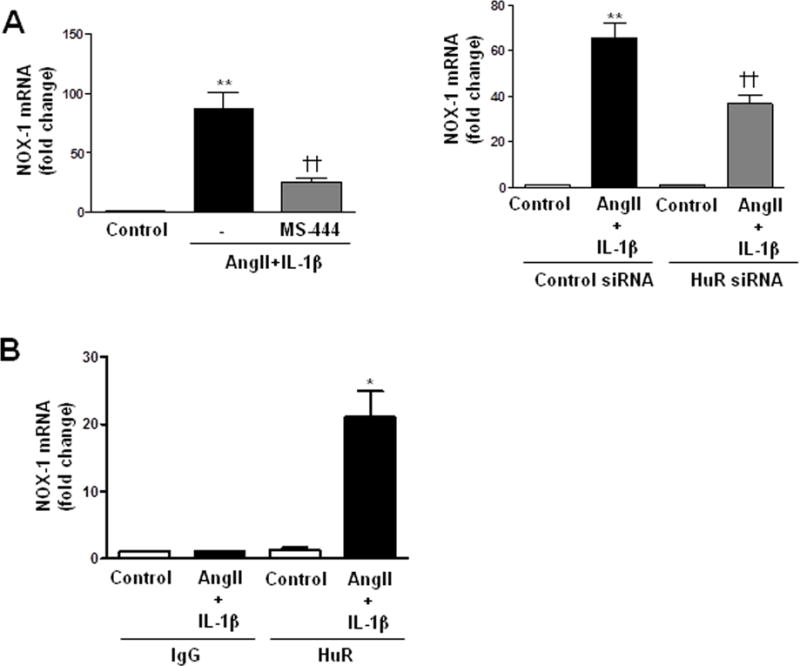
AngII potentiates IL-1β-induced NOX-1 expression in VSMC through HuR-dependent NOX-1 mRNA stability. (A) Effects of MS-444 and HuR siRNA on NOX-1 mRNA levels in VSMC stimulated with AngII+IL-1β for 24 h. (B) NOX-1 mRNA binding to HuR by ribonucleoprotein immunoprecipitation in VSMC stimulated with AngII+IL-1β for 24 h. Data are expressed as mean±SEM. *P<0.05, **P<0.01 vs Control; ††P<0.01 vs AngII+IL-1β. n=4–7.
IL-1β decreases NOX-4 through transcriptional repression
In VSMC, IL-1β did not modify NOX-4 mRNA decay (Figure 5A) supporting the involvement of a transcriptional mechanism. Accordingly, transient transfection assays with a luciferase reporter construct containing NOX-4 promoter [9], evinced that IL-1β reduced NOX-4 transcriptional activity (Figure 5B). Interestingly, the decrease in NOX-4 promoter activity triggered by IL-1β was partially reversed with U0126, SP600125 and LY294002 (Figure 5B) which were the same inhibitors that partially restored the IL-1β-mediated NOX-4 mRNA decrease. Serial deletion analysis of NOX-4 promoter region narrowed IL-1β-dependent down-regulation to the 90 bp proximal region (Figure 5C).
Figure 5.
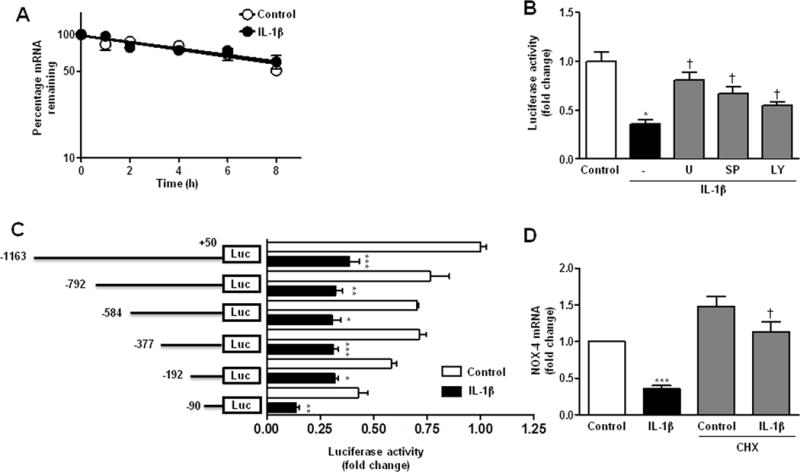
NOX-4 down-regulation induced by IL-1β is mediated by a transcriptional mechanism in VSMC. (A) NOX-4 mRNA stability after 24 h stimulation with IL-1β. NOX-4 mRNA levels were measured by qPCR at indicated times after actinomycin D addition (t=0). (B) Effect of MAPKs and PI3K inhibitors (U: U0126; SP: SP600125; LY: LY294002) on luciferase activity of NOX-4 promoter in cells incubated with IL-1β for 24 h. (C) Luciferase activity of rat VSMC transfected with luciferase reporter constructs containing different lengths of the human NOX-4 promoter and stimulated with IL-1β for 24 h. Scheme of the constructs is also shown. (D) Effect of cycloheximide (CHX) on NOX-4 mRNA levels in VSMC stimulated with IL-1β for 8 h. Data are expressed as mean±SEM. *P<0.05, **P<0.01, ***P<0.001 vs Control; †P<0.05 vs IL-1β. n=5–12.
