

Abstract
Introduction
Many antidepressants are indicated for the treatment of major depression. Two network meta-analyses have provided the most comprehensive assessments to date, accounting for both direct and indirect comparisons; however, these reported conflicting interpretation of results. Here, we present a protocol for a systematic review and network meta-analysis aimed at updating the evidence base and comparing all second-generation as well as selected first-generation antidepressants in terms of efficacy and acceptability in the acute treatment of major depression.
Methods and analysis
We will include all randomised controlled trials reported as double-blind and comparing one active drug with another or with placebo in the acute phase treatment of major depression in adults. We are interested in comparing the following active agents: agomelatine, amitriptyline, bupropion, citalopram, clomipramine, desvenlafaxine, duloxetine, escitalopram, fluoxetine, fluvoxamine, levomilnacipran, milnacipran, mirtazapine, nefazodone, paroxetine, reboxetine, sertraline, trazodone, venlafaxine, vilazodone and vortioxetine. The main outcomes will be the proportion of patients who responded to or dropped out of the allocated treatment. Published and unpublished studies will be sought through relevant database searches, trial registries and websites; all reference selection and data extraction will be conducted by at least two independent reviewers. We will conduct a random effects network meta-analysis to synthesise all evidence for each outcome and obtain a comprehensive ranking of all treatments. To rank the various treatments for each outcome, we will use the surface under the cumulative ranking curve and the mean ranks. We will employ local as well as global methods to evaluate consistency. We will fit our model in a Bayesian framework using OpenBUGS, and produce results and various checks in Stata and R. We will also assess the quality of evidence contributing to network estimates of the main outcomes with the GRADE framework.
Ethics and dissemination
This review does not require ethical approval.
PROSPERO registration number
CRD42012002291.
Keywords: Major depression, Systematic review, Network meta-analysis, Antidepressants
Strengths and limitations of this study.
We will conduct a random effects network meta-analysis to synthesise all available evidence (either published or unpublished) for each pre-specified outcome, and obtain a comprehensive ranking of all treatments.
We will employ local as well as global methods to evaluate consistency and we will explore whether treatment effects are robust in network meta-regression.
This will be the largest network meta-analysis (in terms of number of studies and patients) ever conducted in psychiatry and the most comprehensive analysis for the greatest number of antidepressants in major depression. The findings from this study have the potential to guide treatment decisions and guideline development.
The risk of publication bias and the risk of selection bias are high in antidepressant trials, in particular with placebo-controlled trials.
The limitations of primary studies will be addressed with the Cochrane risk of bias tool and the quality of evidence for network estimates of the main outcomes will be assessed with the GRADE framework.
Background
Major depressive disorder (MDD) is the most prevalent psychiatric disease in the general population, affecting more than 16% of adults during their lifetime.1 In 2000, the economic burden of depressive disorders in the USA was estimated to be around 80 billion dollars, with more than 30% of these costs being attributable to direct medical expenses.2 Pharmacotherapy plays an important role in the management of major depression.
Before the late 1980s, pharmacological treatment was limited to tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs). TCAs and MAOIs sometimes are referred to as traditional or first-generation antidepressants. These drugs are often accompanied by multiple side effects that many patients find intolerable. TCAs tend to cause anticholinergic effects including dry mouth and eyes, urinary hesitancy or and sometimes even retention, and constipation, and MAOIs have the potential to produce hypertensive crises if taken along with certain foods or dietary supplements containing tyramine. However, even though first-generation antidepressants are no longer agents of choice in many circumstances, TCAs are still used worldwide, especially in low and middle income countries; according to the list of essential medicines issued by the WHO, amitriptyline is one of the two available treatment options for major depression, along with an selective serotonin reuptake inhibitors (SSRI) fluoxetine.3
Newer antidepressants include SSRIs, serotonin and norepinephrine reuptake inhibitors (SNRIs), and other second-generation drugs. The first of the second-generation drugs was introduced to the US market in 1985, when bupropion was approved for the treatment of major depressive disorders. In 1987, the US Food and Drug Administration (FDA) approved the first SSRI, fluoxetine. Since then, five other SSRIs have been introduced into the market between 1991 and 2002: sertraline, paroxetine, citalopram, fluvoxamine and escitalopram. The SNRIs were first introduced in 1993 with the approval of venlafaxine. In 1994, nefazodone, which is essentially an SSRI with additional 5-hydroxytryptamine-2 (5-HT2) and 5-hydroxytryptamine-3 (5-HT3) antagonist properties, was FDA approved. Mirtazapine, a drug that exhibits both noradrenergic and serotonergic activity with central autoreceptors, was added in 1996 and duloxetine, an SNRI, was approved for the treatment of MDD (and diabetic peripheral neuropathic pain) in 2004. The latest second-generation antidepressants approved for the treatment of MDD in adults include desvenlafaxine, the major active metabolite of venlafaxine; agomelatine, a melatonergic agonist with 5-HT2 antagonism; and vortioxetine, a serotonin modulator and stimulator.i Several systematic reviews have assessed the comparative efficacy and safety of second-generation antidepressants, but two recent comparative effectiveness reviews have provided the most comprehensive assessments to date, notwithstanding conflicting interpretation of results.4 5
Network meta-analysis (NMA) is a statistical technique that allows both direct and indirect comparisons to be undertaken, even when pairs of the treatments have not been compared directly (head-to-head) in the same trial.6–8 NMA can summarise randomised controlled trials (RCTs) of several different treatments by providing point estimates for their association, with a given end point as well as an estimate of inconsistency (ie, a measure of how well the entire network fits together, with small values suggesting better internal agreement of the model). NMA has already been used successfully in other fields of medicine9 and psychiatry.4 10–12
The objective of this systematic review and NMA is to compare all second-generation as well as selected first-generation antidepressants (refer Types of interventions section) in terms of efficacy and acceptability in the acute treatment of major depression in adults to better inform clinical practice and mental health policies. The project is called Group of Researchers Investigating Specific Efficacy of individuaL Drugs for Acute depression (GRISELDA) and will be based on our previous NMA on antidepressants;4 however the present review differs in that it will enlarge the number of antidepressants under investigation, add new and clinically informative outcome measures, and particularly include placebo-controlled trials.
