

Abstract
Introduction
γ-amino butyric acid (GABA) is not only the major inhibitory neurotransmitter in the central nervous system (CNS), but it also plays an important role in the lung, mediating airway smooth muscle relaxation and mucus production. As kinases such as protein kinase A (PKA) are known to regulate the release and reuptake of GABA in the CNS by GABA transporters, we hypothesized that β-agonists would affect GABA release from airway epithelial cells through activation of PKA.
Methods
C57/BL6 mice received a pretreatment of a β-agonist or vehicle (PBS), followed by methacholine or PBS. Bronchoalveolar lavage (BAL) was collected and the amount of GABA was quantified using HPLC mass spectrometry. For in vitro studies, cultured BEAS-2B human airway epithelial cells were loaded with 3H-GABA. 3H-GABA released was measured during activation and inhibition of PKA and tyrosine kinase signaling pathways.
Results
β-agonist pretreatment prior to methacholine challenge attenuated in vivo GABA release in mouse BAL and 3H-GABA release from depolarized BEAS-2B cells. GABA release was also decreased in BEAS-2B cells by increases in cAMP but not by Epac or tyrosine kinase activation.
Conclusion
β-agonists decrease GABA release from airway epithelium through the activation of cAMP and PKA. This has important therapeutic implications as β-agonists and GABA are important mediators of both mucus production and airway smooth muscle tone.
Keywords: Bronchoalveolar lavage, 3H-GABA release, Protein kinase A, Tyrosine receptor kinase, BEAS-2B, β-agonist
Introduction
γ-amino butyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system (CNS), but recent studies in our laboratory and others’ demonstrate a physiologic importance of GABA outside of the CNS [1]. Both the ionotropic GABAA channel and metabotropic GABAB receptor are expressed on airway smooth muscle (ASM) [2, 3], and we have shown that GABA agonists relax ASM [2, 4–6]. These studies clearly demonstrate that GABA plays a pro-relaxant role in ASM and that activating the GABAA channel is a promising novel target in the treatment of bronchoconstrictive diseases such as asthma or COPD.
In this study, we demonstrate for the first time the role of β-adrenergic receptors in peripheral, non-neuronal, GABA release by epithelial cells. The airway epithelium serves as the initial barrier to inhaled respiratory particles and has a critical role in the defense of the airway to toxins and pathogens [7]. Similarly, airway epithelium is the first cell layer exposed to inhaled medications used to treat bronchoconstrictive diseases and is exposed to the highest concentration of inhaled medications. Therefore, understanding the biochemical and physiologic interplay between the β-adrenoceptor and GABAergic receptor system at the level of the epithelial cell may provide insights into pathophysiologic as well as novel therapeutic mechanisms.
In the CNS, GABA is classically released by neuronal vesicular synaptic fusion and its action is terminated by reuptake via GABA transporters (GATs) present on synaptic neurons and glia [8]. A GABAergic system including GABAA channels, GABAB receptors, the GABA synthetic enzyme glutamic acid decarboxylase (GAD), and GABA transporters (GATs) are expressed on the airway epithelium [3, 9–11]. We have recently demonstrated in the airway that GABA is predominantly released from the airway epithelium and GABA release is regulated by GATs [12], (namely GAT2 and GAT4) [10]. We have also shown that depolarization increases GAT-mediated release of GABA in epithelial cells [10], mimicking a known neuronal mechanism of depolarization-induced reversal of GAT function causing GABA efflux from cells [13, 14].
Of central interest to the current study, it is known that depolarization is not the only mechanism of GAT regulation. GAT activity is known to be influenced by several families of protein kinases in the CNS and peripheral T lymphocytes [15–21], including protein kinase A (PKA). This led us to question whether β-agonists which have direct effects on G-protein-mediated activation of PKA [22, 23] would also have PKA-mediated effects on GAT function. We demonstrate for the first time a decreased GABA release by epithelial cells through cAMP/PKA-mediated effects of β-agonists, uncovering a complex interaction between β-agonists effects and the GABAergic system in the lung.
Methods
Reagents
Reagents obtained from Sigma (St. Louis, MO) unless otherwise stated.
In Vivo β-Agonist/Methacholine Challenge
Animal protocols were approved by the institutional IACUC. Male C57/BL6 mice (20–25 g) anesthetized with intraperitoneal pentobarbital (50 mg/kg) were tracheotomized and ventilated (150 breaths/min, 10 ml/kg tidal volume) via a Flexivent FX-1 module (SciReq®). Animals received a nebulized treatment of 100 μM terbutaline or vehicle (PBS) followed by methacholine (25 mg/ml) or PBS 15 min later. Animals were sacrificed and lungs were lavaged with 2 ml of PBS. Bronchoalveolar lavage (BAL) fluid was centrifuged (200×g, 10 min, 4 °C).