To gain further insights into mechanisms involved in the IL-1β-mediated NOX-4 decrease, VSMC were treated with a protein synthesis inhibitor (cycloheximide; CHX, 25 μg/ml) before stimulation. We found that CHX abolished the effect of IL-1β (Figure 5D), suggesting that the new synthesis of a repressor is needed for the IL-1β-induced NOX-4 decrease. Thereafter we studied the expression of some transcription factors that could act as repressors by binding to the 90 bp proximal promoter. After IL-1β stimulation, PPAR-γ mRNA levels were decreased (Supplementary Figure S7A), while Egr-1 expression was increased (Supplementary Figure S7B). However, Egr-1 overexpression increased NOX-4 expression (Supplementary Figure S7C), suggesting no implication of these transcription factors in the IL-1β-dependent NOX-4 decrease.
NOX-1-derived O2•− and NOX-4-derived H2O2 participate in cell migration
NOX-1 has been described to produce O2•−, while H2O2 is produced, at least in part, by NOX-4 [4]. AngII or IL-1β induced NADPH oxidase activity and O2•− production that were further increased by the combination of both stimuli (Figure 6A, B). AngII+IL-1β-induced NADPH oxidase activation and O2•− production, were blocked by an unspecific NOX-1 inhibitor (ML171, 0.5 μmol/l) and by a NOX-1 siRNA (Figure 6A–D and Supplementary Figure S6B). In addition, HuR knockdown by siRNA partially decreased NADPH oxidase activity and