

Abstract
Purpose
To identify pathogenic mutations responsible for autosomal recessive retinitis pigmentosa (arRP) in consanguineous familial cases.
Methods
Seven large familial cases with multiple individuals diagnosed with retinitis pigmentosa were included in the study. Affected individuals in these families underwent ophthalmic examinations to document the symptoms and confirm the initial diagnosis. Blood samples were collected from all participating members, and genomic DNA was extracted. An exclusion analysis with microsatellite markers spanning the TULP1 locus on chromosome 6p was performed, and two-point logarithm of odds (LOD) scores were calculated. All coding exons along with the exon–intron boundaries of TULP1 were sequenced bidirectionally. We constructed a single nucleotide polymorphism (SNP) haplotype for the four familial cases harboring the K489R allele and estimated the likelihood of a founder effect.
Results
The ophthalmic examinations of the affected individuals in these familial cases were suggestive of RP. Exclusion analyses confirmed linkage to chromosome 6p harboring TULP1 with positive two-point LOD scores. Subsequent Sanger sequencing identified the single base pair substitution in exon14, c.1466A>G (p.K489R), in four families. Additionally, we identified a two-base deletion in exon 4, c.286_287delGA (p.E96Gfs77*); a homozygous splice site variant in intron 14, c.1495+4A>C; and a novel missense variation in exon 15, c.1561C>T (p.P521S). All mutations segregated with the disease phenotype in the respective families and were absent in ethnically matched control chromosomes. Haplotype analysis suggested (p<10−6) that affected individuals inherited the causal mutation from a common ancestor.
Conclusions
Pathogenic mutations in TULP1 are responsible for the RP phenotype in seven familial cases with a common ancestral mutation responsible for the disease phenotype in four of the seven families.
Introduction
Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous group of hereditary retinal disorders that primarily affect the ocular retina, with a prevalence of 1:4,000 [1,2]. RP is characterized by progressive degeneration of rod photoreceptors, leading to night blindness and constriction of the visual field, followed by the degeneration of cone photoreceptors, resulting in a total loss of vision [3]. The clinical manifestation of the disease includes pigmentary deposits in the retina, waxy disc pallor, and attenuation of retinal blood vessels [3]. Affected individuals often have severely abnormal or undetectable electroretinography responses, even in the early stage of the disease [3].
RP is a genetically heterogeneous disorder that manifests as an autosomal dominant, autosomal recessive, or X-linked trait. To date, a total of 73 genes have been implicated in the pathogenesis of RP. Of these, 27 genes have been associated with autosomal dominant RP (adRP) [4-30], while mutations in 50 genes have been identified in patients with autosomal recessive RP (arRP) [31-77]. Mutations in RHO (Gene ID: 6010; OMIM: 180380), RP1 (Gene ID: 6101; OMIM: 603937), NRL (Gene ID: 4901; OMIM: 162080), RPE65 (Gene ID: 6121; OMIM: 180069), BEST1 (Gene ID: 7439; OMIM: 607854), NR2E3 (Gene ID: 10,002; OMIM: 604485), and IMPDH1 (Gene ID: 3614; OMIM: 146690) have been identified in familial cases of adRP and arRP. Likewise, causal mutations in OFD1 (Gene ID: 8481; OMIM: 300170), RP2 (Gene ID: 6102; OMIM: 300757), and RPGR (Gene ID: 6103; OMIM: 312610) have been identified in RP cases with an X-linked inheritance pattern [78-80].
The tubby-like protein 1 (TULP1) gene consists of 15 coding exons spanning a 15 kb region and encodes for a 542 amino acid protein that has been associated with the transport of rhodopsin from its site of synthesis in the inner segments through the connecting cilium to the outer segments [81]. North and colleagues previously reported that TULP1 is expressed in many tissues, specifically in the rod and cone photoreceptor cells, and is involved in the transport of rhodopsin [82]. TULP1 has been associated with retinal degeneration, and pathogenic mutations in TULP1 have been identified in patients with arRP, rod-cone dystrophy, and Leber congenital amaurosis (LCA).
We previously reported five familial cases of arRP harboring mutations in TULP1 [83]. Since Iqbal et al. published their study, we have ascertained more than 200 familial cases of arRP. To investigate the genetic load of TULP1 in our familial cohort, we performed an exclusion linkage analysis that identified seven additional intermarried familial cases with multiple consanguineous marriages, diagnosed with early-onset RP. Clinical records available to us suggest an early, probably congenital onset, while exclusion analysis localized the retinal phenotype in all seven families to chromosome 6p harboring TULP1. Sanger sequencing of TULP1 identified causal mutations that segregated with the disease phenotype in the respective families and were absent in ethnically matched controls and genome-variant databases.
