

Abstract
We are currently in the midst of an epidemic of metabolic disorders, which may, in part, be explained by excess fructose intake. This theory is supported by epidemiological observations as well as experimental studies in animals and humans. Rising consumption of fructose has been matched with growing rates of hypertension, leading to concern from public health experts. At this stage, the mechanisms underlying fructose-induced hypertension have not been fully characterized and the bulk of our knowledge is derived from animal models. Animal studies have shown that high-fructose diets up-regulate sodium and chloride transporters, resulting in a state of salt overload that increases blood pressure. Excess fructose has also been found to activate vasoconstrictors, inactivate vasodilators, and over-stimulate the sympathetic nervous system. Further work is required to determine the relevance of these findings to humans and to establish the level at which dietary fructose increases the risk of developing hypertension
Keywords: blood pressure, endothelium, fructose, hypertension, salt, sympathetic nervous system
INTRODUCTION
Fructose consumption has been escalating over the past several decades and is believed to play a role in the rising epidemic of metabolic disorders [1]. Fructose is a simple monosaccharide that occurs naturally in fruit, though the two main sources of dietary fructose in the Western diet are sucrose (table sugar) and high-fructose corn syrup (HFCS) [1]. Sucrose is cleaved enzymatically during digestion to produce one fructose molecule and one glucose molecule. HFCS, on the contrary, contains free fructose and glucose in varying ratios. A popular type of HFCS that is used to sweeten beverages in the United States – HFCS-55 – contains 55% fructose, 42% glucose and 3% oligosaccharides [2]. The 1999–2004 data from the National Health and Nutrition Examination Survey (NHANES) show that the average daily intake of fructose in the United States is now approximately 49 g, which equates to 9.1% of total energy intake [3]. In comparison, the average daily intake of fructose during 1977–1978 was 37 g [3]. The highest consumers of fructose are 19–22-year-olds, largely due to excess consumption of sugar-sweetened beverages. Fructose consumption as a percentage of total energy intake amongst male and female 19–22-year-olds in the 95th percentile is 17.5 and 17.9%, respectively [3].
The rise in fructose intake has been paralleled by a rise in hypertension. A study of the US population during 2007–2008 found that 29% of adults were hypertensive, compared to 11–13% in 1939 and 24% during 1988–1994 [4,5]. Epidemiological studies have hinted at a link between fructose consumption and hypertension. Jalal et al.[6] reported that excess dietary fructose (≥74 g/day) in the form of added sugar was associated with higher blood pressure (BP) values in US adults who did not have a history of hypertension. Similarly, a study of 4867 adolescents found that SBP rose by 2 mmHg from the lowest to the highest category of sugar-sweetened beverage intake [7]. In a prospective study of US adults, Chen et al.[8] found that drinking one less sugar-sweetened beverage per day was associated with a 1.8 mmHg reduction in SBP and a 1.1 mmHg reduction in DBP over 18 months. These studies controlled for BMI and dietary factors (amongst other things) to remove potential confounders. High intake of sugar-sweetened beverages is associated with poor diet and obesity, which have clear links with hypertension [9,10], but fructose appears to have an independent effect on BP.
Fructose metabolism differs to that of glucose due to almost complete first-pass extraction by the liver, where it is rapidly converted to fructose 1-phosphate [11]. As a result, plasma fructose levels do not normally exceed the micromolar range, even after a high fructose meal. At this stage, only one study in humans has measured plasma fructose levels at regular intervals following fructose ingestion. Le et al.[12] monitored the changes in plasma fructose that occurred after healthy volunteers consumed a 24 ounce bottle of a commercially available beverage (Dr Pepper) containing 39 g of fructose. The average fasting plasma fructose concentration was 5 μmol/l. The maximum level following consumption of the Dr Pepper was 300 μmol/l on average. To put this into perspective, this value was still approximately 15 times lower than the average fasting glucose concentration (80 mg/dl or 4440 μmol/l).
For reasons that are still incompletely understood, experimental studies in human volunteers have shown that fructose consumption has an acute effect on BP. For example, when healthy young adults consumed drinks containing 60 g of fructose or glucose, the fructose drink raised BP significantly above baseline (average increase of 6.2 mmHg), whereas the glucose drink had no effect on BP [13]. Similarly, Perez-Pozo et al.[14] found that ambulatory SBP and DBP increased significantly in 74 healthy men after they consumed 200 g of fructose daily for 2 weeks. Although these studies demonstrate the acute effects of dietary fructose on BP, the chronic effects have not yet been established.
In animals, high-fructose diets have been used for decades to generate models of hypertension and insulin resistance [15] (for a review of the mechanisms underlying fructose-induced insulin resistance, refer to the study by Tappy and Le [11]). In 1987, Hwang et al. reported that the SBP of rats fed a 66% fructose diet for 2 weeks rose from 124 to 145 mmHg, paving the way for dozens of studies investigating the mechanisms of fructose-induced hypertension [15,16]. Similarly, Martinez et al.[17] found that the mean arterial pressure of dogs subsisting on a 60% fructose diet increased from 100.4 to 122.6 mmHg after 28 days, whereas there was no significant increase in the group fed a 60% glucose diet. While most animal studies have examined the physiological effects of high-fructose diets (usually 60% of total energy intake), there is evidence that lower levels of fructose can also induce hypertension. For instance, Glushakova et al.[18] found that a 20% fructose diet significantly increased SBP in rats after 33 weeks. Most animal studies have not found a significant association between short-term fructose-feeding and weight gain, suggesting that fructose-induced hypertension is not related to obesity in these models [16,17].
Studies to date show that the mechanisms by which excess fructose increases BP fall into three broad categories: increased salt absorption, endothelial dysfunction and chronic stimulation of the sympathetic nervous system. In this review, we will attempt to summarize the evidence for each of these theories and highlight future avenues for research.
INCREASED SALT ABSORPTION
Several animal studies have demonstrated that dietary fructose stimulates sodium and chloride absorption, leading to a state of salt overload that raises BP [19–24]. There are three transporters that are up-regulated by fructose in the small intestine and proximal renal tubule. The first is the major fructose transporter GLUT5, which facilitates fructose absorption [19–21]. The second is the sodium-hydrogen exchanger 3 (NHE3), a sodium transporter, and the third is the putative anion transporter 1 (PAT1), a chloride transporter (Fig. 1) [19–21].
FIGURE 1.