O2•− production induced by AngII+IL-1β (Figure 7). On the other hand, H2O2 production was decreased in the presence of both AngII and IL-1β (Figure 8A). The effect of AngII+IL-1β was similar to IL-1β alone (Figure 8A). In agreement, NOX-4 overexpression abolished the decrease in IL-1β-induced H2O2 production (Figure 8B).
Figure 6.
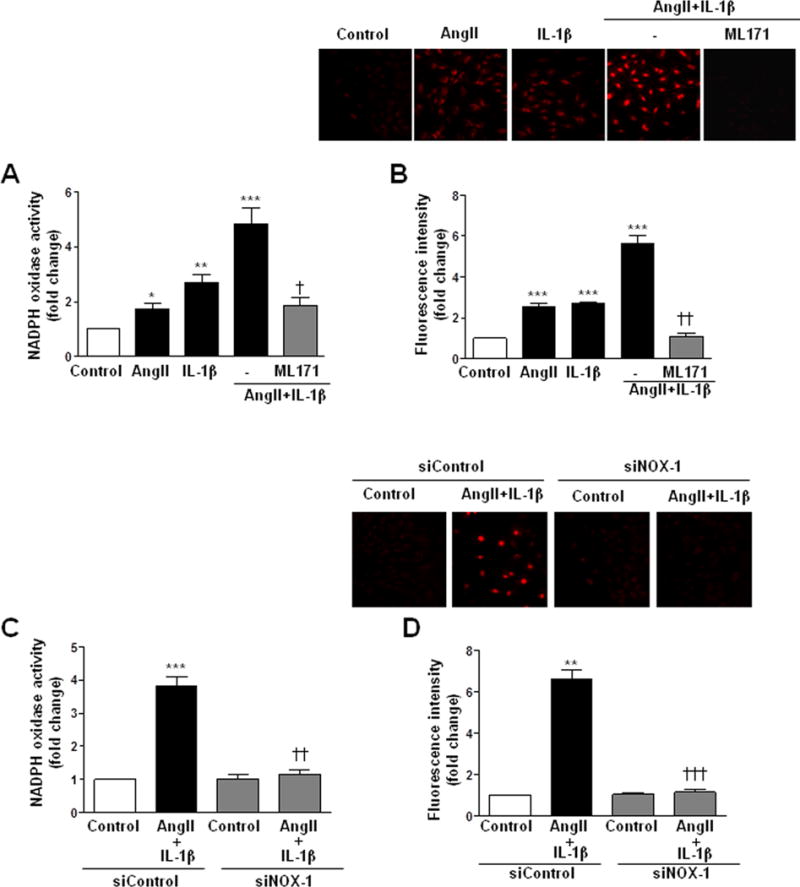
NOX-1 is responsible for AngII+IL-1β-induced O2•− production. Effect of ML171 and NOX-1 siRNA on NADPH oxidase activity (A, C) and ROS production (B, D) induced by AngII+IL-1β for 24 h in rat VSMC. Effects of AngII and IL-1β are also shown. Data are expressed as mean±SEM. *P<0.05, **P<0.01, ***P<0.001 vs Control. ††P<0.01, †††P<0.001 vs AngII+IL-1β. n=3–7.
Figure 7.
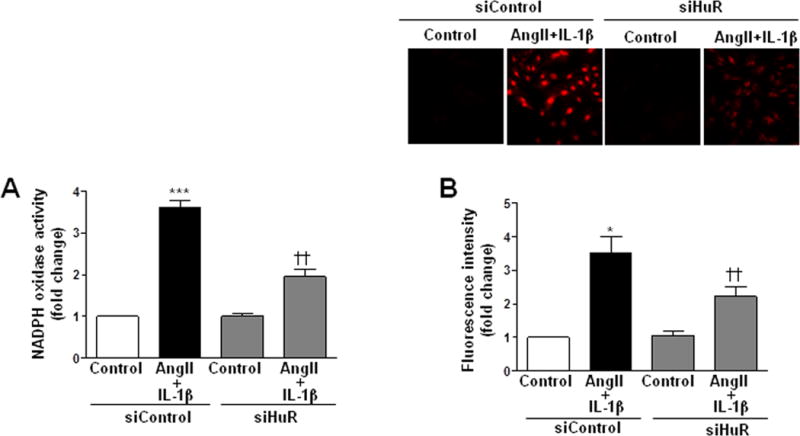
HuR is responsible for AngII+IL-1β-induced O2•− production. Effects of HuR siRNA on NADPH oxidase activity (A) and ROS production (B) in rat VSMC stimulated with AngII+IL-1β for 24 h. Data are expressed as mean±SEM. *P<0.05, ***P<0.001 vs Control. ††P<0.01 vs AngII+IL-1β. n=3–6.
Figure 8.
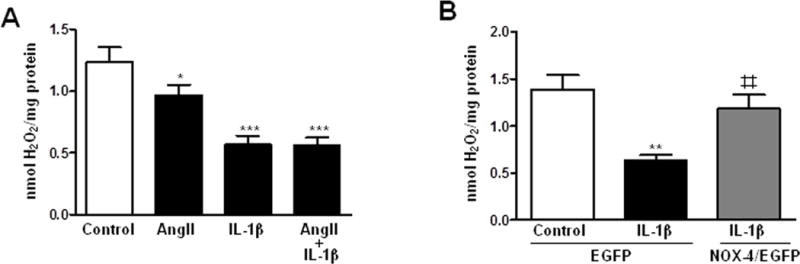
NOX-4 participates of IL-1β-dependent H2O2 production. H2O2 production in VSMC stimulated with AngII, IL-1β or AngII+IL-1β (A) or transfected with EGFP or NOX-4/EGFP and stimulated with IL-1β (B). Data are expressed as mean±SEM. *P<0.05, **P<0.01, ***P<0.001 vs Control. ‡‡P<0.01 vs EGFP-IL-1β. n=5–6.