Methods
Clinical ascertainment
A total of more than 350 consanguineous Pakistani families with non-syndromic retinal dystrophies were recruited to identify new disease loci responsible for inherited visual diseases. The Institutional Review Boards (IRBs) of the National Centre of Excellence in Molecular Biology (Lahore, Pakistan), the National Eye Institute (Bethesda, MD), and Johns Hopkins University (Baltimore, MD) approved the study. All participating family members provided informed written consent that was endorsed by the respective IRBs and is consistent with the tenets of the Declaration of Helsinki.
A detailed clinical and medical history was obtained by interviewing the family members. Funduscopy was performed at the Layton Rehmatulla Benevolent Trust (LRBT) Hospital (Lahore, Pakistan). Electroretinography (ERG) measurements were recorded by using equipment manufactured by LKC (Gaithersburg, MD). Dark-adapted rod responses were determined through incident flash attenuated by −25 dB, whereas rod–cone responses were measured at 0 dB. The 30 Hz flicker responses were recorded at 0 dB to a background illumination of 17 to 34 cd/m2. All participating members voluntarily provided a sample of approximately 10 ml of blood that was stored in 50 ml Sterilin® falcon tubes containing 400 μl of 0.5 M EDTA. The blood samples were stored at −20 °C for long-term storage.
Genomic DNA extraction
Genomic DNA was extracted from white blood cells using a non-organic modified procedure as described previously [84]. The concentration of the extracted genomic DNA was estimated with a SmartSpec Plus Spectrophotometer (Bio-Rad, Hercules, CA).
Exclusion and linkage analysis
PCR was performed in a 5 μl mixture containing 40 ng of genomic DNA, 0.5 μl of 10 μM fluorescent-labeled primer pairs, 0.5 μl of 10X PCR Buffer (100 mM Tris HCl (pH 8.4), 400 mM NaCl, 15 mM MgCl2, 2.5 mM spermidine), 2 mM dNTP mix, and 0.2 U Taq DNA Polymerase (New England BioLabs Inc., Ipswich, MA). Initial denaturation was performed for 5 min at 95 °C, followed by ten cycles of 15 s at 94 °C, 15 s at 55 °C, and 30 s at 72 °C and then 20 cycles of 15 s at 89 °C, 15 s at 55 °C, and 30 s at 72 °C. The final extension was performed for 10 min at 72 °C. PCR products were mixed with a loading cocktail containing HD-400 size standards (Applied Biosystems, Foster City, CA) and resolved in an ABI PRISM 3100 Genetic Analyzer. Genotypes were assigned using Gene Mapper software from Applied Biosystems.
Linkage analysis was performed with alleles obtained through exclusion analysis using the FASTLINK version of MLINK from the LINKAGE Program Package [85,86]. Maximum LOD scores were calculated using ILINK from the LINKAGE Program Package. Autosomal recessive RP was investigated as a fully penetrant disorder with an affected allele frequency of 0.001. The marker order and distances between the markers were obtained from the National Center for Biotechnology Information chromosome 6 sequence maps.
Mutation screening
Individual exons of TULP1 were amplified with PCR using primer pairs designed by the primer3 program (Appendix 1). PCR reactions were completed in 10 μl volumes containing 20 ng of genomic DNA, 1 μl of the forward and reverse primers at 10 µM, 1 μl of 10X PCR Buffer (100 mM Tris HCl (pH 8.4), 400 mM NaCl, 15 mM MgCl2, 2.5 mM spermidine), 2 mM dNTP mix, 500 mM betaine, and 0.2 U Taq DNA Polymerase. PCR amplification consisted of a denaturation step at 95 °C for 5 min followed by a two-step touchdown procedure. The first step of ten cycles consisted of denaturation at 95 °C for 30 s, followed by a primer set-specific annealing for 30 s (annealing temperature decreased by 1 °C per cycle) and elongation at 72 °C for 45 s. The second step of 30 cycles consisted of denaturation at 95 °C for 30 s, followed by annealing (10 °C below the annealing temperature used in the first step) for 30 s and elongation at 72 °C for 45 s, followed by a final elongation at 72 °C for 5 min.