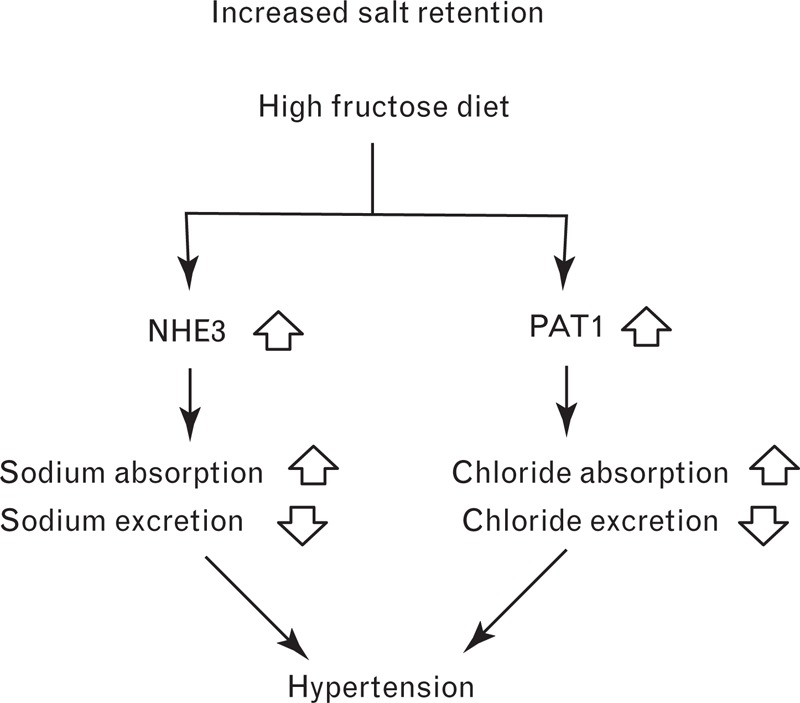
The proposed role of salt transporters in the development of hypertension following a high fructose diet. NHE3, sodium-hydrogen exchanger 3, PAT1, putative anion transporter 1.
Fructose-induced hypertension has been shown to be attenuated in both GLUT5 and PAT1 knockout mice [21,22]. Singh et al.[21] found that the SBP of wild-type mice fed a 60% fructose diet for 12 weeks was 9 mmHg higher than those fed 60% starch. The average plasma fructose concentration in the fructose-fed mice was 1550 μmol/l, compared to 230 μmol/l in the starch-fed mice. In contrast, the SBP of PAT1 null mice on the same high-fructose diet did not increase significantly compared to the control group. To confirm that fructose increases BP by enhancing salt absorption, the researchers perfused the jejuna of the mice with sodium chloride (130 mmol/l) and fructose (40 mmol/l) or NaCl (150 mmol/l) alone. Fluid absorption in wild-type mice was approximately 1.7 times higher in the presence of 40 mmol/l fructose, suggestive of greater sodium absorption. Singh et al.[21] also revealed that a 60% fructose diet reduces kidney renin expression by approximately 50% in rats after 2 weeks. This is consistent with previous studies showing that increased dietary NaCl reduces renin expression and activity [25,26]. Finally, fructose feeding has been shown to lower urinary excretion of sodium chloride in rats to less than half of the control value after 24 h. The increased salt absorption by the intestine and decreased salt excretion by the kidney observed in fructose-fed rodents are believed to contribute to hypertension (Fig. 1).
The synergistic action of dietary fructose and salt on BP has been further illustrated by the work of Cabral et al.[23]. When rats were fed a 20% fructose diet for 7 days, no significant rise in SBP was observed, but when rats were fed a 20% fructose/high-salt diet for the same length of time, SBP increased steadily from 118 to 132 mmHg. This suggests that the high sugar/high salt combination in the Western diet may provide fertile ground for the development of hypertension. Cabral et al.[23] also showed that a 5 mmol/l fructose solution stimulated NHE3 activity in proximal renal tubule segments isolated from rats (likely via a protein kinase C mechanism), and sensitized the tubules to angiotensin II. The 5 mmol/l concentration of fructose used in this experiment was higher than that which is found in human plasma (up to ∼0.3 mmol/l after a high-fructose meal), but is closer to that which is found in the hepatic portal vein of rats (1.1–2.2 mmol/l following a large fructose meal) [27]. At this stage, the typical range of fructose concentrations in the proximal tubule in rats and humans is unknown, making it difficult to assess the relevance of these results.
Catena et al.[28] have investigated the relationship between salt retention, insulin and hypertension in fructose-fed rats. Rats were fed a 66% fructose diet containing either low (0.07%), normal (0.3%) or high (7.5%) levels of NaCl. SBP increased significantly in rats receiving the normal diet and high-salt diet, but not in those receiving the low-salt diet. In control rats that did not consume fructose, the high-salt diet decreased the density of insulin messenger RNA (mRNA) and receptors in the kidney. This feedback mechanism normally limits insulin-induced sodium reabsorption when high levels of salt are consumed. However, in fructose-fed rats, the high-salt diet did not reduce the renal insulin receptor number or the mRNA levels, and urinary sodium excretion was significantly lower than in the control group. This may explain why SBP is higher in the fructose-fed rats than in control rats receiving the same high-salt diet.
ENDOTHELIAL DYSFUNCTION
The vascular endothelium provides an intricate interface between the blood and the vessel wall. Endothelial cells play a vital role in the regulation of vascular tone through synthesis and release of contracting and relaxing factors. There is evidence that excess levels of circulating insulin and fructose metabolites that result from high-fructose diets can alter endothelial activity, thereby raising BP. Katakam et al.[29] have reported that hyperinsulinemia emerges in rats after 3 days of fructose feeding, whereas endothelial dysfunction is observed after 18 days, and hypertension after 28 days. The interplay between insulin, fructose metabolites, and contracting and relaxing factors in the endothelium appears to be a complex web that is yet to be fully characterized. The proposed pathways are shown in Fig. 2 and are summarized below.
FIGURE 2.
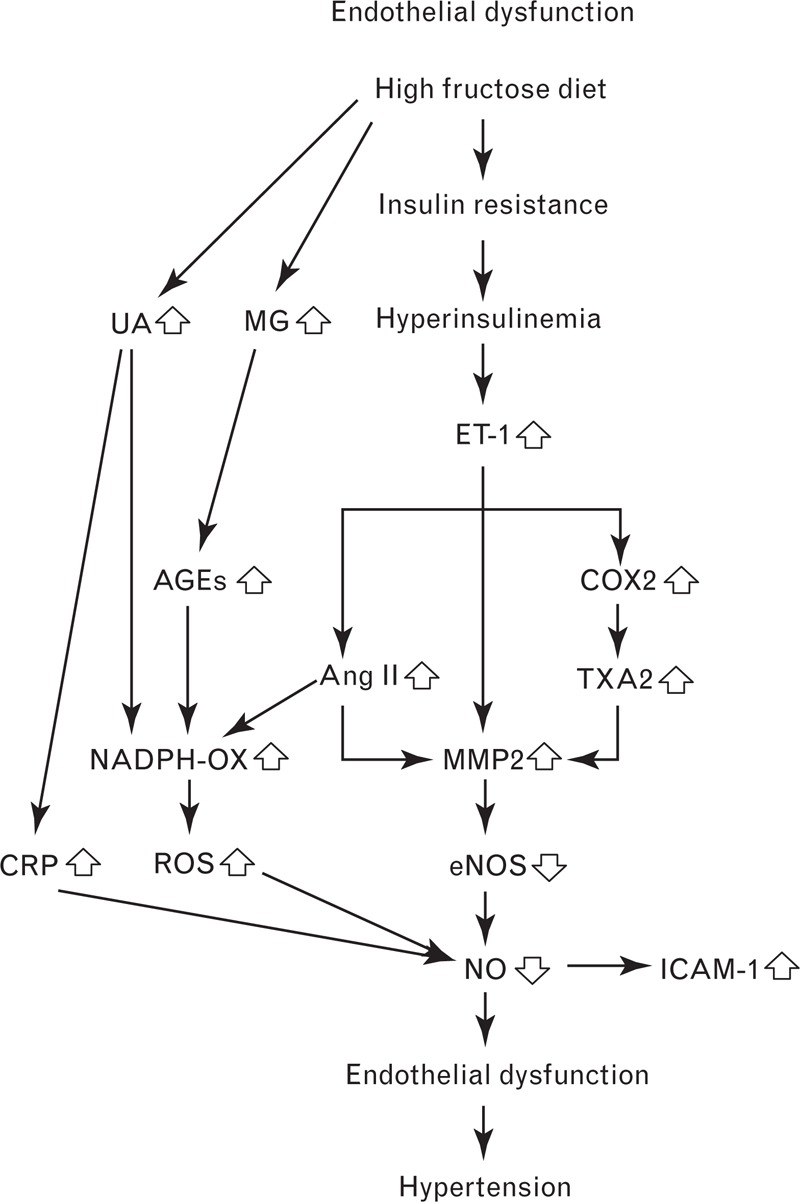
Fructose consumption can lead to endothelial dysfunction via several different pathways, ultimately leading to hypertension. AGEs, advanced glycation end products; Ang II, angiotensin II; COX2, cyclooxygenase-2; CRP, C-reactive protein; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; ICAM-1, intercellular adhesion molecule 1; MG, methylglyoxal; MMP2, matrix metalloproteinase-2; NADPH-OX, nicotinamide adenine dinucleotide phosphate oxidase; NO, nitric oxide; ROS, reactive oxygen species; TXA2, thromboxane A2; UA, uric acid.