HPLC-MS-MS Measurement of BAL GABA
400 μl of lyophilized BAL and GABA standards, 10 μl of internal standard (U-13C L-Alanine), and 200 μl of 100 mM borate buffer (pH 9.5) were incubated with 90 μl H2O, 100 μl of 10 mM 5-N-succinimidoxy-5-oxopentyl)triphenylphosphonium bromide (SPTPP) in acetonitrile [24]. The solution was heated (40 °C for 10 min), and 1600 μl of H2O/Acetonitrile (80:20) containing 0.1 vol.% formic acid was added and cooled to room temperature. A 10 μl aliquot was injected into the LC-MS/MS system (Agilent 1200 series liquid chromatograph, 6430 triple quadrupole mass spectrometer with a turbo spray ionization (ESI) source). Reversed-phase LC was performed using LS Poroshell 120 EC-C18 (2.1 mm × 50 mm, 2.7 um, Agilent Technologies, Santa Clara, CA). The column temp 50 °C, flow rate 0.125 ml/min, and mobile phase were H2O/Acetonitrile (80:20) containing 0.1 vol.% formic acid. Nitrogen gas was used as the collision gas in multiple reaction monitoring mode. Data were analyzed using Agilent Masshunter Quantitative Analysis software version 6.0.
Cell Culture
The immortalized human bronchial epithelial cell line (BEAS-2B) (ATCC, Manassas VA) was cultured as described previously [10]. Immortalized cultures of human airway smooth muscle (ASM) cells (gift from Dr. William Gerthoffer, U of South Alabama) [25] were cultured as described previously [10].
3H-GABA Release Assay
3H-GABA release assay has been described previously [10]. Briefly, BEAS-2B cells were incubated in 190 μl GABA assay buffer containing 10 μM phaclofen and 200 μM gabazine (to block 3H-GABA binding to GABAB receptors and GABAA channels, respectively) at 37 °C for 15 min. Cells were loaded with 3H-GABA ((8 uCi/ml, MP Biomedicals (Irvine, CA) 30 min 37 °C). Cells were treated for 15 min with: forskolin 10 μM (adenylyl cyclase activator), terbutaline 100 μM (β-agonist), PDGF 10 ng/mL (platelet-derived growth factor, receptor tyrosine kinase activator, Calbiochem (Darmstadt, Germany)), herbimycin A 1 μM (receptor tyrosine kinase inhibitor, Tocris (Ellisville, MO)), propranolol 100 μM (β-adrenoceptor antagonist), PKI 14-22 amide 1 μM (PKA inhibitor, Tocris), and Epac activator 100 μM (8-(4-chlorophenylthio)-2′-O-methyladenosine3′,5′-cyclic monophosphate monosodium hydrate). Given that Na+ and Cl− ions are unidirectionally co-transported with GABA, the external buffer was changed to reduce the external Cl− which favors release of 3H-GABA via GABA transporters. Cells were incubated (15 min) with and without the presence of 80 mM potassium gluconate (depolarizing agent that does not alter the extracellular concentration of Cl−). Depolarization has been shown to reverse GAT function, favoring GABA efflux [13, 14]. Supernatant and cell lysate were collected, and fractional release of 3H-GABA was calculated.
Quantitative Polymerase Chain Reaction for GAD65 and GAD67
RNA was extracted as described previously [10]. Using the Super Script® VILO™ cDNA synthesis kit (Invitrogen, Carlsbad, CA), 2 μg RNA was reverse transcribed. Quantitative PCR was performed as described previously with primers for corresponding GAD isoforms [12]. ΔCt was calculated as (Ct of GAD67 or GAD65) − (Ct of GAPDH).
Immunoblot Analysis of PDGF-Induced Akt Phosphorylation in BEAS-2B Cells
BEAS-2B cells were incubated with or without 10 ng/ml PDGF for 30 min. Lysates prepared as previously described [26] with the addition of phosphatase inhibitor cocktail 2 and 3 (1:100) and subjected to immunoblot analysis with antibodies against total AKT or phosphorylated (Ser473) AKT (both rabbit polyclonal, 1:5000, Cell Signaling Technology, Danvers, MA). Signal was detected as described [26].
Statistics
Data were analyzed using one-way ANOVA with selected Bonferroni post-tests using Prism 4.0 software (GraphPad, San Diego, CA) with repeated measures where appropriate and presented as mean ± standard error of the mean (SEM). p < 0.05 was accepted as statistically significant.
Results
Terbutaline Treatment Attenuates In Vivo GABA Release in Mouse Bronchoalveolar Lavage (BAL)
Utilizing an in vivo mouse model, pretreated with either nebulized PBS (vehicle) or the β-agonist terbutaline, we quantified GABA levels in the BAL samples following methacholine challenge. Terbutaline pretreatment attenuated GABA release, demonstrating a greater than threefold decrease (1.83 ± 0.58 nM (PBS/methacholine), versus 0.55 ± 0.10 nM (terbutaline/methacholine), n = 5, p < 0.05) (Fig. 1) (PBS/PBS and terbutaline/PBS controls were also performed (data not shown)). Since β-adrenoceptor activation results in activation of PKA [22], and most GABA in the airway is synthesized/released by airway epithelium [12], we hypothesized that PKA modulates GABA release in airway epithelium and investigated this hypothesis in airway epithelial cells.
Fig. 1.