The PCR primers for each amplicon were used for bidirectional sequencing using the BigDye Terminator Ready Reaction mix according to the manufacturer’s instructions. The sequencing products were resolved on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems), and results were analyzed with Applied Biosystems SeqScape software.
In silico analysis
The degree of evolutionary conservation of c.1495+4A>C in other TULP1 orthologs was examined using the UCSC Genome browser. The effect of the c.1495+4A>C mutation on TULP1 mRNA splicing was predicted with an online bioinformatics tool, Human Splicing Finder 3.0 (HSF3). The possible impact of an amino acid change in the structure of TULP1 was examined with the SIFT and PolyPhen-2 tools available online.
Estimating the likelihood of a common founder effect
A total of five single nucleotide polymorphisms (SNPs) within 11 kb of TULP1 were selected, and one affected individual from each family was genotyped to construct the causal haplotype. SNP genotypes of 96 individuals of Pakistani descent were obtained from the 1000 Genomes database and used to construct ethnically matched control haplotypes. The haplotype frequencies were estimated to calculate the likelihood of a common founder effect.
Results
We ascertained a large cohort of highly intermarried familial cases of retinal dystrophies to investigate the genetic basis of arRP. We previously reported five familial cases of arRP harboring pathogenic mutations in TULP1 [83]. Since Iqbal and colleagues [83] published their study, we have ascertained more than 200 additional familial cases of arRP, and therefore, we reexamined our expanded cohort for mutations in TULP1 with closely spaced fluorescently labeled short tandem repeat (STR) markers spanning the TULP1 locus. These analyses identified seven additional intermarried families (PKRP259, PKRP268, PKRP301, PKRP309, PKRP356, PKRP364, and PKRP367) linked to TULP1 (Figure 1).
Figure 1.

Pedigree drawings with haplotype formed from alleles of chromosome 6p microsatellite markers. A: PKRP259. B: PKRP268. C: PKRP301. D: PKRP309. E: PKRP356. F: PKRP364. G: PKRP367. The alleles forming the risk haplotype are shaded black, and the alleles that do not cosegregate with retinitis pigmentosa (RP) are shown in white. Squares = males; circles = females; filled symbols = affected individuals; double line between individuals = consanguineous marriage; diagonal line through a symbol = deceased family member.
Affected individuals in these families fulfilled the diagnostic criteria of RP (Table 1). Fundus photographs of affected individuals revealed typical symptoms of RP, including attenuated retinal arteries, a waxy, pale optic disc, and bone spicule–like pigment deposits in the lateral and mid-periphery of the retina (Figure 2). Likewise, scotopic ERG recordings measured at −25 dB and photopic responses at 0 dB (30 Hz flicker) were undetectable in affected individuals, suggestive of compromised rod and cone photoreceptor cells, while unaffected individuals exhibited rod and cone responses in the normal range (Figure 3).
Table 1. Clinical characteristics of the patients screened for TULP1 mutations.
| Family | ID | C-Age (Yr.) | D-age (Yr.) | First symptoms | Night blindness | Fundus examination |
Electroretinography |
Visual acuity |
||
|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | |||||||
| PKRP259 |
10 |
28 |
6 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/36 |
6/40 |
| PKRP259 |
15 |
22 |
7 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/24 |
6/24 |
| PKRP268 |
12 |
17 |
7 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/20 |
6/20 |
| PKRP268 |
13 |
14 |
6 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/20 |
6/20 |
| PKRP301 |
14 |
20 |
5 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/18 |
6/20 |
| PKRP301 |
17 |
14 |
5 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/20 |
6/24 |
| PKRP309 |
11 |
34 |
6 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/30 |
6/28 |
| PKRP309 |
15 |
25 |
7 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/25 |
6/25 |
| PKRP356 |
10 |
10 |
5 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/12 |
6/12 |
| PKRP356 |
12 |
8 |
5 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/20 |
6/20 |
| PKRP364 |
10 |
58 |
7 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/50 |
6/50 |
| PKRP364 |
20 |
21 |
8 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/24 |
6/24 |
| PKRP367 |
11 |
31 |
6 |
Night blindness |
progressive |
MD, Art. Atten, Pig.dep, PD |
NAB, NF |
NAB, NF |
6/24 |
6/28 |
| PKRP367 | 12 | 20 | 7 | Night blindness | progressive | MD, Art. Atten, Pig.dep, PD | NAB, NF | NAB, NF | 6/20 | 6/20 |
MD: macular degeneration; Art. Atten: artery attenuation; Pig.dep: pigment deposit; PD: Pale optic disc; NAB: no ‘a’ or ‘b’ wave response; NF: no flicker response; C-Age: current age; D-Age: age at first diagnosis of the retinal dystrophy.