Nitric oxide and endothelial nitric oxide synthase
Nitric oxide (Fig. 2) is a powerful BP-lowering vasodilator that is generated by endothelial nitric oxide synthase (eNOS). Fructose feeding has been shown to diminish the production of nitric oxide by lowering the activity and expression of eNOS [18,30,31]. Miatello et al.[32] have shown that eNOS activity decreases by approximately 30% in mesenteric resistance vessels isolated from fructose-fed rats, while Palanisamy and Venkataraman [30] have reported that eNOS expression in fructose-fed rats is approximately half that of control rats.
Endothelin-1
Endothelin-1 (ET-1) (Fig. 2) is a potent vasoconstrictor that is produced by vascular endothelial cells. Verma et al.[33] have shown that the ET-1 content of mesenteric vascular tissue isolated from rats fed a 66% fructose diet is 1.5 times higher than in control rats. This increased ET-1 expression is thought to arise from fructose-induced hyperinsulinemia, since insulin has been shown to stimulate the production and secretion of ET-1 in vitro and in vivo[34,35]. For instance, Juan et al.[35] have reported that chronic insulin infusion increases plasma ET-1 levels by approximately 1.5 times and induces hypertension in rats.
Endothelin-1 exerts its vasoconstrictive effects by binding to ETA and ETB receptors [33]. The discovery that treatment with bosentan – a dual ETA and ETB receptor blocker – prevents elevations in BP in fructose-fed rats provides further evidence that ET-1 contributes to fructose-induced hypertension [33].
Thromboxane A2
Fructose-fed hypertensive rats also display higher levels of the vasoconstrictor thromboxane A2 (TXA2) [36]. Jiang et al.[36] have revealed that plasma levels of TXB2 (a stable metabolite of TXA2) are significantly higher in fructose-fed rats than in control rats (769 vs. 467 pg/ml). Interestingly, the researchers found that TXB2 levels and SBP did not increase above control levels when fructose-fed rats were co-treated with the thromboxane synthase inhibitor, dazmegrel, or the endothelin receptor antagonist, bosentan. This suggests that hyperinsulinemia increases ET-1 production, which in turn increases TXA2 levels (Fig. 2). There is evidence that ET-1 stimulates TXA2 production by up-regulating cyclooxygenase-2 (COX2), an enzyme involved in thromboxane synthesis [36,37]. Protein expression of COX2 is elevated in fructose-fed hypertensive rats – an effect that is blocked by bosentan [36].
Angiotensin II
Angiotensin II (Ang II) (Fig. 2) is a vasoconstrictor that is up-regulated in fructose-fed hypertensive rats [38,39]. Ang II performs its actions by binding to the angiotensin type I and II receptors (AT1 and AT2), though most of its well known functions occur through interactions with AT1 [40]. AT1 receptors are known to be up-regulated in fructose-fed rats, and the effects of fructose on SBP are mitigated by co-treatment with the AT1 receptor antagonists L-158,809 and losartan [38,39]. The role of Ang II in fructose-induced hypertension is supported by the work of Erlich and Rosenthal [41], who demonstrated that three different angiotensin-converting enzyme (ACE) inhibitors are able to normalize BP in fructose-fed rats.
Interestingly, Tran et al.[38] found that chronic bosentan treatment completely blocked the increase of plasma Ang II during fructose feeding in rats, suggesting that insulin-induced overexpression of ET-1 may be responsible for up-regulating Ang II. Additionally, Ang II is known to stimulate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase through its actions on AT1 receptors. This promotes generation of reactive oxygen species (ROS), including superoxide and hydrogen peroxide, which inflict oxidative stress on the vascular endothelium [42]. Shinozaki et al.[39] have shown that treatment of aortic segments from fructose-fed rats with inhibitors of NADPH oxidase significantly reduces superoxide production.
There is some evidence that impaired nitric oxide production in fructose-fed rats is due to elevated Ang II levels. Researchers have found that eNOS activity in the vascular smooth muscle cells of fructose-fed rats can be restored by lowering Ang II levels though chronic ACE inhibition or AT1 receptor antagonism [32,43]. This may be related to the fact that Ang II stimulates release of superoxide, which is known to inactivate nitric oxide [44–46].
Matrix metalloproteinase-2
Ang II, ET-1 and TXA2 are able to up-regulate matrix metalloproteinases (MMPs), a family of zinc-dependent endopeptidases that can degrade membrane proteins [47–49]. Since eNOS is a membrane-bound protein, it has been hypothesized that fructose feeding increases Ang II, ET-1 and TXA2, leading to higher levels of MMPs that degrade eNOS, lower nitric oxide production, and drive BP upwards (Fig. 2) [49].
Nagareddy et al.[49] have shown that dietary fructose enhances the activity and expression of MMP2, and that treatment with the MMP2 inhibitor doxycycline completely prevents the development of hypertension in fructose-fed rats. Additionally, when endothelial cells were incubated with MMP2, nitric oxide production declined in a dose-dependent manner. Finally, it was shown that MMP2 colocalizes with both eNOS and its cofactor heat shock protein 90 (HSP90) in endothelial cells, and that MMP2 is able to cleave and degrade HSP90.
Intercellular adhesion molecule 1
There is evidence that fructose feeding raises levels of intercellular adhesion molecule 1 (ICAM-1) – an inflammatory molecule expressed by endothelial cells (Fig. 2). Glushakova et al.[18] have shown that addition of fructose to human aortic endothelial cells increases ICAM-1 expression in a dose and time-dependent manner. The authors attributed this phenomenon to the lower levels of nitric oxide and eNOS observed in the fructose-exposed aortic cells, since nitric oxide is known to have anti-inflammatory properties [50]. Further experiments revealed that serum ICAM-1 concentrations and BP were significantly higher in fructose-fed rats compared to those fed starch [18].