Methods and analysis
Criteria for considering studies for this review
Types of studies
All RCTs reported as double-blind comparing one active drug with another or with placebo in the acute phase treatment of major depression will be included. Only monotherapy studies will be included; thus RCTs in which antidepressants were used as an augmentation strategy will be excluded. Quasi-randomised trials (such as those allocating by using alternate days of the week) will be excluded. Cross-over and cluster randomised trials will be included. We will not include studies where sequence generation was at high risk of bias, or where the allocation was clearly not concealed.
Types of participants
Patients aged 18 years or older, of both sexes, with a primary diagnosis of major depression will be included. Studies adopting any standard operationalised diagnostic criteria to define patients suffering from unipolar major depression will be included, such as Feighner criteria, Research Diagnostic Criteria, DSM-III, DSM-III-R, DSM-IV, DSM-5 and ICD-10. Studies in which 20% or more of the participants may be suffering from bipolar or psychotic depression will be excluded. A concurrent secondary diagnosis of another psychiatric disorder will not be considered as exclusion criterion, but RCTs in which all participants have a concurrent primary diagnosis of another mental disorder will be excluded. Studies in which all participants have a diagnosis of resistant depression will be excluded. Antidepressant trials in depressive patients with a serious concomitant medical illness will be excluded. RCTs of women with postpartum depression will be also excluded, because postpartum depression appears to be clinically different from major depression.13 Trials which allow rescue medications will be included so long as these are equally provided among the randomised arms.
Types of interventions
We are interested in comparing the following active agents: agomelatine, amitriptyline, bupropion, citalopram, clomipramine, desvenlafaxine, duloxetine, escitalopram, fluoxetine, fluvoxamine, levomilnacipran, milnacipran, mirtazapine, nefazodone, paroxetine, reboxetine, sertraline, trazodone, venlafaxine, vilazodone and vortioxetine. We will include all the second generation antidepressants, and of older agents, we have selected the two tricyclics included in the WHO3 list of essential medicine: (1) amitriptyline, recommended for major depression and (2) clomipramine, although a typical tricyclic antidepressant, as it has a different biochemical, mainly serotonergic, action. We also selected trazodone and nefazodone because these are believed to have very distinct effect and tolerability profiles.14 We will include only studies randomising patients to the drug within its licensed dose range.4 If a study included arms with both unapproved and approved doses, we include the study but only the arms that used the therapeutic doses.15
We will obtain information about the interventions of interest either from head-to-head or placebo controlled trials. Hence the synthesis comparator set consists of all the interventions listed above and placebo controlled trials. Figure 1 shows the network of all possible pairwise comparisons between the eligible interventions. We anticipate that any patient who meets all inclusion criteria is, in principal, equally likely to be randomised to any of the interventions in the synthesis comparator set.
Figure 1.

Network of all possible pairwise comparisons between the eligible interventions.
Outcome measures
Considering that clinical trials of antidepressant drugs are usually small and that data distribution is difficult to assess for studies with small samples, in this review priority will be given to the use and analysis of dichotomous variables both for efficacy and acceptability.
- Primary outcomes
- (1) Efficacy (as dichotomous outcome)—response
- Measured by the total number of patients who had a reduction of at least 50% on the total score between baseline and week 8 (range 4–12 weeks) on a standardised observer-rating scale for depression. We will employ Hamilton Depression Rating Scale (HDRS) or, if HDRS was not used, another standardised and validated observer-rating scale. Any version of HDRS will be accepted.
- (2) Acceptability of treatment
- Treatment discontinuation (acceptability) is defined as the proportion of patients who leave the study early for any reason during the first 8 weeks of treatment (range 4–12 weeks).
- Secondary outcomes
- (3) Efficacy (as continuous outcome)
- Measured by the end point score on the HDRS or Montgomery-Åsberg Depression Rating Scale (MADRS), if HDRS was not used, after 8 weeks (range 4–12 weeks). If none of the former scales is used, we will consider other standardised rating scales. When end point scores are not reported but change scores are, we will use the latter scores.16 See figure 2 for full details about the data extraction process (decision tree).
- (4) Efficacy (as dichotomous outcome)—remission
- Measured by the total number of patients who had a remission of depressive symptoms between baseline and week 8 (range 4–12 weeks) on a standardised rating scale for depression (HDRS or another standardised rating scale, if HDRS was not used). Remission will be defined as score of less or equal to 7 or 8 on the 17-item HDRS (or the corresponding threshold for longer versions of HDRS),17 or of less or equal to 10 or 11 on the MADRS scale at week 8 (range 4–12 weeks).18
- (5) Tolerability of treatment
- The proportion of patients who leave the study early due to adverse events during the first 8 weeks of treatment (range 4–12 weeks).
Figure 2.