Figure 2.
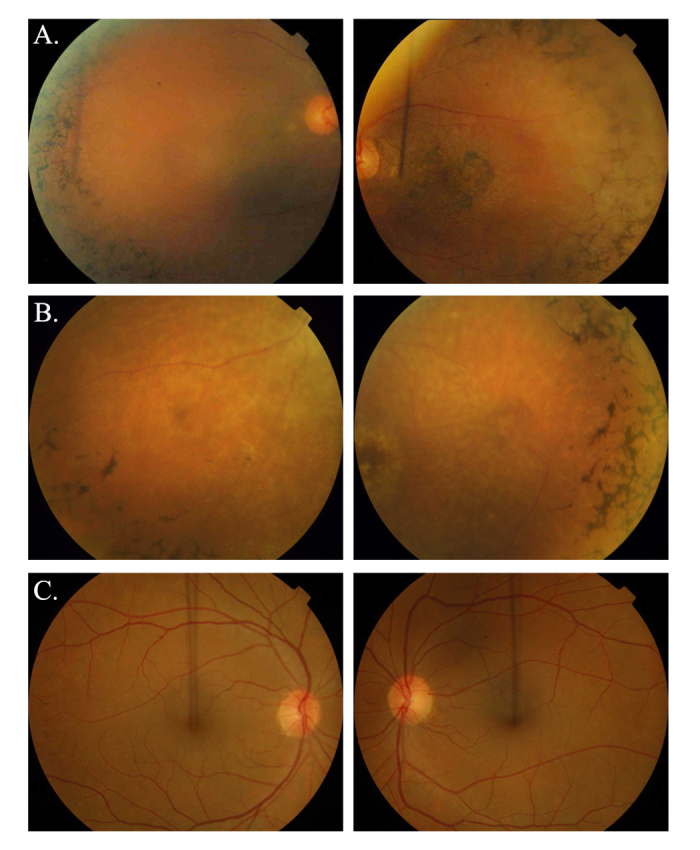
Fundus photographs of affected individuals illustrating symptoms of retinitis pigmentosa. A: OD and OS of individual 10 (affected: 30 years) of family PKRP259. B: OD and OS of individual 11 (affected: 18 years) of family PKRP309. C: OD and OS of individual 8 (unaffected: 52 years) of family PKRP259. Fundus photographs of affected individuals show bone spicule-like pigmentation in the mid-periphery of the retina, attenuated retinal arterioles, severe maculopathy, and disc pallor. OD = oculus dexter; OS = oculus sinister.
Figure 3.

Electroretinography responses of PKRP259 family members. In the stimulus conditions, scotopic 0 dB bright flashes elicit rod responses (left column of each pair), and a photopic 0 dB, 30 Hz flicker elicits cone responses (right column of each pair). Responses are of A) OD and B) OS of individual 10 (affected: 30 years); C) OD and D) OS of individual 14 (affected: 25 years); and E) OD and F) OS of individual 8 (unaffected: 52 years). The affected individuals exhibit undetectable electroretinography responses whereas the unaffected individual exhibits normal a- and b-waves suggestive of normal rod and cone function. OD = oculus dexter; OS = oculus sinister.
All seven families yielded positive two-point LOD scores for chromosome 6p markers flanking TULP1 (Table 2). We sequenced all coding exons and the exon–intron boundaries of TULP1, which identified four different causal mutations. They included a novel missense variation in exon 15, c.1561C>T (p.P521S), in PKRP259 (Figure 4A); a homozygous splice site variant in intron 14, c.1495+4A>C, in PKRP268 that affects the conserved splice donor site (Figure 4B); a single base pair substitution in exon 14, c.1466A>G (p.K489R), in four families, PKRP301 (Figure 4C), PKRP309 (Figure 4D), PKRP356 (Figure 4E), and PKRP367 (Figure 4G); and a two-base deletion in exon 4, c.286_287delGA (p.E96Gfs77*), in PKRP364 (Figure 4F). These variants segregated in their respective families: Affected individuals were homozygous whereas unaffected individuals were heterozygous carriers or homozygous for the wild-type allele. These mutations were absent in ethnically matched control chromosomes and were not present in the 1000 Genomes database.