Methylglyoxal
Methylglyoxal (Fig. 2) is a highly reactive dicarbonyl molecule that is formed as a by-product of fructose and glucose metabolism [51]. Dhar et al.[52] and Wang et al.[53] have shown that aortic levels of methylglyoxal and BP are significantly higher in fructose-fed rats. In contrast, no significant increases in aortic methylglyoxal levels or BP were observed when fructose-fed rats received simultaneous treatment with the methylglyoxal scavenger and antidiabetic drug metformin.
Methylglyoxal has been established as an important precursor to advanced glycation end products (AGEs) [51,52,54]. AGEs are a heterogeneous class of molecules that are formed when carbonyl groups react with free amino groups in proteins, lipids and nucleic acids. In the endothelium, binding of AGEs to the receptor for AGEs (RAGE) activates NADPH oxidase, which in turn increases levels of ROS and inflicts oxidative stress [55,56]. Dhar et al.[52] have reported that levels of RAGE are elevated in the aortas of rats fed a high-fructose diet. It should be noted that the aorta is not a resistance vessel and as such, is not necessarily representative of the resistance vessels that influence BP. Future work will be required to confirm that RAGE levels are also increased in the resistance vessels of fructose-fed rats. Finally, methylglyoxal is able to exacerbate oxidative stress by reducing levels of the antioxidant glutathione, as well as diminishing the activity of the antioxidant enzymes glutathione reductase and glutathione peroxidase [57].
Uric acid
The first step in the metabolism of fructose is phosphorylation to fructose 1-phosphate by fructokinase, which uses ATP as a phosphate donor [58]. Unlike glucose, whose phosphorylation is tightly regulated so that intracellular ATP levels are never depleted, there is no feedback mechanism regulating the phosphorylation of fructose [59]. As a result, intracellular phosphate levels continue to fall and AMP deaminase is activated [59]. This enzyme converts AMP to inosine monophosphate (IMP), which is further metabolized to hypoxanthine [60]. Xanthine oxidase then converts hypoxanthine to xanthine to uric acid [61].
Nguyen et al.[7] have used NHANES data to investigate the relationship between sugar-sweetened beverage intake and serum uric acid in US adolescents. The authors found that serum uric acid increased by 0.18 mg/dl (11 μmol/l) from the lowest to the highest category of sugar-sweetened beverage intake, a statistically significant rise that was paralleled by an increase in BP. Since glucose metabolism does not enhance uric acid production, it can be assumed that the higher uric acid levels were associated with the fructose content of the drinks. Several decades ago, Macdonald et al.[62] showed that oral fructose raises serum uric acid significantly in normal individuals, whereas oral glucose has no effect.
In rats, plasma uric acid is also found to increase during high fructose diet regimes [63–65]. Moreover, xanthine oxidase inhibitors that lower uric acid have been shown to decrease BP. Allopurinol and febuxostat have both been reported to lower SBP in fructose-fed rats, though the reduction was not statistically significant for febuxostat [63,64]. Allopurinol has also been reported to normalize the effects of fructose on BP in healthy adult men [14]. The men consumed 200 g of fructose daily for 2 weeks with or without allopurinol. In the group that did not receive allopurinol, serum uric acid increased by 21.2%, whereas a 31.7% decrease from baseline was observed in the allopurinol group. SBP rose by 5.5% in the nonallopurinol group, compared to 1.5% in the allopurinol group, whereas DBP rose by 6.3% in the nonallopurinol group and 1% in the allopurinol group. Thus, both animal and human studies indicate that allopurinol attenuates the development of fructose-induced hypertension by lowering uric acid.
Hyperuricemia is believed to cause endothelial dysfunction by inflicting oxidative stress and reducing levels of eNOS and nitric oxide (Fig. 2) [60,66–70]. Uric acid has been widely reported to possess antioxidant properties in the extracellular environment, but there is growing evidence that it acts as a pro-oxidant once inside cells [71]. Ejaz et al.[72] have reported that uric acid stimulates production of ROS via its action on NADPH oxidase, and that nitric oxide and eNOS are subsequently inactivated. Similarly, Hong et al.[73] have revealed that stimulation of endothelial cells with 600 μmol/l of uric acid significantly reduces eNOS expression and inhibits release of nitric oxide. This was accompanied by significant elevations in intracellular ROS and mitochondrial superoxide, which may have been due to calcium overload in the mitochondria. Levels of mitochondrial superoxide were higher than intracellular ROS, suggesting that the mitochondria play an important role in uric acid-mediated oxidative stress. The finding that co-treatment with a ROS scavenger normalized eNOS and nitric oxide levels reinforces the theory that uric acid inflicts endothelial damage by increasing levels of ROS. ROS production may also be due to the actions of xanthine oxidase during the metabolism of fructose to uric acid [74].
Finally, uric acid is also able to induce the expression of C-reactive protein (CRP) in the vascular endothelium. Kang et al.[69] have shown that uric acid increases CRP in endothelial cells in a dose-dependent manner, and that CRP is able to inhibit nitric oxide release. Moreover, co-treatment with an anti-CRP antibody was found to normalize nitric oxide levels, suggesting that CRP may play a role in uric acid-mediated endothelial dysfunction. Then again, a study in 107 healthy adults found no relationship between serum uric acid and endothelial dysfunction, despite there being a significant correlation between serum uric acid and levels of CRP [75]. The authors speculated that CRP may only inflict damage on endothelial cells when in the presence of other adverse factors, such as aldosterone, meaning that the results may have been different in a hypertensive population. Further work will be required to clarify the link between uric acid, endothelial dysfunction and hypertension.
Relative contributions of the pathways leading to endothelial dysfunction
On the basis of the experimental models described above, there are many different ways in which excess dietary fructose can cause endothelial dysfunction. Untangling these pathways and determining their relative importance is not a simple task. As Fig. 2 illustrates, there are multiple pathways branching off ET-1 and uric acid, suggesting that these factors play the most prominent roles in fructose-induced hypertension. In rats fed high-fructose diets, ET-1 has been shown to increase by approximately 1.5 times in mesenteric resistance vessels [33], while blood uric acid levels have been shown to increase by 1.3–2.8 times [63–65]. The fact that these experiments were performed over different timeframes and measured ET-1 and uric acid in different contexts makes it difficult to compare them in any meaningful way. It is tempting to conclude that increased uric acid has the largest impact on endothelial function because it exhibits the biggest increase in response to fructose and there are epidemiological studies and human experiments to support the role of uric acid in fructose-induced hypertension [7,14], but further validation is required.
Finally, it should be noted that endothelial dysfunction can be caused by high BP itself [76,77], meaning that other pathways leading to fructose-induced hypertension, such as increased salt retention and chronic activation of the sympathetic nervous system, have the potential to impair endothelial function.
OVERACTIVATION OF THE SYMPATHETIC NERVOUS SYSTEM
Excess dietary fructose leads to chronic stimulation of the sympathetic nervous system, primarily as a result of increased insulin levels (Fig. 3) [15]. In turn, overactivation of the sympathetic nervous system is believed to exacerbate insulin resistance, thereby setting up a positive feedback loop [15].