Decision-tree for data extraction of continuous efficacy outcome. HDRS: Hamilton Depression Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale.
Search strategy and study selection
Searches for published RCTs will be undertaken in the following electronic databases: CENTRAL, CINAHL, EMBASE, LiLACS, MEDLINE, MEDLINE In-Process and PSYCINFO. The electronic search will be supplemented with manual searches for published, unpublished and ongoing RCTs in the following drug-approval agencies: the Food and Drug Administration (FDA) in the USA, the Medicines and Healthcare products Regulatory Agency in the UK, the European Medicines Agency (EMA) in the European Union, the Medicines Evaluation Board in the Netherlands, the Medical Products Agency in Sweden, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, and the Therapeutic Goods Administration (TGA) in Australia. We will also undertake searches for published, unpublished and ongoing studies in a range of research registries (see online supplementary appendix for the full list of resources). It is important to include unpublished data, since publication bias leads to exaggerated effect sizes15 and reporting bias can bias NMA-based estimates of treatments efficacy and modify ranking.17 Studies will be identified using search terms for depression (depress* or dysthymi* or adjustment disorder* or mood disorder* or affective disorder or affective symptoms) appended to the list of antidepressants under review. No data limits or language restrictions will be applied to any of the searches.
bmjopen-2015-010919supp_appendix.pdf (262.4KB, pdf)
The reference lists of included studies will be searched for additional studies. Where eligible studies are found, unpublished data will be requested from the investigators. We will also contact the National Institute for Health and Care Excellence (NICE, UK), the Institut für Qualität und Wirtschaftlichkeit intramuscular Gesundheitswesen (IQWiG, Germany), and any other relevant organisations and individuals for any additional information not already identified. We are aware that there are many RCTs published in Chinese journals. However, in many of these studies only incomplete or conflicting information is available, and it has been reported that many of them do not use appropriate randomisation procedures.19 In an effort to avoid the potential biases that may be introduced by including these trials without further information, we will not search the Chinese databases. However, to be consistent in our selection procedure, we will include all studies, irrespective of their country of origin, identified in the international databases listed above and satisfying our eligibility criteria.
Two persons will independently review references and abstracts retrieved by the search. If both reviewers agree that a trial does not meet eligibility criteria, it will be excluded. We will obtain the full text of all remaining articles and use the same eligibility criteria to determine which, if any, to exclude at this stage. Any disagreements will be resolved via discussion with a third member of the review team.
Data extraction
Two reviewers will then independently read each article/study report, evaluate the completeness of the data abstraction and confirm the quality rating (see details below). We will design and use a structured data extraction form to ensure consistency of information and appraisal for each study. Information extracted will include study characteristics (such as lead author, publication year and journal), participant characteristics (such as diagnostic criteria for depression, age, sex, setting and severity of depression), intervention details (such as drug dose and dosing schedule (fixed vs flexible)), and outcome measures. Two review authors will ascertain that the data are entered correctly into the final data set. When published and unpublished studies provide different values, we will prioritise the unpublished data.15
Dichotomous outcomes
We opt for the number of successes and failures per treatment arm as defined in Outcome measures section. When these numbers are not reported but baseline mean and end point mean and SDs of the depression rating scales (such as HDRS or MADRS) are provided, we will calculate the number of responding patients at 8 weeks (range 4–12 weeks) by employing a validated imputation method.20 Below we also discuss our strategy when means and/or SDs are not reported in the articles.
Continuous outcomes
We will extract means, SDs, and numbers of patients randomised in each study arm. When means and their SDs are not recorded, authors will be asked to supply the data. When SEs, t-statistics or p values are reported, these will be transformed to SDs. If SDs are not reported and not provided by the authors, the mean value of known SDs will be calculated from the group of included studies according to Furukawa and colleagues.21 When mixed method repeated measures or other appropriate imputation methods are used,22 we will prefer these results. When data on dropouts are carried forward and included in the evaluation (Last Observation Carried Forward, LOCF), these will be analysed according to the primary studies.
Missing outcome data
Outcomes of patients who leave the study early are typically imputed by the trialists, often using LOCF.23 It is very rare for an article to report the outcome separately for fully observed and imputed data, and the summary statistics that we will collect are bound to refer to both completers and patients who dropped out. The appropriateness of the imputation method to account for early dropouts will be considered in the Risk of bias assessment section. During the protocol development process, we have carried out some exploratory analyses to assess the comparability between studies with placebo arm and studies with only active treatments. Considering that the number of dropouts is usually higher in placebo controlled trials,24 we anticipate that the imputation of missing outcome data using LOCF can be problematic when comparing head-to-head with placebo trials within the same network of treatments. In case of material differences between these types of studies, we will carefully investigate this methodological issue, and try to address it properly from a statistical point of view (refer Risk of bias assessment section).
After imputations at the individual participant level by the original authors, the outcome might be unknown (and not imputed by the original authors) for a very small proportion of study participants. For the dichotomous efficacy outcome, we will first assume that participants with an unknown outcome are non-responders. Although this corresponds to naive imputations24 an extensive sensitivity analysis using more appropriate methods to account for missing outcome data in antidepressant trials has shown that imputing outcomes for a very small percentage of patients (as in our case) has no material impact on the results.25 For continuous outcomes, participants with missing outcome data will be excluded from the analysis.