Transwell and wound healing assays were used to study the implication of O2•− and H2O2 in VSMC migration. AngII or IL-1β induced cell migration that was further increased by the combination of both stimuli (Figure 9A). AngII+IL-1β-induced cell migration was blocked by ML171 and NOX-1 silencing (Figure 9B, C and Supplementary Figure S8) as well as by MS-444 and HuR siRNA (Figure 9B, D and Supplementary Figure S8). In addition, overexpression of NOX-4 increased IL-1β-induced cell migration (Supplementary Figure S9). Altogether these results suggest the implication of the HuR/NOX-1 axis and NOX-4 on VSMC migration in response to inflammatory stimuli.
Figure 9.
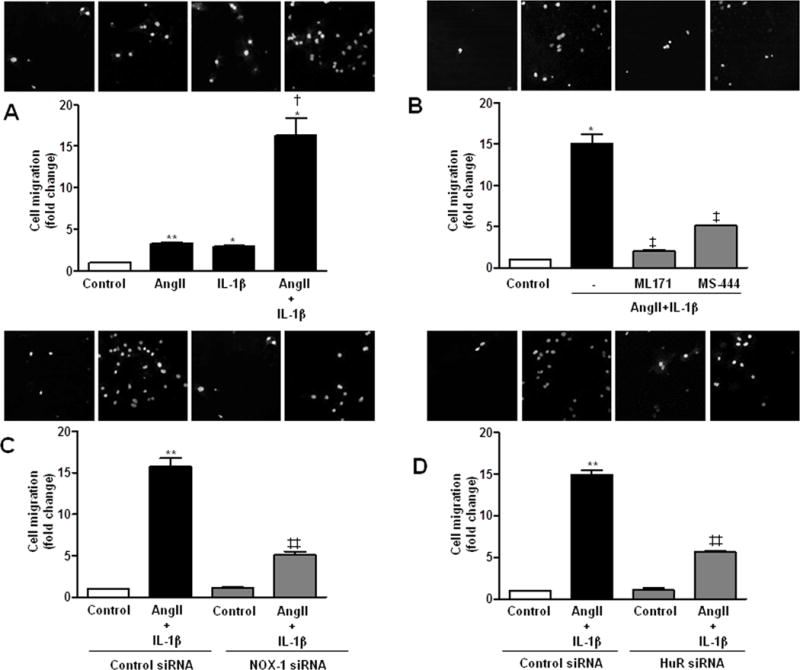
NOX-1 and HuR participate in cell migration induced by AngII+IL-1β. (A) Transwell assay performed in rat VSMC unstimulated and stimulated with AngII, IL-1β or AngII+IL-1β for 24 h. (B) Effect of ML171 and MS-444 on cell migration induced by AngII+IL-1β. Effect of NOX-1 (C) or HuR (D) siRNA on cell migration induced by AngII+IL-1β. Data are expressed as mean±SEM. *P<0.05, **P<0.01 vs Control. †P<0.05 vs AngII or IL-1β; ‡P<0.05, ‡‡P<0.01 vs AngII+IL-1β. n=3–4.
DISCUSSION
NADPH oxidase-derived ROS are implicated in several cardiovascular pathologies associated with vascular remodeling such as hypertension [24,25]. The present study provides evidence for the first time that in VSMC, AngII and IL-1β synergistically induce NOX-1 expression, at least in part, through a HuR-mediated increase in NOX-1 mRNA stability, subsequently leading to increased NADPH oxidase activity and O2•− production. In contrast, IL-1β decreases NOX-4 expression and NOX-4-derived H2O2 through a transcriptional mechanism involving response elements located in the 90 bp proximal promoter. Regulation of NOX-1 and NOX-4 contribute to cell migration triggered by inflammatory mediators and could be implicated in the vascular remodeling associated with cardiovascular diseases.