FIGURE 3.
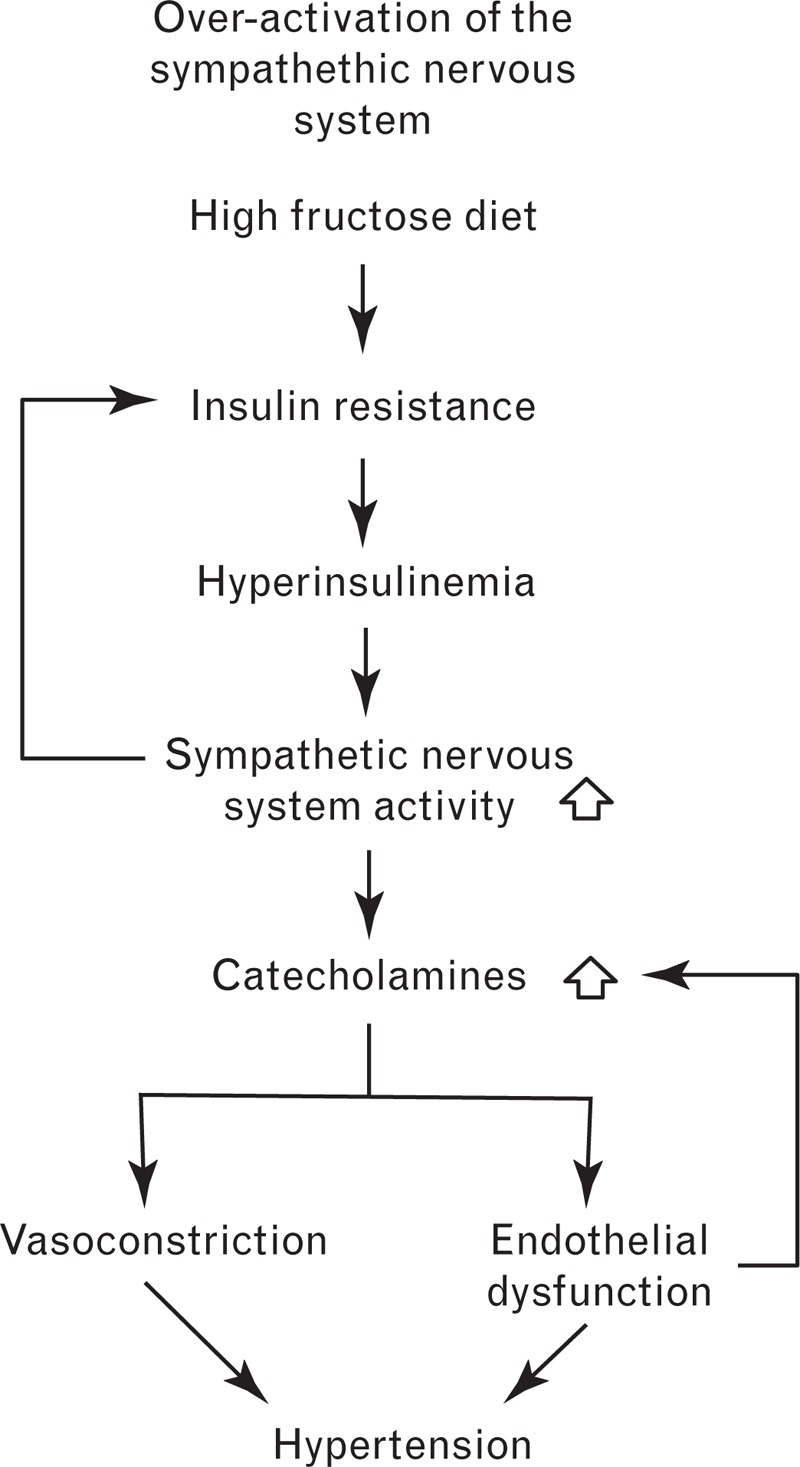
The proposed role of the sympathetic nervous system in the development of hypertension following a high fructose diet.
There are several pieces of evidence that implicate chronic activation of the sympathetic nervous system in fructose-induced hypertension. Insulin resistance, hyperinsulinemia and hypertension are prevented in fructose-fed rats that receive treatment with moxonidine and rilmenidine – imidazoline receptor agonists that reduce sympathetic outflow [78,79]. In addition, a study by Verma et al.[80] revealed that chemical sympathectomy prevents increases in BP and insulin in the fructose-fed rat model, suggesting that a functional sympathetic nervous system is necessary for the development of hyperinsulinemia and hypertension. More recently, Tran et al.[81] reported that fructose-fed rats exhibit significantly higher adrenergic activity than control rats, as reflected by higher plasma norepinephrine levels. Interestingly, they showed that chronic treatment with the sympatholytic agent prazosin blocks the development of hypertension in fructose-fed rats [81]. Prazosin prevents norepinephrine-induced vasoconstriction through selective antagonism of α1-adrenoreceptors [82].
By stimulating catecholamine release, excess dietary fructose is believed to indirectly impair endothelial function. Fu et al.[83] have shown that norepinephrine induces apoptosis in endothelial cells derived from neonatal rat hearts in a dose and time-dependent manner, though it is unknown whether this type of endothelial dysfunction leads to higher BP. The finding that norepinephrine increases the production of ROS in rat endothelial cells suggests that norepinephrine may cause hypertension via the ROS pathway shown in Fig. 2, which involves a down-regulation of nitric oxide [84].
The effect of excess norepinephrine on endothelial function has been studied in patients with norepinephrine-secreting tumors known as pheochromocytomas. Higashi et al. demonstrated that endothelium-dependent vasodilation in the forearm arteries was impaired to a greater extent in patients with pheochromocytoma than in patients with essential hypertension and normotensive individuals. Surgical removal of the norepinephrine-secreting tumors improved endothelium-dependent vasodilation in the pheochromocytoma patients, with the size of this improvement correlating significantly with reductions in urinary norepinephrine levels. In healthy people, endothelium-dependent vasodilation has been found to be inversely related to plasma norepinephrine levels when measured by brachial artery flow-mediated dilation (FMD), though the precise reasons for this are unclear [85,86].
Finally, it is possible that the reverse process also occurs – high-fructose diets may indirectly raise catecholamine levels by impairing endothelial function. Several studies have shown that nitric oxide inhibits the release of norepinephrine, thereby enhancing its own direct vasodilatory action [87–89]. As a result, reduced nitric oxide production following excess fructose consumption is likely to increase norepinephrine levels, raising BP as a result. Future work will be required to better understand the relationship between endothelial dysfunction and sympathetic nervous system activity.
In conclusion, the epidemiological and experimental studies conducted to date suggest a strong link between excess fructose consumption and hypertension. The causal effects of fructose on BP elevation appear to stem from complex and myriad metabolic pathways that are still being explored. The relative clinical and prognostic importance of each of these pathways and the timeline of events between fructose ingestion and high BP are still poorly understood. Further studies will be required to characterize the relationships between increased salt retention, endothelial dysfunction and the sympathetic nervous system in fructose-induced hypertension.