Unit of analysis issues
We will extract data from cross-over studies using only the first period because carry-over effects can be important in antidepressant trials.26 In cluster randomised trials, we will extract data that account for the clustering in the results (eg, from multilevel models). If such adjusted results are not available, we will extract unadjusted data and will adjust the sample size (in the continuous outcomes) and both the sample size and number of events (in the dichotomous outcomes) by dividing it with the design effect.27
Length of trial
Clinically, whether efficacy is assessed after 8 weeks of treatment or after 16–24 weeks or more may lead to differences in terms of assessed treatment outcome. Clinicians need to know whether (and to what extent) treatments work within a clinically reasonable period of time. Unfortunately, there is no consensus on what the appropriate duration of an acute phase trial is. In the present review, acute treatment will be defined as an 8-week treatment in both the efficacy and acceptability analyses.4 If 8-week data are not available, we will use data as close to 8 weeks as possible (ranging between 4 and 12 weeks). Longer term studies will be included in the systematic review, but excluded from the statistical synthesis of data if they do not provide data for the 4–12 weeks period.
Comparability of dosages
We will include only study arms randomising patients to drugs within the licensed dose. Both fixed-dose and flexible-dose designs will be allowed.4 There is a possibility that some trials compare one agent at the upper limit of its therapeutic range with another agent at the lower limit of its therapeutic range within the same study. We plan to capture this study characteristic by adding a dichotomous variable indicating whether dosages are comparable, and use this information for a sensitivity analysis.
Risk of bias assessment
We will assess risk of bias in the included studies using the tool described in the Cochrane Collaboration Handbook as a reference guide.28 The assessment will be performed by two independent raters. If the raters disagree, the final rating will be made by consensus with the involvement (if necessary) of another member of the review group. We will evaluate the risk of bias in the following domains: generation of allocation sequence, allocation concealment, blinding of study personnel and participants, blinding of outcome assessor, attrition, selective outcome reporting and other domains, including sponsorship bias. Where inadequate details of allocation concealment and other characteristics of trials are provided, the trial authors may be contacted in order to obtain further information.
Selective outcome reporting will be rated with regard to the two primary outcomes in the systematic review. It will be rated at low risk of bias if the number of responders is reported (or if the continuous outcome measures of depression severity are reported in enough details to enable imputation of the number of responders), and if the number of total dropouts is reported. It will be rated at high risk of bias if neither is reported, and will be rated as unclear risk of bias otherwise.
Losses to follow-up are typically associated with the outcome and the treatment received. Patients tend to leave a trial early because of early response, side effects or lack of response. Consequently, missingness is typically informative in antidepressant trials. Inappropriate methods to impute data (such as the LOCF approach) are often applied and are known to produce biased results.22 However, even appropriate methods (such as multiple imputations) when applied in practice often use the missing at random assumption, which is often difficult to defend. Consequently, we will classify the studies with respect to attrition bias as being: (1) at low risk if an appropriate imputation method has been employed that accounts for the different reasons for dropout between arms (especially in placebo-controlled trials, where the lack of active comparator can affect dropout rates in a specific way), or if the percentage of missing outcome data is 20% or less overall and is balanced between arms (ie, absolute difference in dropouts <5% for active comparison and <10% for placebo comparison); (2) at high risk of bias if dropout is unbalanced between the arms, and an inappropriate imputation method (eg, LOCF) has been used to impute dropouts. All other cases will be classified as unclear risk of bias. Studies will be classified as having low risk of bias if none of the domains above was rated as high risk of bias and three or less were rated as unclear risk; moderate if one was rated as high risk of bias or none was rated as high risk of bias but four or more were rated as unclear risk, and all other cases will be assumed to pertain to high risk of bias.
Statistical synthesis of study data
Characteristics of included studies and information flow in the network
We will generate descriptive statistics for the trial, and study population characteristics across all eligible trials, describing the types of comparisons and some important variables, either clinical or methodological (such as year of publication, age, severity of illness, sponsorship and clinical setting).
The available evidence will be presented in the network diagram. The size of the nodes will reflect the amount of evidence accumulated for each treatment (total number of patients), the breadth of each edge will be proportional to the inverse of the variance of the summary effect of each direct treatment comparison, and the colour of each edge will represent risk of bias (low, moderate or high, refer Risk of bias assessment section). To understand which are the most influential comparisons in the network and how direct and indirect evidence influences the final summary data, we will use the contribution matrix that describes the percentage contribution of each direct meta-analysis to the entire body of evidence.29 30
Pairwise meta-analyses
For each pair-wise comparison, we will synthesise data to obtain summary standardised mean differences (SMD, Cohen's d) for continuous outcomes or ORs for dichotomous outcomes, both with 95% Credible Intervals (CrI). We will use a random effects model to incorporate the assumption that the different studies are estimating different, yet related, treatment effects.27 For each outcome, we will first assume that each pairwise meta-analysis comparing treatments X and Y has its own heterogeneity variance parameter 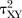 and then assume that there are two heterogeneity parameters; one common for all placebo-controlled trials
and then assume that there are two heterogeneity parameters; one common for all placebo-controlled trials 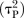 and one for all active versus active comparisons
and one for all active versus active comparisons 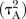 . Visual inspection of the forest plots and monitoring of the posterior distributions of
. Visual inspection of the forest plots and monitoring of the posterior distributions of 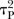 ,
, 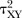 and
and 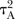 will be used to investigate the possibility of statistical heterogeneity. The posterior distributions of the heterogeneity parameters will be compared to their predictive distributions, as described elsewhere.31
32 Finally the I2 statistic and its 95% CrI will be calculated to convey the amount of heterogeneity.
will be used to investigate the possibility of statistical heterogeneity. The posterior distributions of the heterogeneity parameters will be compared to their predictive distributions, as described elsewhere.31