AngII and cytokines, including IL-1β, play a key role in hypertension, atherosclerosis and restenosis through the generation of an inflammatory environment associated with the induction of pro-inflammatory enzymes such as NADPH oxidase at vascular level [2, 3, 26]. We observed that IL-1β and AngII induced NOX-1 expression in VSMC, which is in agreement with previous studies [8,10,27]. We also found that the combination of AngII and IL-1β potentiated NOX-1 induction suggesting that amplifying mechanisms occur in the presence of several pro-inflammatory stimuli. The synergistic effect was dependent on transcriptional and post-transcriptional mechanisms. Specifically, we demonstrate that a post-transcriptional regulatory mechanism activated, at least in part, by MAPK and mediated by the RNA binding protein HuR, participates in the exacerbated NOX-1 induction in VSMC. This is based on the following findings: 1) actinomycin D pretreatment did not fully abolish the AngII+IL-1β-mediated NOX-1 induction; 2) the increase in NOX-1 3′UTR luciferase activity triggered by AngII+IL-1β was reduced by ERK1/2, JNK and p38 MAPK inhibitors; 3) AngII+IL-1β interfered with the decay of NOX-1 mRNA; 4) AngII+IL-1β increased HuR binding to NOX-1 mRNA; 5) HuR silencing and treatment with the HuR inhibitor MS-444 reduced NOX-1 mRNA stability and AngII+IL-1β-induced NOX-1 mRNA levels. HuR-dependent mRNA stabilization has been suggested to be involved in the post-transcriptional regulation of various pro-inflammatory genes [14] which in turn could help to the deleterious environment in cardiovascular disease. However, to our knowledge this is the first report demonstrating that HuR is involved in NOX-1 expression. HuR is mainly located in the nucleus and shuttles between the nucleus and the cytoplasm. Cytoplasmic HuR seems to contribute in promoting cell growth, proliferation, angiogenesis and survival [28], through its capacity of stabilizing ARE-containing mRNAs [28]. HuR subcellular localization can be modified by post-translational modifications such as serine and threonine phosphorylation [29]. Specifically, ERK1/2 can phosphorylate HuR modifying its activity in a lung cancer cell line [30] or its location in hepatic stellate [31]. In addition, we have previously shown that AngII+IL-1β increased HuR cytoplasmic localization and this effect was abolished by ERK1/2 inhibition [32]. More importantly, this regulation has a functional role on enzyme activity, since we demonstrate that the synergistically increased NADPH oxidase activity and O2•− production induced by AngII plus IL-1β, were abolished by NOX-1 and HuR blockade using pharmacological strategies. Moreover, knockdown approaches were also used because the specificity of ML171 and MS-444 are under debate [21,33]. Since NOX-1 has been described to be responsible for O2•− production and redox signaling in pathological conditions including atherosclerosis, diabetes, and hypertension [34,35], a limitation of our study is that we do not provide in vivo models that determine the potential role of HuR/NOX-1 regulation. Future studies are warranted to address this issue.
Previous studies suggested that NOX-4 is constitutively active and might be a housekeeping gene due to the presence of GC rich regions in its promoter [36]. However, modulation of NOX-4 by different stimuli has been described [7]. TGF-β and IFN-γ induce NOX-4 expression in VSMC [9,37], while the effects of AngII and IL-1β on NOX-4 expression are controversial with NOX-4 being up- and down-regulated by these agents in vascular cells [8,10,18,38]. We found that IL-1β substantially decreased NOX-4 gene expression with AngII having no additional effect probably because of the low levels of NOX-4 observed in the presence of IL-1β. Our results also indicate that IL-1β-mediated NOX-4 decrease is due to a transcriptional repression, involving ERK1/2, JNK and PI3K, and factor/s binding to a 90-bp region in NOX-4 proximal promoter. Thus, we found that cycloheximide, a protein synthesis inhibitor abolished IL-1β-dependent NOX-4 mRNA decrease. In addition, serial deletion analysis of NOX-4 promoter region enclosed IL-1β-dependent down-regulation to the 90-bp proximal region. In silico analysis of the 90-bp promoter region using MatInspector software indicated some candidate repressors (PPAR-γ and Egr-1) that may contribute to IL-1β-dependent NOX-4 down regulation. However, our data exclude the participation of these transcription factors to the down-regulation of NOX-4 expression induced by IL-1β. Very few studies have examined transcriptional mechanisms involved in NOX-4 down-regulation. Recently JunD, a member of the AP-1 family of transcription factors, has been described as a repressor of NOX-4 expression at the vascular level [39]; however, the 90-bp proximal region lacks an AP-1 binding site. Further examination is needed to identify and characterize which factor is responsible for this effect. Moreover, we cannot discard that other post-translational mechanisms such as protein stability could be also involved in NOX-4 down-regulation. Of interest is the fact that the effects of IL-1β on NOX-4 expression had a functional consequence in ROS production. Thus, IL-1β decreased H2O2 production and this effect was reversed by NOX-4 overexpression suggesting that in VSMC NOX-4 might function as a H2O2 generating enzyme as has been previously suggested by other authors [4,40].