At this stage, most of our insights into the mechanisms of fructose-induced hypertension are derived from animal studies, particularly rodents. One drawback of animal models is that fructose is usually considered in isolation, meaning that its interactions with other nutrients are overlooked. A small number of animal studies have revealed that fructose and salt have a synergistic effect on BP, highlighting the importance of studying fructose in the context of other dietary components.
It would be simplistic to blame the upwards trend in hypertension solely on fructose; however, the growing levels of this sweetener in our diets make it imperative that we investigate its health effects further. The evidence presented in this study highlights the need to establish how much fructose can be safely consumed on a daily basis without increasing the risk of developing hypertension and other metabolic disorders.
ACKNOWLEDGEMENTS
All authors contributed to the writing and editing of the manuscript and approved the final version submitted for publication.
The manuscript has not been published or submitted elsewhere.
Source of funds: The salaries of the authors are provided by the Cardiac Health Institute and Macquarie University. No additional funding was obtained for this review.
Conflicts of interest
The authors have no conflicts of interest.
Reviewers’ Summary Evaluations
Referee 1
This article examines the mechanisms that might underlie the contribution of dietary fructose intake hypertension development. The strength is that there is a comprehensive review of the role of local factors in the generation of endothelial and vascular dysfunction as a consequence of increased fructose intake which relies on an extensive body of in vitro studies. The weakness of the review is that there is relatively little information on the actual circulating levels of fructose existing as a result of the increased dietary intake, or the action of acutely increased fructose intake on autonomic control of the cardiovascular system.
Referee 2
This is an interesting review looking at the link between hypertension and fructose consumption. Considering the recent increase in the use of artificial sweeteners, this review was certainly warranted. As such, this paper nicely reviews the literature which has suggested that fructose ingestion can favor hypertension development. In addition, it suggested potential mechanisms implicated. Interestingly, they suggest that dietary salt may compound the effect of fructose intake further highlighting the deleterious effect of the typical North-American diet.
Footnotes
Abbreviations: ACE, angiotensin-converting enzyme; AGEs, advanced glycation end products; AMP, adenosine monophosphate; Ang II, angiotensin II; AT1, angiotensin type I receptor; AT2, angiotensin type II receptor; ATP, adenosine triphosphate; BP, blood pressure; COX2, cyclooxygenase-2; CRP, C-reactive protein; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; ETA, endothelin receptor type A; ETB, endothelin receptor type B; HFCS, high-fructose corn syrup; HSP90, heat shock protein 90; ICAM-1, intercellular adhesion molecule 1; IMP, inosine monophosphate; MMPs, matrix metalloproteinases; mRNA, messenger ribonucleic acid; NADPH, nicotinamide adenine dinucleotide phosphate; NHANES, National Health and Nutrition Examination Survey; NHE3, sodium-hydrogen exchanger 3; PAT1, putative anion transporter 1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; TXA2, thromboxane A2; TXB2, thromboxane B2
REFERENCES
- 1.Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang D-H, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr 2007; 86:899–906. [DOI] [PubMed] [Google Scholar]
- 2.Hanover LM, White JS. Manufacturing, composition, and applications of fructose. Am J Clin Nutr 1993; 58:724S–732S. [DOI] [PubMed] [Google Scholar]
- 3.Marriott BP, Cole N, Lee E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J Nutr 2009; 139:1228S–1235S. [DOI] [PubMed] [Google Scholar]
- 4.Egan BM, Zhao Y, Axon RN. Us trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 2010; 303:2043–2050. [DOI] [PubMed] [Google Scholar]
- 5.Robinson SC, Brucer M. Range of normal blood pressure. A statistical and clinical study of 11,383 persons. Arch Intern Med 1939; 64:409–444. [Google Scholar]
- 6.Jalal DI, Smits G, Johnson RJ, Chonchol M. Increased fructose associates with elevated blood pressure. J Am Soc Nephrol 2010; 21:1543–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen S, Choi HK, Lustig RH, Hsu C-y. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr 2009; 154:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Caballero B, Mitchell DC, Loria C, Lin P-H, Champagne CM, et al. Reducing consumption of sugar-sweetened beverages is associated with reduced blood pressure: aprospective study among United States adults. Circulation 2010; 121:2398–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 2010; 285:17271–17276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dustan HP. Obesity and hypertension. Diabetes Care 1991; 14:488–504. [DOI] [PubMed] [Google Scholar]
- 11.Tappy L, Le K-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev 2010; 90:23–46. [DOI] [PubMed] [Google Scholar]
- 12.Le MT, Frye RF, Rivard CJ, Cheng J, McFann KK, Segal MS, et al. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metab Clin Exp 2012; 61:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown Clive M, Dulloo Abdul G, Yepuri G, Montani J-P. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Regul Integr Comp Physiol 2008; 294:R730–737. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Pozo SE, Schold J, Nakagawa T, Sanchez-Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes 2010; 34:454–461. [DOI] [PubMed] [Google Scholar]
- 15.Tran LT, Yuen VG, McNeill JH. The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol Cell Biochem 2009; 332:145–159. [DOI] [PubMed] [Google Scholar]
- 16.Hwang IS, Ho H, Hoffman BB, Reaven GM. Fructose-induced insulin resistance and hypertension in rats. Hypertension 1987; 10:512–516. [DOI] [PubMed] [Google Scholar]
- 17.Martinez FJ, Rizza RA, Romero JC. High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension 1994; 23:456–463. [DOI] [PubMed] [Google Scholar]
- 18.Glushakova O, Kosugi T, Roncal C, Mu W, Heinig M, Cirillo P, et al. Fructose induces the inflammatory molecule ICAM-1 in endothelial cells. J Am Soc Nephrol 2008; 19:1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soleimani M. Dietary fructose, salt absorption and hypertension in metabolic syndrome: towards a new paradigm. Acta Physiol 2011; 201:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soleimani M, Alborzi P. The role of salt in the pathogenesis of fructose-induced hypertension. Int J Nephrol 2011; 392708, 392708pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh AK, Amlal H, Haas PJ, Dringenberg U, Fussell S, Barone SL, et al. Fructose-induced hypertension: essential role of chloride and fructose absorbing transporters PAT1 and GLUT5. Kidney Int 2008; 74:438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, et al. Slc2a5 (GLUT5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem 2009; 284:5056–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabral PD, Hong NJ, Khan MAH, Ortiz PA, Beierwaltes WH, Imig JD, et al. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension 2014; 63:e68–e73. [DOI] [PubMed] [Google Scholar]