Table 2. Two-point LOD scores of chromosome 6p markers for families A) PKRP259, B) PKRP268, C) PKRP301, D) PKRP309, E) PKRP356, F) PKRP364, and G) PKRP367.
|
Markers |
cM |
Mb |
0.00 |
0.01 |
0.05 |
0.09 |
0.10 |
0.20 |
0.30 |
Zmax |
θmax |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | |||||||||||
| D6S439 |
48.26 |
35.18 |
2.21 |
2.16 |
1.95 |
1.74 |
1.68 |
1.15 |
0.65 |
2.21 |
0.00 |
| D6S1611 |
47.71 |
35.40 |
2.79 |
2.73 |
2.50 |
2.26 |
2.20 |
1.57 |
0.93 |
2.79 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 2.02 | 1.97 | 1.77 | 1.57 | 1.51 | 1.00 | 0.51 | 2.02 | 0.00 |
| B | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6S439 |
48.26 |
35.18 |
2.30 |
2.26 |
2.06 |
1.86 |
1.80 |
1.27 |
0.72 |
2.30 |
0.00 |
| D6S1611 |
47.71 |
35.40 |
2.48 |
2.43 |
2.22 |
2.00 |
1.94 |
1.39 |
0.85 |
2.48 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 3.33 | 3.26 | 2.99 | 2.72 | 2.65 | 1.94 | 1.22 | 3.33 | 0.00 |
| C | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6S439 |
48.26 |
35.18 |
-∞ |
−5.70 |
−3.82 |
−2.96 |
−2.40 |
−2.00 |
−1.83 |
−2.00 |
0.20 |
| D6S1611 |
47.71 |
35.40 |
1.14 |
1.12 |
1.07 |
1.02 |
0.97 |
0.93 |
0.90 |
1.14 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 1.44 | 1.40 | 1.33 | 1.25 | 1.18 | 1.11 | 1.07 | 1.44 | 0.00 |
| D | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6S439 |
48.26 |
35.18 |
1.55 |
1.52 |
1.46 |
1.39 |
1.33 |
1.27 |
1.23 |
1.55 |
0.00 |
| D6S1611 |
47.71 |
35.40 |
1.58 |
1.55 |
1.48 |
1.42 |
1.35 |
1.28 |
1.25 |
1.58 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 0.66 | 0.64 | 0.61 | 0.58 | 0.55 | 0.52 | 0.50 | 0.66 | 0.00 |
| E | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6S439 |
48.26 |
35.18 |
2.19 |
2.15 |
2.06 |
1.98 |
1.89 |
1.80 |
1.76 |
2.19 |
0.00 |
| D6S1611 |
47.71 |
35.40 |
0.49 |
0.47 |
0.44 |
0.41 |
0.38 |
0.35 |
0.33 |
0.49 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 2.19 | 2.15 | 2.06 | 1.98 | 1.89 | 1.80 | 1.76 | 2.19 | 0.00 |
| F | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6S439 |
48.26 |
35.18 |
-∞ |
−0.81 |
0.01 |
0.33 |
0.50 |
0.59 |
0.62 |
0.62 |
0.30 |
| D6S1611 |
47.71 |
35.40 |
2.03 |
1.98 |
1.89 |
1.79 |
1.69 |
1.59 |
1.54 |
2.03 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 3.93 | 3.86 | 3.71 | 3.56 | 3.41 | 3.25 | 3.17 | 3.93 | 0.00 |
| G | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6S439 |
48.26 |
35.18 |
0.73 |
0.71 |
0.64 |
0.45 |
0.10 |
0.20 |
0.30 |
0.73 |
0.00 |
| D6S1611 |
47.71 |
35.40 |
0.43 |
0.42 |
0.37 |
0.46 |
0.55 |
0.37 |
0.19 |
0.43 |
0.00 |
| D6S1645 | 48.26 | 35.61 | 0.43 | 0.42 | 0.37 | 0.46 | 0.32 | 0.20 | 0.10 | 0.43 | 0.00 |
Figure 4.