32 Finally the I2 statistic and its 95% CrI will be calculated to convey the amount of heterogeneity.
Assessment of the transitivity assumption
Transitivity, which is the key underlying assumption of NMA, will be investigated carefully. Joint analysis of treatments can be misleading if the network is substantially intransitive. We will need to investigate the distribution of clinical and methodological variables that can act as effect modifiers across treatment comparisons.33 The clinical features, which have been demonstrated to date to moderate efficacy of antidepressants include bipolarity,34 psychotic features,35 and subthreshold depression.36 We have assured transitivity in our network with regard to these variables by limiting our samples to participants with non-psychotic unipolar major depression. Other clinical or methodological variables that may influence our primary outcomes of antidepressant efficacy or acceptability include: age, depressive severity at baseline,37 38 and the dosing schedule.39 We will investigate if these variables are similarly distributed across studies grouped by comparison. The inclusion of placebo and concerns about its potential to violate the transitivity assumption have been highlighted in general7 8 and particularly in depression studies.40 41 Consequently, the comparability of placebo-controlled studies with those that provide head-to-head evidence will be examined carefully.
Network meta-analyses
We assume that patients who fulfil the inclusion criteria outlined in Criteria for considering studies for this review section are equally likely to be randomised to any of the antidepressants that we plan to compare. If the collected studies appear to be sufficiently similar with respect to the distribution of effect modifiers (refer Assessment of transitivity assumption section), we will conduct a random effects NMA to synthesise all evidence for each outcome, and obtain a comprehensive ranking of all treatments. We will use arm-level data and the binomial likelihood for dichotomous outcomes. We will account for the correlations induced by multiarm studies by employing multivariate distributions. We will assume a single heterogeneity parameter for each network. We will present the summary ORs or SMD for all pairwise comparisons in a league table. We will also estimate the prediction intervals to assess how much the common heterogeneity affects the relative effect with respect to the extra uncertainty anticipated in a future study. To rank the various treatments for each outcome, we will use the surface under the cumulative ranking curve (SUCRA) and the mean ranks.42
Assessment of inconsistency
The strategical and conceptual evaluation of transitivity will be supplemented with a statistical evaluation of consistency, the agreement between direct and indirect evidence. We will employ local as well as global methods to evaluate consistency.43 Local methods detect ‘hot spots’ of inconsistency, evidence loops that are inconsistent or comparisons for which direct and indirect evidence disagree. We will employ the loop-specific approach to evaluate inconsistency within each loop of evidence,44 and a method that separates direct evidence from indirect evidence provided by the entire network.45 We will also evaluate consistency in the entire network by calculating the I2 for network heterogeneity, inconsistency, and for both.46 47
Tests for inconsistency are known to have low power,48 and empirical evidence has suggested that 10% of evidence loops published in the medical literature are expected to be inconsistent.49 Therefore, interpretation of the statistical inference about inconsistency will be carried out with caution and possible sources of inconsistency will be explored even in the absence of evidence for inconsistency.
Exploring heterogeneity and inconsistency and sensitivity analyses
We expect small amounts of heterogeneity and inconsistency to be present given the variety of study settings we plan to include. We will explore whether treatment effects for the two primary outcomes are robust in subgroup analyses and network meta-regression using the following characteristics: (1) study year; (2) sponsorship; (3) depressive severity at baseline; (4) dosing schedule; (5) response to placebo; (6) proportion of participants allocated to placebo; number of recruiting centres (single-centre vs multicentric studies).50 51 The sensitivity of our conclusions for the two primary outcomes will be evaluated by analysing (1) only studies with reported SD rather than imputed; (2) only studies with balanced doses in all arms (ie, we will exclude studies with unfair dose comparisons); (3) only studies with unpublished data (ie, we will exclude studies providing published data only); (4) only studies with low risk of bias (as defined in Risk of bias assessment section); (5) only head-to-head studies.
Selection bias
The risk of selection bias is high in antidepressants trials, in particular with placebo-controlled trials.15 We will use the comparison-adjusted30 and contour-enhanced52 funnel plots to investigate whether results in imprecise trials differ from those in more precise trials. We will also run network meta-regression models to detect associations between study size and effect size.53 If an important association is found and publication bias is suspected, we will attempt to explore the possibility that funnel plot asymmetry is due to publication bias by employing a selection model.54
Model implementation
We will fit our model using OpenBUGS55 and Stata (StataCorp. 2015. Stata Statistical Software: Release 14. College Station, TX: StataCorp LP). For the Bayesian implementation we will employ the binomial likelihood for dichotomous outcomes and will use uninformative prior distributions for the treatment effects, that is, N(0,1000), and a minimally informative prior distribution for the common heterogeneity SD depending on the outcome, that is, U(0,5). Also, we will assume uninformative priors, that is, N(0,1000) for all meta-regression coefficients. To check convergence, we will run multiple chains and monitor their mixing; we will use the Brooks-Gelman-Rubin diagnostic.
Analyses for statistical evaluation of the inconsistency and production of network graphs and result figures will be carried out in Stata using the mvmeta command56 and a collection of routines described elsewhere.30 All analyses of the primary outcomes will be duplicated using the netmeta package in R.57
GRADE quality assessment of all comparisons in the network
We will also assess the quality of evidence contributing to network estimates of the main outcomes with the GRADE framework, which characterises the quality of a body of evidence on the basis of the study limitations, imprecision, inconsistency, indirectness and publication bias.43 The starting point for confidence in each network estimate is high, but will be downgraded according to the assessments of these five domains.