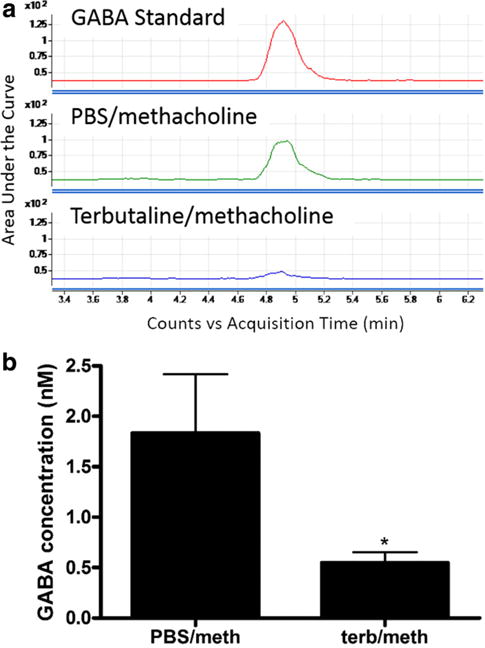
Terbutaline pretreatment attenuated GABA release after methacholine challenge in an in vivo mouse model. a Representative HPLC-MS-MS tracing of a GABA standard (15 nM), PBS/methacholine-treated BAL and terbutaline/methacholine BAL demonstrating an attenuation of GABA with pretreatment with terbutaline. b Pretreatment with nebulized terbutaline (100 μM), a β-adrenergic agonist, before challenge with nebulized methacholine (25 mg/ml) attenuated GABA release in bronchoalveolar lavage. n = 5, *p < 0.05
Quantitative RT-PCR Demonstrates Increased mRNA of GAD67 in Cultured Human Airway Epithelium Compared to Smooth Muscle Cells
We have previously shown in human airway tissue that GAD67, the enzyme responsible for synthesizing GABA, is present in greater amounts in the airway epithelium compared to airway smooth muscle (ASM) [12]. Since we used BEAS-2B human epithelial cells to investigate PKA’s effect on GABA release, we measured mRNA expression of GAD67 and GAD65 and compared it to human ASM cells to see if this relationship was preserved in culture. Quantitative RT-PCR revealed more mRNA encoding GAD67 in epithelial cells compared to ASM cells. After correcting for the expression of the housekeeping gene GAPDH, the ΔCt (Ct of GAD67 – Ct of GAPDH) for BEAS-2B cells was 8.3 ± 0.42 and 18.3 ± 1.28 for ASM cells (n = 7, p < 0.001) (Fig. 2). Since a lower ΔCt value represents more abundant expression of GAD67 mRNA, this difference corresponds to a 1000-fold increase in expression of GAD67 in epithelial cells compared to ASM cells. GAD65 was expressed at much lower levels in both cell types. Therefore, GAD67, an important enzyme for the synthesis of GABA, is present in greater abundance in airway epithelial cells as compared to airway smooth muscle cells.
Fig. 2.
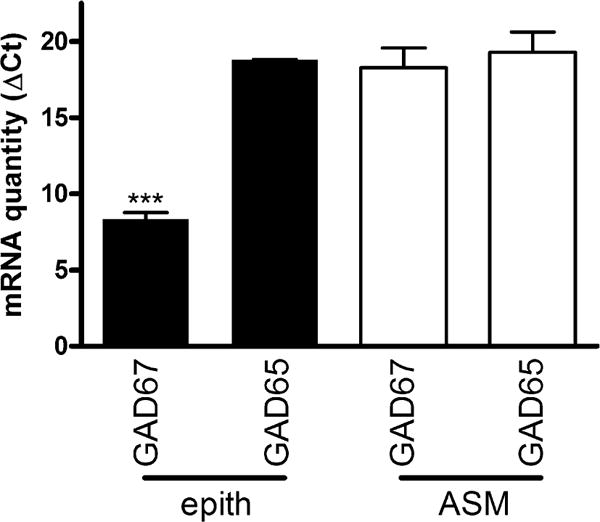
Glutamic acid decarboxylase 67 (GAD67) and GAD65 mRNA quantified by qRT-PCR in cultured human bronchial airway epithelial (epith) and airway smooth muscle (ASM) cells demonstrates GAD67 is the predominant GAD isoform and is expressed in much greater abundance in airway epithelial cells. Note abundance of GAD67 mRNA in epithelium compared to ASM (reflected in lower ΔCt values). ΔCt was calculated as the (Ct of GAD67 or GAD65) −(the Ct of GAPDH) for each sample. n = 3–7, ***p < 0.001 when comparing GAD67 in human airway epithelium to GAD65 in epithelium or to GAD67 in human ASM
Down Regulation of GABA Release by Protein Kinase A
Treatment with terbutaline (100 μM), a β-adrenoceptor agonist which results in elevations of cyclic AMP and PKA activation, significantly decreased depolarization (i.e., potassium gluconate)-induced 3H-GABA release from airway epithelial cells (Fig. 3). This attenuation also occurred with forskolin (10 μM), a direct adenylyl cyclase activator. Studies were repeated in the presence of propranolol (β-adrenoceptor antagonist) in combination with terbutaline to rule out any non-specific effects (Fig. 4). Propranolol reversed the decrease in depolarization-induced 3H-GABA release observed with terbutaline, confirming β-adrenoceptor activation as the specific mechanism by which terbutaline decreases 3H-GABA release. To incriminate a role for PKA in terbutaline-mediated inhibition of 3H-GABA release, studies were done in the presence of PKI 14–22, a PKA inhibitor [27–29]. PKI 14–22 also reversed the decrease in depolarization-induced 3H-GABA release observed with terbutaline, confirming this decrease is mediated through PKA (Fig. 5).
Fig. 3.