Sequence chromatograms of TULP1 variations identified in this study. A: Unaffected individual 8 is a heterozygous carrier, and affected individual 10 is homozygous for the single base pair substitution c.1561C>T in family PKRP259. B: Unaffected individual 19 is a heterozygous carrier, and affected individual 12 is homozygous for the splice region variant c.1495+4A>C in family PKRP268. C: Unaffected individual 12 is a heterozygous carrier, and affected individual 17 is homozygous for the single base pair substitution c.1466A>G in family PKRP301. D: Unaffected individual 7 is a heterozygous carrier, and affected individual 15 is homozygous for the single base pair substitution c.1466A>G in family PKRP309. E: Unaffected individual 8 is a heterozygous carrier, and affected individual 10 is homozygous for the single base pair substitution c.1466A>G in family PKRP356. F: Unaffected individual 18 is a heterozygous carrier, and affected individual 10 is homozygous for the two-base deletion c.286_287delGA in family PKRP364. G: Unaffected individual 9 is a heterozygous carrier, and affected individual 12 is homozygous for the single base pair substitution c.1466A>G in family PKRP367.
We examined the evolutionary conservation of amino acid Pro521 and nucleotide c.1495+4A and found that Pro521 and c.1495+4A are completely conserved in TULP1 orthologs (Figure 5). We examined the possible impact of the Pro521Ser substitution on the TULP1 protein using the PolyPhen-2 algorithm, which suggested that the serine substitution at position 521 would probably be damaging. Subsequently, we evaluated the effect of the c.1495+4A>C variation on TULP1 mRNA splicing using Human Splice Finder 3 (HSF3). HSF3 generated consensus values of 82.12 and 73.32 for the wild-type (c.1495+4A) and mutant (c.1495+4C) nucleotides, respectively (Figure 6A,B). The predicted consensus value deviation of −10.72 for c.1495+4A>C suggests that the wild-type splice donor site will be broken. Loss of the wild-type splice site will result in the retention of intron 14 of TULP1 (Figure 6B), resulting in a frame shift and is likely to produce aberrant TULP1 (p.P499Rfs104*).
Figure 5.

Sequence conservation of amino acid Pro521 and nucleotide 1495+4A in TULP1 orthologs. Primates are green, placental mammals are blue, and vertebrates are purple. The arrow points to amino acid residue Pro521 and nucleotide 1495+4A, which were mutated in individuals with retinitis pigmentosa.
Figure 6.

In silico analysis of the splice donor site mutation in TULP1. The HSF3 algorithm predicted a consensus value (CV) of A) 82.12 for the wild-type (c.1495+4A) and B) 73.32 (c.1495+4C) for the mutant splice donor site. The CV deviation of −10.72 suggests that the loss of the wild-type splice site will result in the retention of intron 14 of TULP1, resulting in a frame shift likely to produce aberrant TULP1 (p.P499Rfs104*).
All four families (PKRP301, PKRP309, PKRP356, and PKRP367) harboring the K489R allele were recruited from the Punjab province of Pakistan; they reside in different cities with no known relationship between them. We previously reported four families (PKRP084, PKRP111, PKRP122, and PKRP171) harboring the same missense variation, and SNP analysis suggested a common ancestor who transmitted the causal allele [83]. The presence of a common causal mutation in eight familial cases of our cohort prompted us to investigate the ancestral relationships among the cases. We used single nucleotide polymorphisms in the immediate neighborhood of the causal mutation, which identified a haplotype (CTGT/CC) common to all four families harboring the K489R allele (Table 3) suggestive of a common founder effect. To confirm the effect, we retrieved the genotype information of ethnically matched controls from the 1000 Genomes database and estimated the respective population haplotype frequencies (four of the five SNPs, including rs12665445, rs7770128, rs12215920, and rs7764472, were to construct the haplotype). The CTGC haplotype had an allele frequency of 0.04 in the Punjabi population of Pakistani decent, which suggested a high probability (p>2.56×10−6) that affected individuals in these four families inherited the causal mutation from a common ancestor. Interestingly, these odds increased significantly (p>6.5×10−12) when PKRP084, PKRP111, PKRP122, and PKRP171 (harboring the K489R allele reported by Iqbal et al. [83]) were included in the analysis.