Ethics and dissemination
This review does not require ethical approval. We will publish findings from this systematic review in a peer-reviewed scientific journal, and data set will be made freely available. The completed review will be disseminated electronically in print and on social media, where appropriate.
Footnotes
Twitter: Follow Andrea Cipriani at @And_Cipriani
Contributors: AC and TAF devised the study and drafted the protocol; will assist with the data extraction and analysis, and draft the results and discussion sections. LZA, SL, HGR, EHT and JRG revised the protocol, assisted with study design and data extraction, and will help draft the final manuscript. YO, NT, YH, HI, KS, AS and NW revised the protocol and will carry out most of the data collection. SS designed and conducted the search strategy, and provided input on the working of the manuscript. GS and ACh provided input on the protocol, designed the analysis plan, and will carry out the statistical analyses.
Funding: None.
Competing interests: TAF has received lecture fees from Eli Lilly, Janssen, Meiji, Mochida, MSD, Otsuka, Pfizer and Tanabe-Mitsubishi, and consultancy fees from Sekisui Chemicals and Takeda Science Foundation. He has received royalties from Igaku-Shoin, Seiwa-Shoten and Nihon Bunka Kagaku-sha publishers. He has received grant or research support from the Japanese Ministry of Education, Science, and Technology, the Japanese Ministry of Health, Labour and Welfare, the Japan Society for the Promotion of Science, the Japan Foundation for Neuroscience and Mental Health, Mochida and Tanabe-Mitsubishi. He is diplomate of the Academy of Cognitive Therapy. SL has received honoraria for consulting/advisory boards from Alkermes, Eli Lilly, Janssen, Johnson & Johnson, Lundbeck, MedAvante, Roche, Otsuka and Teva; lecture honoraria from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Janssen, Johnson & Johnson, Lundbeck (Institute), Pfizer, Sanofi-Aventis, ICON, AbbVie, AOP Orphan and Servier; for the preparation of educational material and publications from Lundbeck Institute and Roche; and Eli Lilly has provided medication for a trial with SL as the primary investigator. NW has research funds from the Japanese Ministry of Health, Labor and Welfare, and the Japanese Ministry of Education, Science, and Technology. He has also received royalties from Sogensha and Paquet, and speaking fees and research funds from Asahi Kasei, Dai-Nippon Sumitomo, Eli Lilly, GlaxoSmithKline, Janssen, Meiji, MSD, Otsuka and Pfizer. JRG is an NIHR Senior Investigator. AC is supported by the NIHR Oxford Cognitive Health Clinical Research Facility, and was expert witness for Accord Healthcare for a patent issue about quetiapine extended release.
Provenance and peer review: Not commissioned; externally peer reviewed.
The mechanism of the antidepressant effect of vortioxetine is not fully understood, but is thought to be related to its enhancement of serotonergic activity in the central nervous system through inhibition of the re-uptake of serotonin (5-HT). It also has several other activities, including 5-HT3 receptor antagonism and 5-HT1A receptor agonism. The contribution of these activities to vortioxetine's antidepressant effect has not been established.
References
- 1.Kessler RC, Berglund P, Demler O et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS- R). JAMA 2003;289:3095–105. 10.1001/jama.289.23.3095 [DOI] [PubMed] [Google Scholar]
- 2.Greenberg PE, Kessler RC, Birnbaum HG et al. The economic burden of depression in the United States: how did it change between 1990 and 2000? J Clin Psychiatry 2003;64:1465–75. 10.4088/JCP.v64n1211 [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization (WHO). WHO Model List of Essential Medicines, 2011. http://whqlibdoc.who.int/hq/2011/a95053_eng.pdf.26240823
- 4.Cipriani A, Furukawa TA, Salanti G et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple treatment meta-analysis. Lancet 2009;373:746–58. 10.1016/S0140-6736(09)60046-5 [DOI] [PubMed] [Google Scholar]
- 5.Gartlehner G, Hansen RA, Morgan LC et al. Comparative benefits and harms of second-generation antidepressants for treating major depressive disorder: an updated meta-analysis. Ann Intern Med 2011;155:772–85. 10.7326/0003-4819-155-11-201112060-00009 [DOI] [PubMed] [Google Scholar]
- 6.Caldwell DM, Ades AE, Higgins JP. Simultaneous comparison of multiple treatments: combining direct and indirect evidence. BMJ 2005;331:897–900. 10.1136/bmj.331.7521.897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salanti G, Marinho V, Higgins JP. A case study of multiple-treatments meta-analysis demonstrates that covariates should be considered. J Clin Epidemiol 2009;62:857–64. 10.1016/j.jclinepi.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 8.Cipriani A, Higgins JP, Geddes JR et al. Conceptual and technical challenges in network meta-analysis. Ann Intern Med 2013;159:130–7. 10.7326/0003-4819-159-2-201307160-00008 [DOI] [PubMed] [Google Scholar]
- 9.Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007;369:201–7. 10.1016/S0140-6736(07)60108-1 [DOI] [PubMed] [Google Scholar]
- 10.Cipriani A, Barbui C, Salanti G et al. Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple-treatments meta-analysis. Lancet 2011;378:1306–15. 10.1016/S0140-6736(11)60873-8 [DOI] [PubMed] [Google Scholar]