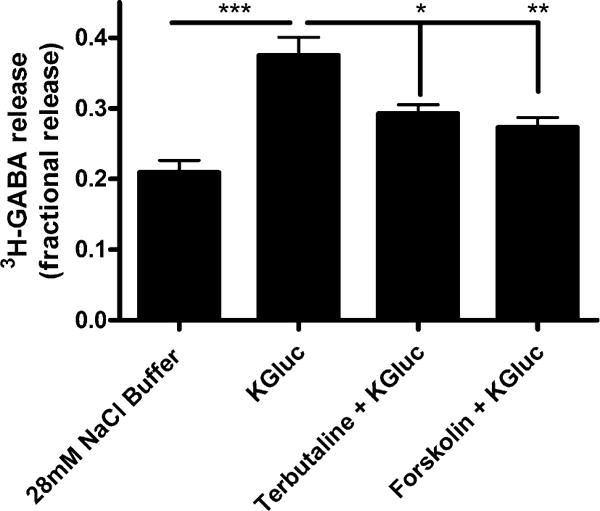
Protein kinase A modulates 3H-GABA release in cultured human airway epithelial (BEAS-2B) cells. Terbutaline (100 μM), a β-adrenergic agonist, and forskolin (10 μM), an activator of adenylyl cyclase, both decrease depolarization-induced fractional 3H-GABA release in cultured human airway epithelial cells. n = 6–8, ***p < 0.001, **p < 0.01, *p < 0.05
Fig. 4.
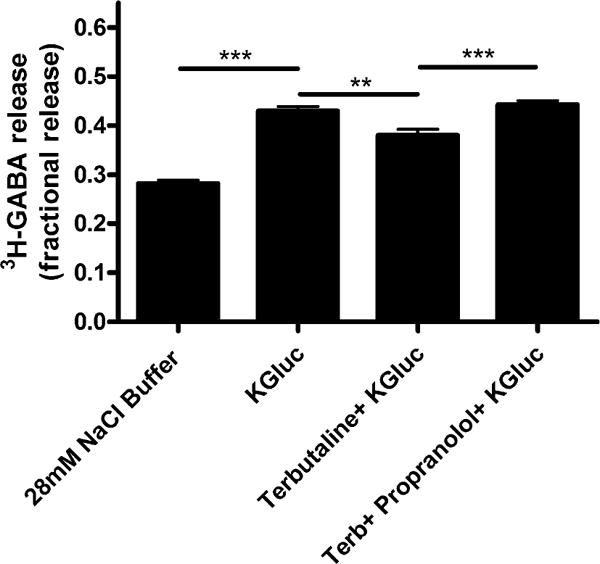
Propranolol completely reverses terbutaline-induced decrease in fractional 3H-GABA release in cultured human airway epithelial (BEAS-2B) cells. Terbutaline (100 μM) treatment decreased fractional 3H-GABA release, and this decrease was completely reversed by co-treatment with propranol. n = 6–8, ***p < 0.001, **p < 0.01
Fig. 5.
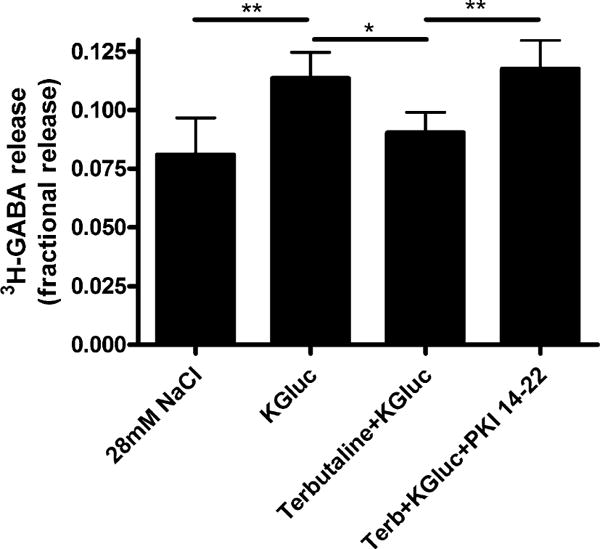
PKI 14-22 (PKA inhibitor) completely reverses terbutaline-induced decrease in fractional 3H-GABA release in cultured human airway epithelial (BEAS-2B) cells. Terbutaline (100 μM) treatment decreased fractional 3H-GABA release, and this decrease was completely reversed by co-treatment with the PKA inhibitor PKI 14-22 (1 μM). n = 4, **p < 0.01, *p < 0.05 ANOVA with repeated measures
While elevations of cAMP are classically understood to activate PKA, additional targets of cAMP are known (e.g., exchange protein directly activated by cAMP (Epac)) in airway epithelial cells [23, 30]. To determine if Epac signaling was a mechanism by which cAMP elevations modulate GABA release, we directly activated Epac, which had no effect on GABA release (Fig. 6). Therefore, elevations of cAMP leading to PKA activation by β-agonists leads to an attenuation of GABA release independent of Epac activation.
Fig. 6.