Both O2•− and H2O2 are able to induce cell migration [6,41–43] through actin cytoskeleton reorganization [44]. We demonstrate that NOX-1-derived O2•− presumably under the influence of HuR, participates in cell migration since the potentiation induced by AngII+IL-1β on VSMC migration was inhibited by pharmacological inhibition and knockdown of both NOX-1 and HuR. This might have a role in end organ damage in cardiovascular pathologies associated with increased local levels of NOX-1 and/or HuR including AngII-induced hypertension [32,45], carotid ligation mouse model [32] and other vascular injuries like arterialized saphenous vein or atherosclerotic plaques [46]. Moreover, NOX-4 overexpression further increased IL-1β-induced VSMC migration, suggesting that both NOX-1 and NOX-4 participate in cell migration in inflammatory conditions. Interestingly, as discussed above, we observed that IL-1β decreased NOX-4 expression in VSMC suggesting that NOX-4 might not significantly contribute to IL-1β induced vascular damage. Alternatively, NOX-4 down-regulation might favor a dedifferentiated VSMC phenotype which is an important process involved in cell migration [47,48]. The role of NOX-1 in vascular remodeling has been previously demonstrated since NOX-1 knockout mice were protected against vascular remodeling induced by wire injury [49]. However, the role of NOX-4 in vascular damage is controversial. Thus, depending on the pathology or the vascular bed studied, increased, decreased or unchanged NOX-4 expression can be found and some studies suggest NOX-4 as a deleterious protein whereas others demonstrate that NOX-4 is a vascular protective enzyme [7,50,51]. Future studies are warranted to further clarify the role of NOX-4 in vascular diseases.
In conclusion, we demonstrate that transcriptional and post-transcriptional mechanisms dependent on HuR-mediated mRNA stabilization are responsible for AngII plus IL-1β-dependent NOX-1 expression in VSMC. We also demonstrate that IL-1β decreases NOX-4 expression in VSMC through transcriptional mechanisms involving the proximal promoter. Regulation of NOXs is important since NOX-1-derived O2•− and NOX-4-derived H2O2 participate in cell migration.
Limitations and perspectives
In this study we focused on NOX-1 and NOX-4 because these isoforms appear to be the key vascular NOXs involved in VSMC migration [52]. However, it is well known that activation of other NOXs (NOX-2 and NOX-5) also contribute to O2•− production in rodent and/or human VSMC [52]. As such, we cannot exclude the possibility that these NOXs may also play a role in the results observed here. In depth studies to further study NOX-2 and NOX-5 regulation, particularly in human VSMC, will elucidate whether these isoforms also play a role in AngII and/or IL-1β-mediated ROS-dependent migration of VSMC.
Exact reasons for the differential regulation of NOX-1 and NOX-4 remain unclear but may relate to localization of different NOXs in distinct intracellular regions [52]. For example, NOX-1 appears to localize primarily in caveolae whereas NOX-4 localizes to the nucleus and focal adhesions [53]. In VSMC, both NOX-1 and NOX-4 participate in cell migration, although via different mechanisms [52]. Our study further elucidates mechanisms regulating NOX-1 and NOX-4 and indicates how this impacts on functional responses of VSMC. Targeting such mechanisms may provide new approaches to regulate abnormal VSMC migration in vascular injury associated with cardiovascular disease.
Supplementary Material
Acknowledgments
This study was supported by MINECO (SAF2012-36400), ISCIII (RD12/0042/0024, RD12/0042/0053, PI13/01488 and PI12/01952), COST BM1301, and NIH (R01CA134609). AA and AMB were supported by a FPI fellowship and the Ramón y Cajal Program (RYC-2010-06473), respectively. RMT was supported through grants from the Canadian Institutes of Health Research and the British Heart Foundation.
We thank the excellent technical assistance of Laura Garcia Redondo.
Footnotes
Conflict of interest: NONE declared
Part of this work was previously presented at the Congress Frontiers in Cardiovascular Biology 2014.