Table 3. Single nucleotide polymorphism (SNP) haplotypes of affected individuals in PKRP301, PKRP309, PKRP356, and PKRP367 harboring the c.1466A>G (p.K489R) mutation in TULP1.
| Family | Individual |
c.1466A>G |
rs12665445 |
rs7770128 |
rs12215920 |
rs34126023 |
rs7764472 |
|---|---|---|---|---|---|---|---|
| Chr6: 35500039 | Chr6: 35500262 | Chr6: 35505901 | Chr6: 35506296 | Chr6: 35509796 | Chr6: 35511797 | ||
|
PKRP301 |
14 |
G |
C |
T |
G |
T/C |
C |
|
PKRP309 |
15 |
G |
C |
T |
G |
T/C |
C |
|
PKRP356 |
10 |
G |
C |
T |
G |
T/C |
C |
| PKRP367 | 12 | G | C | T | G | T/C | C |
Discussion
Here, we report seven consanguineous families recruited from the Punjab province of Pakistan with multiple members manifesting cardinal symptoms of RP. Exclusion analysis with closely spaced STR markers localized the linkage interval in all seven families to chromosome 6p21.3 harboring TULP1, while bidirectional Sanger sequencing of TULP1 identified a novel missense variation, a splice site variant, a previously reported single base pair substitution, and a two-base deletion. All these variants segregate with the disease phenotype in the respective families. These variations were absent in 190 ethnically matched control chromosomes, and the absence of the variants in the 1000 Genomes database, the NHLBI Exome Variant Server, and the dbSNP database strongly suggests that these variations are responsible for the retinal phenotype of the patients reported in this study.
As shown in Table 4, a total of 50 causal mutations have been reported in TULP1, and mutations in TULP1 account for 1–2% of arRP cases in different ethnic populations worldwide [37,81,83,87-116]. Previously, Gu and colleagues screened a large cohort of patients of German origin with arRP and identified the K489R pathogenic allele in TULP1 [92]. More recently, Maria and colleagues identified the K489R allele in a family of Pakistani descent [113]. We found the same residue, p.K489R, in eight families; therefore, this allele is by far the most abundant RP-associated allele of TULP1 found in the Pakistani population. In our large cohort of more than 350 familial cases of arRP, we identified 12 families harboring causal mutations in TULP1; however, as eight of these families harbor a common ancestral mutation, we estimate that TULP1 contributes nearly 1% of the total genetic load of arRP in our cohort.
Table 4. List of mutations reported in TULP1-associated retinal dystrophies.
| Exon/ Intron | Nucleotide change | Amino acid change | Phenotype | Reference |
|---|---|---|---|---|
| Exon 1 |
c.3G>A |
p.M1I |
arRP |
87 |
| Intron 2 |
c.99+1G>A |
Aberrant splicing |
LCA, arRP |
88, 89 |
| Exon 4 |
c.280G>T |
p.D94Y |
LCA |
90 |
| Intron 4 |
c.350–2delAGA, (IVS4–2delAGA) |
Aberrant splicing |
arRP |
91 |
| Exon 5 |
c.394_417del |
p.E120_D127del |
arRP |
92 |
| Exon 5 |
c.539G>A |
p.R180H |
LCA |
93 |
| Exon 6 |
c.627delC |
p.S210QfsX27 |
LCA |
94 |
| Exon 6 |
c.629C>G |
p.S210* |
RP |
95 |
| Intron 7 |
c.718+2T>C |
Aberrant splicing |
JRP, LCA |
96 |
| Exon 7 |
c.725_728delCCAA |
p.P242Qfs×16 |
LCA |
97 |
| Exon 10 |
c.901C>T |
p.Q301* |
LCA, RCD |
98, 99 |
| Exon 10 |
c.937delC |
p.Q301fs9* |
arRP |
91 |
| Exon 10 |
c.932G>A |
p.R311Q |
arRP |
100 |
| Exon 10 |
c.956G>A |
p.G319D |
RP |
101 |
| Exon 10 |
c.961T>G |
p.Y321D |
LCA |
97 |
| Intron 10 |
c.999+5G>C |
Aberrant splicing |
JRP, LCA |
96 |
| Exon 11 |
c.1025G>A |
p.R342Q |
arRP |
100 |
| Exon 11 |
c.1047T>G |
p.N349K |
arRP |
102 |
| Exon 11 |
c.1064A>T |
p.D355V |
LCA |
97 |
| Exon 11 |
c.1087G>A |
p.G363R |
RCD |
103 |