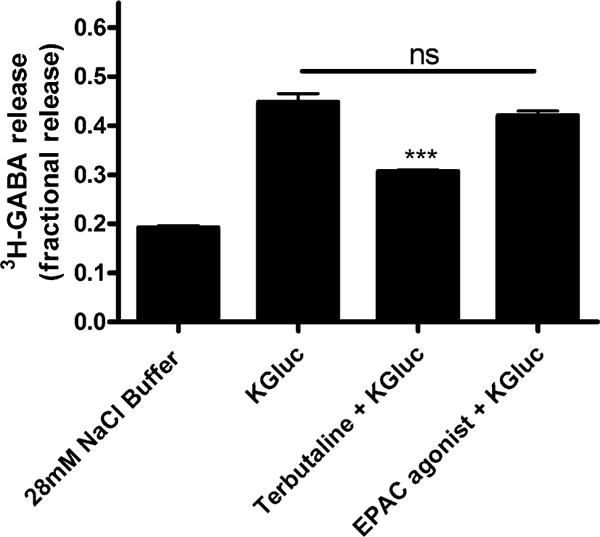
An Epac agonist does not affect depolarization-induced fractional 3H-GABA release in cultured human airway epithelial cells. The EPAC agonist [8-(4-chlorophenylthio)-2′-O-methy-ladenosine 3′,5′-cyclic monophosphate monosodium hydrate] (100 μM) did not affect fractional 3H-GABA release in cultured human airway epithelial cells. n = 4–6, ***p < 0.001 compared to potassium gluconate
Tyrosine Kinase is Not Involved in Release of GABA by GABA Transporters
Although modulation of the PKA pathway clearly affects epithelial GABA release, the tyrosine kinase pathway has been implicated in GABA transport in hippocampal neurons [20]. Given the role of platelet-derived growth factor (PDGF) (receptor tyrosine kinase activator) previously described in BEAS-2B cells [31], we sought to determine if PDGF modulates epithelial GABA release. PDGF had no effect on 3H-GABA release from airway epithelial cells (Fig. 7a). Reduction in tyrosine receptor kinase activity with herbimycin A was also without an effect on 3H-GABA release (Fig. 7b). To confirm that the lack of effect was not due to the inability of our BEAS-2B cells to respond to PDGF, we assessed PDGF-induced phosphorylation of Akt (protein kinase B) which illustrates intact PDGF signaling (Fig. 7c).
Fig. 7.
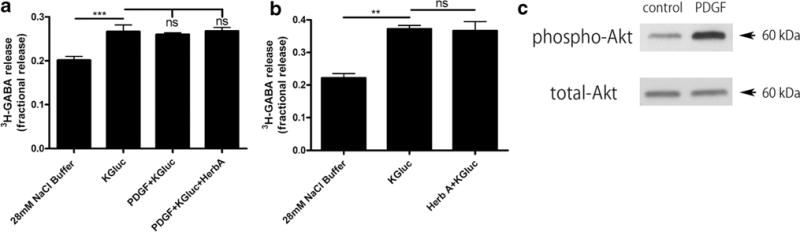
PDGF-induced tyrosine kinase modulation does not affect fractional 3H-GABA release in cultured human airway epithelial (BEAS-2B) cells. Neither a platelet-derived growth factor (PDGF) (10 ng/ml), a receptor tyrosine kinase activator, nor b herbimycin A (1 μM), an inhibitor of receptor tyrosine kinases, had an effect on depolarization-induced fractional 3H-GABA release in cultured human airway epithelial cells. n = 6–7, ***p < 0.001. c Human airway epithelial (BEAS-2B) cells had an intact signaling response to PDGF as confirmed by PDGF-induced phosphorylation of Akt (protein kinase B) detected by immunoblotting
Discussion
In the present study, we demonstrate that protein kinase A (PKA) regulates the release of GABA in both an in vivo and in vitro model. We first demonstrate in vivo that mice pretreated with the β-agonist terbutaline showed a significant decrease in GABA release into bronchoalveolar lavage after methacholine challenge. We next sought to elucidate whether GABA release was regulated by PKA in an in vitro model using BEAS-2B human airway epithelial cells.
Previous studies have confirmed that airway epithelium is the primary source of GABA in human airway tissue [12], guinea pig airway tissue [3], and mouse lung [9]. In this study, we demonstrate that this relationship is preserved in culture by demonstrating that the expression of GAD67, the major enzyme responsible for synthesizing GABA in peripheral airways, is significantly higher in epithelial cells when compared to smooth muscle cells in culture. Given that these results are from a cultured epithelial line, we are confident that GABA synthesis can occur in the absence of a neural contribution.
Utilizing BEAS-2B, we confirmed that GABA release is decreased by terbutaline (β-agonist) and forskolin (adenylyl cyclase activator to increase cAMP). Furthermore, the attenuation of GABA release by terbutaline was reversed by co-treatment with propranolol (β-antagonist); ruling out any non-specific effect of terbutaline. The terbutaline effect was also reversed by PKI 14-22 (PKA inhibitor) demonstrating that this effect is specific to a PKA pathway. Although studies in airway smooth muscle (ASM) have demonstrated that relaxation of ASM mediated by β-agonists occurs through a PKA-dependent pathway, Epac (an exchange protein activated by cAMP) may also contribute to the relaxation achieved by β-adrenoceptor activation [22, 23]. Although PKA is the classic target of cAMP, cAMP is also known to activate Epac [32, 33]. In the current study, we demonstrate that the attenuation of GABA release is not regulated by an Epac agonist. Therefore, the attenuation of GABA release by the β-agonist terbutaline appears to mechanistically involve an increase in cAMP which then activates PKA.