- 11.Leucht S, Cipriani A, Spineli L et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet 2013;382:951–62. 10.1016/S0140-6736(13)60733-3 [DOI] [PubMed] [Google Scholar]
- 12.Miura T, Noma H, Furukawa TA et al. Comparative efficacy and tolerability of pharmacological treatments in the maintenance treatment of bipolar disorder: a systematic review and network meta-analysis. Lancet Psychiatry 2014;1:351–9. 10.1016/S2215-0366(14)70314-1 [DOI] [PubMed] [Google Scholar]
- 13.Cooper PJ, Murray L. Postnatal depression. BMJ 1998;316:1884–6. 10.1136/bmj.316.7148.1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linde K, Kriston L, Rücker G et al. Efficacy and acceptability of pharmacological treatments for depressive disorders in primary care: systematic review and network meta-analysis. Ann Fam Med 2015;13:69–79. 10.1370/afm.1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner EH, Matthews AM, Linardatos E et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med 2008;358:252–60. 10.1056/NEJMsa065779 [DOI] [PubMed] [Google Scholar]
- 16.da Costa BR, Nüesch E, Rutjes AW et al. Combining follow-up and change data is valid in meta-analyses of continuous outcomes: a meta-epidemiological study. J Clin Epidemiol 2013;66:847–55. 10.1016/j.jclinepi.2013.03.009 [DOI] [PubMed] [Google Scholar]
- 17.Furukawa TA, Akechi T, Azuma H et al. Evidence-based guidelines for interpretation of the Hamilton Rating Scale for Depression. J Clin Psychopharmacol 2007;27:531–4. 10.1371/journal.pone.0035219 [DOI] [PubMed] [Google Scholar]
- 18.J Clin Psychiatry. doi: 10.1111/j.1600-0447.2007.01027.x. Bandelow B, Baldwin DS, Dolberg OT, et al. What is the threshold for symptomatic response and remission for major depressive disorder, panic disorder, social anxiety disorder, and generalized anxiety disorder? 2006;67:1428–34. [DOI] [PubMed] [Google Scholar]
- 19.Wu TX, Li YP, Liu GJ et al. Investigation of authenticity of ‘claimed’ randomized controlled trials (RCTs) and quality assessment of RCT reports published in China. Dublin, Ireland: Presented at XIV Cochrane Colloquium, 2006. [Google Scholar]
- 20.Furukawa TA, Cipriani A, Barbui C et al. Imputing response rates from means and standard deviations in meta-analysis. Int Clin Psychopharm 2005;20:49–52. 10.1097/00004850-200501000-00010 [DOI] [PubMed] [Google Scholar]
- 21.Furukawa TA, Barbui C, Cipriani A et al. Imputing missing standard deviations in meta-analyses can provide accurate results. J Clin Epidemiol 2006;59:7–10. 10.1016/j.jclinepi.2005.06.006 [DOI] [PubMed] [Google Scholar]
- 22.Little RJ, D'Agostino R, Cohen ML et al. The prevention and treatment of missing data in clinical trials. N Engl J Med 2012;367:1355–60. 10.1056/NEJMsr1203730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spineli LM, Pandis N, Salanti G. Reporting and handling missing outcome data in mental health: a systematic review of Cochrane systematic reviews and meta-analyses. Res Synth Methods 2015;6:175–87. 10.1002/jrsm.1131 [DOI] [PubMed] [Google Scholar]
- 24.Rutherford BR, Sneed JR, Roose SP. Does differential drop-out explain the influence of study design on antidepressant response? A meta-analysis. J Affect Disord 2012;140:57–65. 10.1016/j.jad.2012.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mavridis D, Chaimani A, Efthimiou O et al. Addressing missing outcome data in meta-analysis. Evid Based Ment Health 2014;17:85–9. 10.1136/eb-2014-101900 [DOI] [PubMed] [Google Scholar]
- 26.Spineli LM, Higgins JP, Cipriani A et al. Evaluating the impact of imputations for missing participant outcome data in a network meta-analysis. Clin Trials 2013;10:378–88. 10.1177/1740774512470317 [DOI] [PubMed] [Google Scholar]
- 27.Caddy C, Amit BH, McCloud TL et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev 2015;9:CD011612 10.1002/14651858.CD011612.pub2 [DOI] [PubMed] [Google Scholar]
- 28.Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, eds Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. http://www.cochrane-handbook.org [Google Scholar]
- 29.Chaimani A, Higgins JP, Mavridis D et al. Graphical tools for network meta-analysis in STATA. PLoS ONE 2013;8:e76654 10.1371/journal.pone.0076654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa TA, Miura T, Chaimani A et al. Using the contribution matrix to evaluate complex study limitations in a network meta-analysis: a case study of bipolar maintenance pharmacotherapy review. BMC Res Notes 2016;9:218 10.1186/s13104-016-2019-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner RM, Jackson D, Wei Y et al. Predictive distributions for between-study heterogeneity and simple methods for their application in Bayesian meta-analysis. Stat Med 2015;34:984–98. 10.1002/sim.6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhodes KM, Turner RM, Higgins JP. Predictive distributions were developed for the extent of heterogeneity in meta-analyses of continuous outcome data. J Clin Epidemiol 2015;68:52–60. 10.1016/j.jclinepi.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res Synth Methods 2012;3:80–97. 10.1002/jrsm.1037 [DOI] [PubMed] [Google Scholar]