References
- 1.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Briones AM, Touyz RM. Oxidative stress and hypertension: Current concepts. Curr Hypertens Rep. 2010;12:135–142. doi: 10.1007/s11906-010-0100-z. [DOI] [PubMed] [Google Scholar]
- 4.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katsuyama M, Miller FJ., Jr An oxidized extracellular oxidation-reduction state increases Nox1 expression and proliferation in vascular smooth muscle cells via epidermal growth factor receptor activation. Arterioscler Thromb Vasc Biol. 2010;30:2234–2241. doi: 10.1161/ATVBAHA.110.207639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al Ghouleh I, Rodríguez A, Pagano PJ, Csányi G. Proteomic analysis identifies an NADPH oxidase 1 (Nox1)-mediated role for actin-related protein 2/3 complex subunit 2 (ARPC2) in promoting smooth muscle cell migration. Int J Mol Sci. 2013;14:20220–20235. doi: 10.3390/ijms141020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen F, Haigh S, Barman S, Fulton DJ. From form to function: the role of Nox4 in the cardiovascular system. Front Physiol. 2012;3:412. doi: 10.3389/fphys.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, et al. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 9.Manea A, Tanase LI, Raicu M, Simionescu M. JAK/STAT Signaling Pathway Regulates Nox1 and Nox4-Based NADPH Oxidase in Human Aortic Smooth Muscle Cells. Arterioscler Thromb Vasc Biol. 2010;30:105–112. doi: 10.1161/ATVBAHA.109.193896. [DOI] [PubMed] [Google Scholar]
- 10.Briones AM, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, et al. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens. 2011;5:137–153. doi: 10.1016/j.jash.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Pérez-Girón JV, Palacios R, Martín A, Hernanz R, Aguado A, Martínez-Revelles S, et al. Pioglitazone reduces angiotensin II-induced COX-2 expression through inhibition of ROS production and ET-1 transcription in vascular cells from spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;306:H1582–1593. doi: 10.1152/ajpheart.00924.2013. [DOI] [PubMed] [Google Scholar]
- 12.Katsuyama M, Fan C, Arakawa N, Nishinaka T, Miyagishi M, Taira K, Yabe-Nishimura C. Essential role of ATF-1 in induction of NOX1, a catalytic subunit of NADPH oxidase: involvement of mitochondrial respiratory chain. Biochem J. 2005;386(Pt 2):255–261. doi: 10.1042/BJ20041180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cevik MO, Katsuyama M, Kanda S, Kaneko T, Iwata K, Ibi M, et al. The AP-1 site is essential for the promoter activity of NOX1/NADPH oxidase, a vascular superoxide-producing enzyme: possible involvement of the ERK1/2-JunB pathway. Biochem Biophys Res Commun. 2008;374:351–355. doi: 10.1016/j.bbrc.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 14.Meisner NC, Filipowicz W. Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression. Adv Exp Med Biol. 2011;700:106–123. doi: 10.1007/978-1-4419-7823-3_10. [DOI] [PubMed] [Google Scholar]
- 15.Diebold I, Petry A, Hess J, Görlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell. 2010;21:2087–2096. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 17.Manea A, Tanase LI, Raicu M, Simionescu M. Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2010;396:901–907. doi: 10.1016/j.bbrc.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res. 2005;65:495–504. doi: 10.1016/j.cardiores.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 19.Aguado A, Galán M, Zhenyukh O, Wiggers GA, Roque FR, Redondo S, et al. Mercury induces proliferation and reduces cell size in vascular smooth muscle cells through MAPK, oxidative stress and cyclooxygenase-2 pathways. Toxicol Appl Pharmacol. 2013;268:188–200. doi: 10.1016/j.taap.2013.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Orriols M, Guadall A, Galán M, Martí-Pàmies I, Varona S, Rodríguez-Calvo R, et al. Lysyl Oxidase (LOX) in vascular remodelling: insight from a new animal model. Thromb Haemost. 2014;112:812–824. doi: 10.1160/TH14-01-0024. [DOI] [PubMed] [Google Scholar]
- 21.Aotani Y, Saitoh Y. Structure determination of MS-444; a new myosin light chain kinase inhibitor. J Antibiot. 1995;48:952–953. doi: 10.7164/antibiotics.48.952. [DOI] [PubMed] [Google Scholar]
- 22.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meisner NC, Hintersteiner M, Mueller K, Bauer R, Seifert JM, Naegeli HU, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol. 2007;3:508–515. doi: 10.1038/nchembio.2007.14. [DOI] [PubMed] [Google Scholar]