β-agonists have many different effects on epithelial cells, but our study is the first to demonstrate that β-adrenoceptor activation can affect GABA release. β-agonists are well known clinically for their capacity to relax ASM in asthmatics, and are often given via inhalation. As such, they are in direct contact with airway epithelium prior to reaching the ASM. Indeed, several effects of β-adrenoceptor activation on airway epithelium have been reported [34]. β-agonists are known to increase ciliary beat frequency through the activation of PKA in airway epithelium [35–38], which leads to enhanced mucociliary clearance [38–40] without a large change in mucus secretion or production [34]. The increase of cAMP by β-agonists also opens the cystic fibrosis transmembrane regulator (CFTR), and may play a role in proper airway and mucus hydration [34]. Therefore, β-agonists have beneficial effects on airway epithelium such as enhanced clearance and improved hydration of mucus.
The GABAergic system in airway epithelium has been shown to play a role in mucus production [9, 41]. Xiang et al. demonstrated that OVA sensitization in mice increased expression of components of the GABAergic system (GAD and GABAA β subunits) in airway epithelial cells and that the GABAA antagonist picrotoxin decreased mucus overproduction [9]. Our study uncovers a potential, yet unrecognized beneficial effect of β-agonist therapy on airway epithelial mucus production. While more studies are required, our results suggest β-agonist therapy may decrease mucus production by epithelial cells via a decreased paracrine effect of GABA.
GABA also has significant effects on the ASM in addition to the epithelium. We have previously demonstrated that the source of airway GABA is predominantly synthesized/released by the airway epithelium in the lung, and this GABA released from the epithelium has a paracrine effect on GABAA channels on ASM [12]. This paracrine signaling from epithelium to smooth muscle leads to a tonic pro-relaxant effect on ASM [42]. We found this effect is functionally important during the maintenance phase of an established contraction, where GABA-mediated activation of endogenous ASM GABAA channels contributes a counterbalancing relaxation effect on ASM tone. However, in the current study, we demonstrate that β-agonist therapy decreased epithelial GABA release. While a reduction in release of GABA following β-agonist treatment could be viewed as potentially pro-contractile, the magnitude of pro-relaxation pathways afforded by β-adrenoceptor activation functionally overcomes the diminished role of GABA-mediated relaxation in this particular scenario. In contrast, however, we do believe further work is merited in this area to see if this effect (the pro-contractile GABA effect as a counter to the β-agonist pro-relaxant effect) may account for the poor clinical outcomes that have been associated with chronic long-acting β-agonist therapy [43, 44]. In addition, the decrease of endogenous GABA by β-agonists may allow for enhanced efficacy by direct GABAA agonists. While not yet investigated, reduced GABA release by β-agonists may theoretically result in less competition for exogenously administered direct GABAA receptor agonists at GABAA receptor-binding sites. Indeed, this study may highlight the potential therapeutic importance of administering a GABAA agonist with β-agonists, and suggests GABAA agonists could serve as important adjunctive therapy in patients who are becoming increasingly refractory to β-agonist treatment.
PKA has been shown to modulate GAD67 function in neuronal cells. The phosphorylation of GAD67 by PKA has been shown to inhibit GAD67 function leading to a decrease in GABA synthesis [45, 46]. However, it is difficult to extrapolate from these studies, the time course over which PKA phosphorylation of GAD67 would affect GABA synthesis, since these studies were performed in broken cell preparations or with purified enzymes. In the current study, animals received nebulized terbutaline 15 min before a methacholine challenge and lavage to measure BAL GABA concentrations. It is possible that this nebulized terbutaline resulted in reduced BAL GABA by both inhibition of GABA transporter function as well as reduced synthesis by GAD67.
In summary, we have shown that β-agonists decrease GABA release by airway epithelial cells in both an in vivo and in vitro model. GABA release is regulated by PKA which also plays important roles in mucociliary clearance and ASM relaxation. This study uncovers a previously unknown mechanistic link of β-agonists and the GABAergic system in the lung which has important clinical implications in mucus production and ASM tone.
Acknowledgments
We thank Dr. William Gerthoffer, University of South Alabama for sharing cultured human airway smooth muscle cells. This work was supported by the Stony Wold-Herbert Fund (JD, SZ), the National Institutes of Health Grants GM065281, HL122340, and GM008464 (CWE), and The Foundation for Anesthesia Education and Research (FAER) (JD).
Footnotes
Conflict of Interest None.