- 34.Taylor DM, Cornelius V, Smith L et al. Comparative efficacy and acceptability of drug treatments for bipolar depression: a multiple-treatments meta-analysis. Acta Psych Scand 2014;130:452–69. 10.1111/acps.12343 [DOI] [PubMed] [Google Scholar]
- 35.Wijkstra J, Lijmer J, Burger H et al. Pharmacological treatment for psychotic depression. Cochrane Database Syst Rev 2015;7:CD004044 10.1002/14651858.CD004044.pub4 [DOI] [PubMed] [Google Scholar]
- 36.Barbui C, Cipriani A, Patel V et al. Efficacy of antidepressants and benzodiazepines in minor depression: systematic review and meta-analysis. Br J Psychiatry 2011;198:11–16. 10.1192/bjp.bp.109.076448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fournier JC, DeRubeis RJ, Hollon SD et al. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA 2010;303:47–53. 10.1001/jama.2009.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibbons RD, Hur K, Brown CH et al. Benefits from antidepressants: synthesis of 6-week patient-level outcomes from double-blind placebo-controlled randomized trials of fluoxetine and venlafaxine. Arch Gen Psychiatry 2012;69:572–9. 10.1001/archgenpsychiatry.2011.2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan A, Kolts RL, Thase ME et al. Research design feature and patient characteristics associated with the outcome of antidepressant clinical trials. Am J Psychiatry 2004;161:2045–9. 10.1176/appi.ajp.161.11.2045 [DOI] [PubMed] [Google Scholar]
- 40.Rutherford BR, Sneed JR, Roose SP. Does study design influence outcome? The effects of placebo control and treatment duration in antidepressant trials. Psychother Psychosom 2009;78:172–81. 10.1159/000209348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinyor M, Levitt AJ, Cheung AH et al. Does inclusion of a placebo arm influence response to active antidepressant treatment in randomized controlled trials? Results from pooled and meta-analyses. J Clin Psychiatry 2010;71:270–9. 10.4088/JCP.08r04516blu [DOI] [PubMed] [Google Scholar]
- 42.Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. J Clin Epidemiol 2011;64:163–71. 10.1016/j.jclinepi.2010.03.016 [DOI] [PubMed] [Google Scholar]
- 43.Salanti G, Del Giovane C, Chaimani A et al. Evaluating the quality of evidence from a network meta-analysis. PLoS ONE 2014;9:e99682 10.1371/journal.pone.0099682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bucher HC, Guyatt GH, Griffith LE et al. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol 1997;50:683–91. 10.1016/S0895-4356(97)00049-8 [DOI] [PubMed] [Google Scholar]
- 45.Dias S, Welton NJ, Caldwell DM et al. Checking consistency in mixed treatment comparison meta-analysis. Stat Med 2010;29:932–44. 10.1002/sim.3767 [DOI] [PubMed] [Google Scholar]
- 46.Higgins JP, Jackson D, Barrett JK et al. Consistency and inconsistency in network meta-analysis: concepts and models for multi-arm studies. Res Synth Methods 2012;3: 98–110. 10.1002/jrsm.1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson D, Barrett JK, Rice S et al. A design-by-treatment interaction model for network meta-analysis with random inconsistency effects. Stat Med 2014;33:3639–54. 10.1002/sim.6188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veroniki AA, Mavridis D, Higgins JP et al. Characteristics of a loop of evidence that affect detection and estimation of inconsistency: a simulation study. BMC Med Res Methodol 2014;14:106 10.1186/1471-2288-14-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veroniki AA, Vasiliadis HS, Higgins JP et al. Evaluation of inconsistency in networks of interventions. Int J Epidemiol 2013;42:332–45. 10.1093/ije/dys222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg RP, Bornstein RF, Greenberg MD et al. A meta-analysis of antidepressant outcome under “blinder” conditions. J Consult Clin Psychol 1992;60:664–9; discussion 670–7 10.1037/0022-006X.60.5.664 [DOI] [PubMed] [Google Scholar]
- 51.Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol 2009;19:34–40. 10.1016/j.euroneuro.2008.08.009 [DOI] [PubMed] [Google Scholar]
- 52.Peters JL, Sutton AJ, Jones DR et al. Contour-enhanced meta-analysis funnel plots help distinguish publication bias from other causes of asymmetry. J Clin Epidemiol 2008;61:991–6. 10.1016/j.jclinepi.2007.11.010 [DOI] [PubMed] [Google Scholar]
- 53.Chaimani A, Salanti G. Using network meta-analysis to evaluate the existence of small-study effects in a network of interventions. Res Synth Methods 2012;3:161–76. 10.1002/jrsm.57 [DOI] [PubMed] [Google Scholar]
- 54.Mavridis D, Welton NJ, Sutton A et al. A selection model for accounting for publication bias in a full network meta-analysis. Stat Med 2014;33:5399–412. 10.1002/sim.6321 [DOI] [PubMed] [Google Scholar]
- 55.Lunn D, Spiegelhalter D, Thomas A et al. The BUGS project: evolution, critique and future directions. Stat Med 2009;28:3049–67. 10.1002/sim.3680 [DOI] [PubMed] [Google Scholar]
- 56.White IR, Barrett JK, Jackson D et al. Consistency and inconsistency in network meta-analysis: model estimation using multivariate meta-regression. Res Synth Methods 2012;3:111–25. 10.1002/jrsm.1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwarzer G. Network meta-analysis. In: Schwarzer G, Carpenter JR, Rücker G, eds. Meta-analysis with R. Berlin, Heidelberg: Springer, 2015:187–216. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2015-010919supp_appendix.pdf (262.4KB, pdf)