- 24.Queiroz-Leite GD, Crajoinas RO, Neri EA, Bezerra CNA, Girardi ACC, Reboucas NA, et al. Fructose acutely stimulates NHE3 activity in kidney proximal tubule. Kidney Blood Pressure Res 2012; 36:320–334. [DOI] [PubMed] [Google Scholar]
- 25.Welch WJ, Ott CE, Lorenz JN, Kotchen TA. Control of renin release by dietary sodium chloride in the rat. Am J Physiol 1987; 253:F1051–F1057. [DOI] [PubMed] [Google Scholar]
- 26.Miller CCJ, Samani NJ, Carter AT, Brooks JI, Brammar WJ. Modulation of mouse renin gene expression by dietary sodium chloride intake in one-gene, two-gene and transgenic animals. J Hypertens 1989; 7:861–863. [DOI] [PubMed] [Google Scholar]
- 27.Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr 1993; 58:754S–765S. [DOI] [PubMed] [Google Scholar]
- 28.Catena C, Cavarape A, Novello M, Giacchetti G, Sechi LA. Insulin receptors and renal sodium handling in hypertensive fructose-fed rats. Kidney Int 2003; 64:2163–2171. [DOI] [PubMed] [Google Scholar]
- 29.Katakam PVG, Ujhelyi MR, Hoenig ME, Miller AW. Endothelial dysfunction precedes hypertension in diet-induced insulin resistance. Am J Physiol 1998; 275:R788–R792. [DOI] [PubMed] [Google Scholar]
- 30.Palanisamy N, Venkataraman AC. Beneficial effect of genistein on lowering blood pressure and kidney toxicity in fructose-fed hypertensive rats. Br J Nutr 2013; 109:1806–1812. [DOI] [PubMed] [Google Scholar]
- 31.Okamura T, Tawa M, Geddawy A, Shimosato T, Iwasaki H, Shintaku H, et al. Effects of atorvastatin, amlodipine, and their combination on vascular dysfunction in insulin-resistant rats. J Pharmacol Sci 2014; 124:76–85. [DOI] [PubMed] [Google Scholar]
- 32.Miatello R, Risler N, Gonzalez S, Castro C, Ruttler M, Cruzado M. Effects of enalapril on the vascular wall in an experimental model of syndrome X. Am J Hypertens 2002; 15:872–878. [DOI] [PubMed] [Google Scholar]
- 33.Verma S, Bhanot S, McNeill JH. Effect of chronic endothelin blockade in hyperinsulinemic hypertensive rats. Am J Physiol 1995; 269:H2017–H2021. [DOI] [PubMed] [Google Scholar]
- 34.Hu RM, Levin ER, Pedram A, Frank HJL. Insulin stimulates production and secretion of endothelin from bovine endothelial cells. Diabetes 1993; 42:351–358. [DOI] [PubMed] [Google Scholar]
- 35.Juan C-C, Shen Y-W, Chien Y, Lin Y-J, Chang S-F, Ho L-T. Insulin infusion induces endothelin-1-dependent hypertension in rats. Am J Physiol Endocrinol Metab 2004; 287:E948–E954. [DOI] [PubMed] [Google Scholar]
- 36.Jiang J, Tran L, Vasudevan H, Xia Z, Yuen VG, McNeill JH. Endothelin-1 blockade prevents COX2 induction and TXA2 production in the fructose hypertensive rat. Can J Physiol Pharmacol 2007; 85:422–429. [DOI] [PubMed] [Google Scholar]
- 37.Song D, Arikawa E, Galipeau D, Battell M, McNeill JH. Androgens are necessary for the development of fructose-induced hypertension. Hypertension 2004; 43:667–672. [DOI] [PubMed] [Google Scholar]
- 38.Tran LT, MacLeod KM, McNeill JH. Endothelin-1 modulates angiotensin II in the development of hypertension in fructose-fed rats. Mol Cell Biochem 2009; 325:89–97. [DOI] [PubMed] [Google Scholar]
- 39.Shinozaki K, Ayajiki K, Nishio Y, Sugaya T, Kashiwagi A, Okamura T. Evidence for a causal role of the renin-angiotensin system in vascular dysfunction associated with insulin resistance. Hypertension 2004; 43:255–262. [DOI] [PubMed] [Google Scholar]
- 40.Widdop RE, Jones ES, Hannan RE, Gaspari TA. Angiotensin AT2 receptors: cardiovascular hope or hype? Br J Pharmacol 2003; 140:809–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erlich Y, Rosenthal T. Effect of angiotensin-converting enzyme inhibitors on fructose induced hypertension and hyperinsulinemia in rats. Clin Exp Pharmacol Physiol 1995; 22:S347–S349. [DOI] [PubMed] [Google Scholar]
- 42.Hitomi H, Kiyomoto H, Nishiyama A. Angiotensin II and oxidative stress. Curr Opin Cardiol 2007; 22:311–315. [DOI] [PubMed] [Google Scholar]
- 43.Miatello R, Risler N, Castro C, Cruzado M, Gonzalez S, Zumino AP. Chronic administration of losartan reverses cardiovascular changes in hypertensive fructose-fed rats. Cell Mol Biol 2003; 49:945–952. [PubMed] [Google Scholar]
- 44.Di Wang H, Pagano PJ, Du Y, Cayatte AJ, Quinn MT, Brecher P, et al. Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ Res 1998; 82:810–818. [DOI] [PubMed] [Google Scholar]
- 45.Cuzzocrea S, Mazzon E, Dugo L, Di Paola R, Caputi AP, Salvemini D. Superoxide: a key player in hypertension. FASEB J 2004; 18:94–101. [DOI] [PubMed] [Google Scholar]
- 46.Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res 2005; 96:818–822. [DOI] [PubMed] [Google Scholar]
- 47.Coker ML, Jolly JR, Joffs C, Etoh T, Holder JR, Bond BR, et al. Matrix metalloproteinase expression and activity in isolated myocytes after neurohormonal stimulation. Am J Physiol 2001; 281:H543–H551. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Tai H-H. Thromboxane A2 receptor-mediated release of matrix metalloproteinase-1 (MMP-1) induces expression of monocyte chemoattractant protein-1 (MCP-1) by activation of protease-activated receptor 2 (PAR2) in A549 human lung adenocarcinoma cells. Mol Carcinog 2014; 53:659–666. [DOI] [PubMed] [Google Scholar]
- 49.Nagareddy PR, Rajput PS, Vasudevan H, McClure B, Kumar U, MacLeod KM, et al. Inhibition of matrix metalloproteinase-2 improves endothelial function and prevents hypertension in insulin-resistant rats. Br J Pharmacol 2012; 165:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tedgui A, Mallat Z. Anti-inflammatory mechanisms in the vascular wall. Circ Res 2001; 88:877–887. [DOI] [PubMed] [Google Scholar]
- 51.Dhar A, Desai K, Kazachmov M, Yu P, Wu L. Methylglyoxal production in vascular smooth muscle cells from different metabolic precursors. Metab Clin Exp 2008; 57:1211–1220. [DOI] [PubMed] [Google Scholar]
- 52.Dhar I, Dhar A, Wu L, Desai KM. Increased methylglyoxal formation with upregulation of renin angiotensin system in fructose fed sprague-dawley rats. PLoS One 2013; 8:e74212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Jia X, Chang T, Desai K, Wu L. Attenuation of hypertension development by scavenging methylglyoxal in fructose-treated rats. J Hypertens 2008; 26:765–772. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Desai K, Clausen JT, Wu L. Increased methylglyoxal and advanced glycation end products in kidney from spontaneously hypertensive rats. Kidney Int 2004; 66:2315–2321. [DOI] [PubMed] [Google Scholar]