References
- 1.Gallos G, Emala CW. Anesthetic effects on gamma-aminobutyric acid A receptors: not just on your nerves. Anesthesiology. 2013;118(5):1013–1015. doi: 10.1097/ALN.0b013e31828e1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizuta K, Xu D, Pan Y, et al. GABAA receptors are expressed and facilitate relaxation in airway smooth muscle. Am J Physiol Lung C. 2008;294(6):L1206–L1216. doi: 10.1152/ajplung.00287.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizuta K, Osawa Y, Mizuta F, et al. Functional expression of GABAB receptors in airway epithelium. Am J Resp Cell Mol. 2008;39(3):296–304. doi: 10.1165/rcmb.2007-0414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallos G, Yim P, Chang S, et al. Targeting the restricted alpha-subunit repertoire of airway smooth muscle GABAA receptors augments airway smooth muscle relaxation. Am J Physiol Lung C. 2012;302(2):L248–L256. doi: 10.1152/ajplung.00131.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallos G, Yocum GT, Siviski ME, et al. Selective targeting of the alpha5-subunit of GABAA receptors relaxes airway smooth muscle and inhibits cellular calcium handling. Am J Physiol Lung C. 2015;308(9):L931–L942. doi: 10.1152/ajplung.00107.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallos G, Gleason NR, Virag L, et al. Endogenous gamma-aminobutyric acid modulates tonic guinea pig airway tone and propofol-induced airway smooth muscle relaxation. Anesthesiology. 2009;110(4):748–758. doi: 10.1097/aln.0b013e31819c44e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies DE. Epithelial barrier function and immunity in asthma. An Am Thorac Soc. 2014;11(Suppl 5):S244–S251. doi: 10.1513/AnnalsATS.201407-304AW. [DOI] [PubMed] [Google Scholar]
- 8.Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol. 2005;94(3):2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- 9.Xiang YY, Wang S, Liu M, et al. A GABAergic system in airway epithelium is essential for mucus overproduction in asthma. Nat Med. 2007;13(7):862–867. doi: 10.1038/nm1604. [DOI] [PubMed] [Google Scholar]
- 10.Zaidi S, Gallos G, Yim PD, et al. Functional expression of gamma-amino butyric acid transporter 2 in human and guinea pig airway epithelium and smooth muscle. Am J Respir Cell Mol Biol. 2011;45(2):332–339. doi: 10.1165/rcmb.2010-0177OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu XW, Wood K, Spindel ER. Prenatal nicotine exposure increases GABA signaling and mucin expression in airway epithelium. Am J Respir Cell Mol Biol. 2011;44(2):222–229. doi: 10.1165/rcmb.2010-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallos G, Townsend E, Yim P, et al. Airway epithelium is a predominant source of endogenous airway GABA and contributes to relaxation of airway smooth muscle tone. Am J Physiol Lung C. 2013;304(3):L191–L197. doi: 10.1152/ajplung.00274.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Y, Wang W, Diez-Sampedro A, et al. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron. 2007;56(5):851–865. doi: 10.1016/j.neuron.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Wang W, Richerson GB. GABA transaminase inhibition induces spontaneous and enhances depolarization-evoked GABA efflux via reversal of the GABA transporter. J Neurosci. 2001;21(8):2630–2639. doi: 10.1523/JNEUROSCI.21-08-02630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez B, Paz F, Floran L, et al. Adenosine A2A receptor stimulation decreases GAT-1-mediated GABA uptake in the globus pallidus of the rat. Neuropharmacology. 2006;51(1):154–159. doi: 10.1016/j.neuropharm.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Gomeza J, Gimenez C, Zafra F. Cellular distribution and regulation by cAMP of the GABA transporter (GAT-1) mRNA. Brain Res Mol Brain Res. 1994;21(1–2):150–156. doi: 10.1016/0169-328x(94)90387-5. [DOI] [PubMed] [Google Scholar]
- 17.Tian Y, Kapatos G, Granneman JG, et al. Dopamine and gamma-aminobutyric acid transporters: differential regulation by agents that promote phosphorylation. Neurosci Lett. 1994;173(1–2):143–146. doi: 10.1016/0304-3940(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 18.Cristovao-Ferreira S, Vaz SH, Ribeiro JA, et al. Adenosine A2A receptors enhance GABA transport into nerve terminals by restraining PKC inhibition of GAT-1. J Neurochem. 2009;109(2):336–347. doi: 10.1111/j.1471-4159.2009.05963.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Luo Q, Xu Y, et al. Gamma-aminobutyric acid transporter 1 negatively regulates T cell activation and survival through protein kinase C-dependent signaling pathways. J Immunol. 2009;183(5):3488–3495. doi: 10.4049/jimmunol.0900767. [DOI] [PubMed] [Google Scholar]
- 20.Law RM, Stafford A, Quick MW. Functional regulation of gamma-aminobutyric acid transporters by direct tyrosine phosphorylation. J Biol Chem. 2000;275(31):23986–23991. doi: 10.1074/jbc.M910283199. [DOI] [PubMed] [Google Scholar]
- 21.Beckman ML, Bernstein EM, Quick MW. Multiple G protein-coupled receptors initiate protein kinase C redistribution of GABA transporters in hippocampal neurons. J Neurosci. 1999;19(11):RC9. doi: 10.1523/JNEUROSCI.19-11-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morgan SJ, Deshpande DA, Tiegs BC, et al. β-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J Biol Chem. 2014;289(33):23065–23074. doi: 10.1074/jbc.M114.557652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Billington CK, Ojo OO, Penn RB, et al. cAMP regulation of airway smooth muscle function. Pulm Pharmacol Ther. 2013;26(1):112–120. doi: 10.1016/j.pupt.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inagaki S, Toyo’oka T. Amino acid analysis via LC-MS method after derivatization with quaternary phosphonium. Methods Mol Biol. 2012;828:47–54. doi: 10.1007/978-1-61779-445-2_5. [DOI] [PubMed] [Google Scholar]