- 24.Xu S, Touyz RM. Reactive oxygen species and vascular remodelling in hypertension: still alive. Can J Cardiol. 2006;22:947–951. doi: 10.1016/s0828-282x(06)70314-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vicenová B, Vopálenský V, Burýsek L, Pospísek M. Emerging role of interleukin-1 in cardiovascular diseases. Physiol Res. 2009;58:481–498. doi: 10.33549/physiolres.931673. [DOI] [PubMed] [Google Scholar]
- 27.Martín A, Pérez-Girón JV, Hernanz R, Palacios R, Briones AM, Fortuño A, et al. Peroxisome proliferator-activated receptor-γ activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J Hypertens. 2012;30:315–326. doi: 10.1097/HJH.0b013e32834f043b. [DOI] [PubMed] [Google Scholar]
- 28.Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meisner NC, Filipowicz W. Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression. Adv Exp Med Biol. 2011;700:106–23. doi: 10.1007/978-1-4419-7823-3_10. [DOI] [PubMed] [Google Scholar]
- 30.Yang X, Wang W, Fan J, Lal A, Yang D, Cheng H, Gorospe M. Prostaglandin A2-mediated stabilization of p21 mRNA through an ERK-dependent pathway requiring the RNA-binding protein HuR. J Biol Chem. 2004;279:49298–49306. doi: 10.1074/jbc.M407535200. [DOI] [PubMed] [Google Scholar]
- 31.Woodhoo A, Iruarrizaga-Lejarreta M, Beraza N, García-Rodríguez JL, Embade N, Fernández-Ramos D, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology. 2012;56:1870–1882. doi: 10.1002/hep.25828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguado A, Rodríguez C, Martínez-Revelles S, Avendaño MS, Zhenyukh O, Orriols M, et al. HuR mediates the synergistic effects of angiotensin II and IL-1β on vascular COX-2 expression and cell migration. Br J Pharmacol. 2015;172:3028–3042. doi: 10.1111/bph.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altenhöfer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid Redox Signal. 2015;23:406–427. doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 35.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 36.Katsuyama M, Matsuno K, Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr. 2012;50:9–22. doi: 10.3164/jcbn.11-06SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 38.Richard D, Wolf C, Barbe U, Kefi K, Bausero P, Visioli F. Docosahexaenoic acid down-regulates endothelial Nox 4 through a sPLA2 signalling pathway. Biochem Biophys Res Commun. 2009;38:516–522. doi: 10.1016/j.bbrc.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 39.Paneni F, Osto E, Costantino S, Mateescu B, Briand S, Coppolino G, et al. Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation. 2013;127:1229–1240. e1–21. doi: 10.1161/CIRCULATIONAHA.112.000826. [DOI] [PubMed] [Google Scholar]
- 40.Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, et al. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Velarde V, de la Cerda PM, Duarte C, Arancibia F, Abbott E, González A, et al. Role of reactive oxygen species in bradykinin-induced proliferation of vascular smooth muscle cells. Biol Res. 2004;37:419–430. doi: 10.4067/s0716-97602004000300007. [DOI] [PubMed] [Google Scholar]
- 42.Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480:109–112. doi: 10.1038/nature10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schröder K. NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid Redox Signal. 2014;20:2043–2058. doi: 10.1089/ars.2013.5633. [DOI] [PubMed] [Google Scholar]
- 44.Stanley A, Thompson K, Hynes A, Brakebusch C, Quondamatteo F. NADPH oxidase complex-derived reactive oxygen species, the actin cytoskeleton, and Rho GTPases in cell migration. Antioxid Redox Signal. 2014;20:2026–2042. doi: 10.1089/ars.2013.5713. [DOI] [PubMed] [Google Scholar]
- 45.Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal. 2013;18:51–65. doi: 10.1089/ars.2011.4335. [DOI] [PubMed] [Google Scholar]
- 46.Pullmann R, Jr, Juhaszova M, López de Silanes I, Kawai T, Mazan-Mamczarz K, Halushka MK, et al. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J Biol Chem. 2005;280:22819–22826. doi: 10.1074/jbc.M501106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45(Suppl A):A25–32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 49.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Touyz RM, Montezano AC. Vascular Nox4: a multifarious NADPH oxidase. Circ Res. 2012;110:1159–1161. doi: 10.1161/CIRCRESAHA.112.269068. [DOI] [PubMed] [Google Scholar]
- 51.Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012;110:1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 52.Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.