- 55.Wautier J-L, Schmidt Ann M. Protein glycation: a firm link to endothelial cell dysfunction. Circ Res 2004; 95:233–238. [DOI] [PubMed] [Google Scholar]
- 56.Wautier M-P, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier J-L. Activation of NADPH oxidase by age links oxidant stress to altered gene expression via rage. Am J Physiol 2001; 280:E685–E694. [DOI] [PubMed] [Google Scholar]
- 57.Wu L, Juurlink BHJ. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension 2002; 39:809–814. [DOI] [PubMed] [Google Scholar]
- 58.Van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J 1977; 162:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdelmalek MF, Lazo M, Horska A, Bonekamp S, Lipkin EW, Balasubramanyam A, et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012; 56:952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jia G, Aroor Annayya R, Whaley-Connell Adam T, Sowers James R. Fructose and uric acid: is there a role in endothelial function? Curr Hypertens Rep 2014; 16:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao H, Pauff JM, Hille R. Substrate orientation and catalytic specificity in the action of xanthine oxidase: the sequential hydroxylation of hypoxanthine to uric acid. J Biol Chem 2010; 285:28044–28053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macdonald I, Keyser A, Pacy D. Some effects, in man, of varying the load of glucose, sucrose, fructose, or sorbitol on various metabolites in blood. Am J Clin Nutr 1978; 31:1305–1311. [DOI] [PubMed] [Google Scholar]
- 63.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol 2006; 290:F625–F631. [DOI] [PubMed] [Google Scholar]
- 64.Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, Soto V, Avila-Casado C, Vega-Campos IP, et al. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am J Physiol 2008; 294:F710–F718. [DOI] [PubMed] [Google Scholar]
- 65.Cavarape A, Feletto F, Mercuri F, Quagliaro L, Damante G, Ceriello A. High-fructose diet decreases catalase mrna levels in rat tissues. J Endocrinol Invest 2001; 24:838–845. [DOI] [PubMed] [Google Scholar]
- 66.Sanchez-Lozada Laura G, Lanaspa Miguel A, Cristobal-Garcia M, Garcia-Arroyo F, Soto V, Cruz-Robles D, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular atp concentrations. Nephron Exp Nephrol 2012; 121:e71–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanbay M, Solak Y, Dogan E, Lanaspa MA, Covic A. Uric acid in hypertension and renal disease: the chicken or the egg? Blood Purif 2010; 30:288–295. [DOI] [PubMed] [Google Scholar]
- 68.Gersch C, Palii SP, Kim KM, Angerhofer A, Johnson RJ, Henderson GN. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucl Acids 2008; 27:967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang D-H, Park S-K, Lee I-K, Johnson RJ. Uric acid-induced c-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 2005; 16:3553–3562. [DOI] [PubMed] [Google Scholar]
- 70.Khosla Uday M, Zharikov S, Finch Jennifer L, Nakagawa T, Roncal C, Mu W, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int 2005; 67:1739–1742. [DOI] [PubMed] [Google Scholar]
- 71.Sautin YY, Johnson RJ. Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucl Acids 2008; 27:608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ejaz AA, Mu W, Kang D-H, Roncal C, Sautin YY, Henderson G, et al. Could uric acid have a role in acute renal failure? Clin J Am Soc Nephrol 2007; 2:16–21. [DOI] [PubMed] [Google Scholar]
- 73.Hong Q, Qi K, Feng Z, Huang Z, Cui S, Wang L, et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012; 51:402–410. [DOI] [PubMed] [Google Scholar]
- 74.Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L, et al. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J Am Soc Nephrol 2009; 20:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jalal DI, Jablonski KL, McFann K, Chonchol MB, Seals DR. Vascular endothelial function is not related to serum uric acid in healthy adults. Am J Hypertens 2012; 25:407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dohi Y, Thiel MA, Buehler FR, Luescher TF. Activation of endothelial l-arginine pathway in resistance arteries. Effect of age and hypertension. Hypertension 1990; 16:170–179. [DOI] [PubMed] [Google Scholar]
- 77.Spieker LE, Noll G, Ruschitzka FT, Maier W, Luscher TF. Working under pressure: the vascular endothelium in arterial hypertension. J Hum Hypertens 2000; 14:617–630. [DOI] [PubMed] [Google Scholar]
- 78.Penicaud L, Berthault MF, Morin J, Dubar M, Ktorza A, Ferre P. Rilmenidine normalizes fructose-induced insulin resistance and hypertension in rats. J Hypertens Suppl 1998; 16:S45–S49. [PubMed] [Google Scholar]
- 79.Rosen P, Ohly P, Gleichmann H. Experimental benefit of moxonidine on glucose metabolism and insulin secretion in the fructose-fed rat. J Hypertens Suppl 1997; 15:S31–S38. [DOI] [PubMed] [Google Scholar]
- 80.Verma S, Bhanot S, McNeill JH. Sympathectomy prevents fructose-induced hyperinsulinemia and hypertension. Eur J Pharmacol 1999; 373:R1–R4. [DOI] [PubMed] [Google Scholar]
- 81.Tran LT, MacLeod KM, McNeill JH. Selective alpha1-adrenoceptor blockade prevents fructose-induced hypertension. Mol Cell Biochem 2014; 392:205–211. [DOI] [PubMed] [Google Scholar]
- 82.Chen DG, Dai XZ, Bache RJ. Postsynaptic adrenoceptor-mediated vasoconstriction in coronary and femoral vascular beds. Am J Physiol 1988; 254:H984–H992. [DOI] [PubMed] [Google Scholar]
- 83.Fu Y-C, Chi C-S, Yin S-C, Hwang B, Chiu Y-T, Hsu S-L. Norepinephrine induces apoptosis in neonatal rat endothelial cells via down-regulation of bcl-2 and activation of β-adrenergic and caspase-2 pathways. Cardiovasc Res 2004; 61:143–151. [DOI] [PubMed] [Google Scholar]
- 84.Fu Y-C, Yin S-C, Chi C-S, Hwang B, Hsu S-L. Norepinephrine induces apoptosis in neonatal rat endothelial cells via a ros-dependent jnk activation pathway. Apoptosis 2006; 11:2053–2063. [DOI] [PubMed] [Google Scholar]
- 85.Kaplon RE, Walker AE, Seals DR. Plasma norepinephrine is an independent predictor of vascular endothelial function with aging in healthy women. J Appl Physiol 2011; 111:1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thijssen Dick HJ, Atkinson Ceri L, Ono K, Sprung Victoria S, Spence Angela L, Pugh Christopher JA, et al. Sympathetic nervous system activation, arterial shear rate, and flow-mediated dilation. J Appl Physiol 2014; 116:1300–1307. [DOI] [PubMed] [Google Scholar]
- 87.Barnes RD, Ward LE, Frank KP, Tyce GM, Hunter LW, Rorie DK. Nitric oxide modulates evoked catecholamine release from canine adrenal medulla. Neuroscience 2001; 104:1165–1173. [DOI] [PubMed] [Google Scholar]
- 88.Addicks K, Bloch W, Feelisch M. Nitric oxide modulates sympathetic neurotransmission at the prejunctional level. Microsc Res Tech 1994; 29:161–168. [DOI] [PubMed] [Google Scholar]
- 89.Iida N. Nitric oxide mediates sympathetic vasoconstriction at supraspinal, spinal, and synaptic levels. Am J Physiol 1999; 276:H918–H925. [DOI] [PubMed] [Google Scholar]