- 25.Gosens R, Stelmack GL, Dueck G, et al. Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung C. 2006;291(3):L523–L534. doi: 10.1152/ajplung.00013.2006. [DOI] [PubMed] [Google Scholar]
- 26.Gallos G, Remy KE, Danielsson J, et al. Functional expression of the TMEM16 family of calcium-activated chloride channels in airway smooth muscle. Am J Physiol Lung C. 2013;305(9):L625–L634. doi: 10.1152/ajplung.00068.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saino T, Watson EL. Inhibition of serine/threonine phosphatase enhances arachidonic acid-induced [Ca2+]i via protein kinase A. Am J Physiol Cell Physiol. 2009;296(1):C88–C96. doi: 10.1152/ajpcell.00281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokoyama U, Minamisawa S, Quan H, et al. Prostaglandin E2-activated Epac promotes neointimal formation of the rat ductus arteriosus by a process distinct from that of cAMP-dependent protein kinase A. J Biol Chem. 2008;283(42):28702–28709. doi: 10.1074/jbc.M804223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno-Delgado D, Gomez-Ramirez J, Torrent-Moreno A, et al. Different role of cAMP dependent protein kinase and CaMKII in H3 receptor regulation of histamine synthesis and release. Neuroscience. 2009;164(3):1244–1251. doi: 10.1016/j.neuroscience.2009.08.068. [DOI] [PubMed] [Google Scholar]
- 30.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 31.Kwon HJ, Kim GE, Lee YT, et al. Inhibition of platelet-derived growth factor receptor tyrosine kinase and downstream signaling pathways by Compound C. Cell Signal. 2013;25(4):883–897. doi: 10.1016/j.cellsig.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 32.de Rooij J, Zwartkruis FJ, Verheijen MH, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 33.Kawasaki H, Springett GM, Mochizuki N, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282(5397):2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 34.Salathe M. Effects of beta-agonists on airway epithelial cells. J Allergy Clin Immunol. 2002;110(6 Suppl):S275–S281. doi: 10.1067/mai.2002.129412. [DOI] [PubMed] [Google Scholar]
- 35.Wyatt TA, Spurzem JR, May K, et al. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am J Physiol. 1998;275(4 Pt 1):L827–L835. doi: 10.1152/ajplung.1998.275.4.L827. [DOI] [PubMed] [Google Scholar]
- 36.Braiman A, Zagoory O, Priel Z. PKA induces Ca2+ release and enhances ciliary beat frequency in a Ca2+-dependent and -independent manner. Am J Physiol. 1998;275(3 Pt 1):C790–C797. doi: 10.1152/ajpcell.1998.275.3.C790. [DOI] [PubMed] [Google Scholar]
- 37.Toledo AC, Arantes-Costa FM, Macchione M, et al. Salbutamol improves markers of epithelial function in mice with chronic allergic pulmonary inflammation. Respir Physiol Neurobiol. 2011;177(2):155–161. doi: 10.1016/j.resp.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Frohock JI, Wijkstrom-Frei C, Salathe M. Effects of albuterol enantiomers on ciliary beat frequency in ovine tracheal epithelial cells. J Appl Physiol. 1985;92(6):2396–2402. doi: 10.1152/japplphysiol.00755.2001. [DOI] [PubMed] [Google Scholar]
- 39.Meyer T, Reitmeir P, Brand P, et al. Effects of formoterol and tiotropium bromide on mucus clearance in patients with COPD. Respir Med. 2011;105(6):900–906. doi: 10.1016/j.rmed.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 40.Hasani A, Toms N, O’Connor J, et al. Effect of salmeterol xinafoate on lung mucociliary clearance in patients with asthma. Respir Med. 2003;97(6):667–671. doi: 10.1053/rmed.2003.1498. [DOI] [PubMed] [Google Scholar]
- 41.Lu WY, Inman MD. Gamma-aminobutyric acid nurtures allergic asthma. Clin Exp Allergy. 2009;39(7):956–961. doi: 10.1111/j.1365-2222.2009.03265.x. [DOI] [PubMed] [Google Scholar]
- 42.Gallos G, Gleason NR, Zhang Y, et al. Activation of endogenous GABAA channels on airway smooth muscle potentiates isoproterenol-mediated relaxation. Am J Physiol Lung C. 2008;295(6):L1040–L1047. doi: 10.1152/ajplung.90330.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spitzer WO, Suissa S, Ernst P, et al. The use of beta-agonists and the risk of death and near death from asthma. N Engl J Med. 1992;326(8):501–506. doi: 10.1056/NEJM199202203260801. [DOI] [PubMed] [Google Scholar]
- 44.McMahon AW, Levenson MS, McEvoy BW, et al. Age and risks of FDA-approved long-acting beta(2)-adrenergic receptor agonists. Pediatrics. 2011;128(5):e1147–e1154. doi: 10.1542/peds.2010-1720. [DOI] [PubMed] [Google Scholar]
- 45.Raghuraman G, Prabhakar NR, Kumar GK. Post-translational modification of glutamic acid decarboxylase 67 by intermittent hypoxia: evidence for the involvement of dopamine D1 receptor signaling. J Neurochem. 2010;115(6):1568–1578. doi: 10.1111/j.1471-4159.2010.07063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei J, Davis KM, Wu H, et al. Protein phosphorylation of human brain glutamic acid decarboxylase (GAD)65 and GAD67 and its physiological implications. Biochemistry. 2004;43(20):6182–6189. doi: 10.1021/bi0496992. [DOI] [PubMed] [Google Scholar]