| Exon 11 |
c.1081C>T |
p.R361* |
LCA |
104 |
| Exon 11 |
c.1102G>T |
p.G368W |
LCA |
89 |
| Intron 11 |
c.1112+2T>C (IVS11 ds T-C +2) |
Aberrant splicing |
arRP |
105 |
| Intron 11 |
c.1113–2A>C (IVS11 as A-C −2) |
Aberrant splicing |
LCA |
97 |
| Exon 12 |
c.1138A>G |
p.T380A |
LCA, arRP |
83, 106, 107 |
| Exon 12 |
c.1145T>C |
p.F382S |
arRP |
108 |
| Exon 12 |
c.1198C>T |
p.R400W |
arRP, LCA, RD |
89, 109, 110 |
| Exon 12 |
c.1199G>A |
p.A400Q |
arRP |
111 |
| Exon 12 |
c.1204G>T |
p.E402* |
LCA |
89 |
| Intron 12 |
c.1224+4A>G, (IVS12+4A>G) |
Aberrant splicing |
arRP |
92 |
| Exon 13 |
c.1246C>T |
p.R416C |
ArRP |
87 |
| Exon 13 |
c.1258C>A |
p.R420S |
RCD |
112 |
| Exon 13 |
c.1259G>C |
p.R420P |
arRP |
88 |
| Exon 13 |
c.1318C>T |
p.R440* |
LCA |
94 |
| Exon 14 |
c.1349G>A |
p.W450* |
LCA |
90 |
| Exon 14 |
c.1376T>A |
p.I459K |
arRP |
37, 88 |
| Exon 14 |
c.1376T>C |
p.I459T |
arRP |
105 |
| Exon 14 |
c.1376_1377delTA |
p.I459Rfs×12 |
LCA |
97 |
| Exon 14 |
c.1381C>G |
p.L461V |
JRP, LCA |
96 |
| Exon 14 |
c.1444C>T |
p.R482W |
arRP |
81, 109 |
| Exon 14 |
c.1445G>A |
p.A482Q |
arRP |
107 |
| Exon 14 |
c.1466A>G |
p.K489R |
arRP |
83, 92, 113 |
| Exon 14 |
c.1472T>C |
p.F491L |
arRP |
88 |
| Intron 14 |
c.1495+1G>A, (IVS14+1G>A) |
Aberrant splicing |
arRP |
37 |
| Intron 14 |
c.1495+2_1495+3insT |
Aberrant splicing |
arRP |
114 |
| Intron 14 |
c.1496–6C>A, (IVS14–6C>A) |
Aberrant splicing |
arRP |
88, 92 |
| Exon 15 |
c.1511_1521delTGCAGTTCGGC |
p.L504fs140* |
arRP |
81 |
| Exon 15 |
c.1518C>A |
p.F506L |
LCA |
94 |
| Exon 15 |
c.1582_1587dupTTCGCC |
p.F528_A529dup |
LCA/arRP |
115 |
| Exon 15 | c.1604T>C | p.F535S | LCA | 116 |
arRP: autosomal recessive RP; RD: Retinal degeneration; LCA: Leber congenital amaurosis; JRP: Juvenile onset RP; RCD: Rod-Cone Dystrophy.
Identification of causal mutations reaffirmed the role of TULP1 in the pathogenesis of autosomal recessive RP and reiterates the heterogeneity associated with the disease phenotype. We compared the clinical phenotype of patients with arRP in PKRP084, PKRP111, PKRP122, and PKRP171 harboring the K489R allele reported by Iqbal et al. [83] with affected individuals in PKRP301, PKRP309, PKRP356, and PKRP367. However, we did not identify any distinction between the clinical phenotypes of affected individuals in these eight familial cases. All affected individuals in these familial cases manifested cardinal symptoms of RP, including attenuated retinal arteries and bone spicule–like pigment deposits accompanied by undetectable scotopic and photopic ERG responses. Identification of causal alleles responsible for arRP will help diagnostic efforts to identify carrier status in intermarried familial cases, and subsequent genetic counseling will help families make educated decisions regarding arranged marriages and screening for the status of newborns. In conclusion, we report seven familial cases harboring causal mutations in TULP1, including a common ancestral mutation that has now been identified in eight apparently unrelated familial cases.
Acknowledgments
We are thankful to all family members for their participation in this study. This study was supported in part by the Higher Education Commission, Islamabad, Pakistan (SR), and by the National Eye Institute Grant R01EY021237–01 (RA and SAR).
Appendix 1. Primer sequences for the amplification of TULP1.
To access the data, click or select the words “Appendix 1.”
References
- 1.Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–76. [PubMed] [Google Scholar]
- 2.Bird AC. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 1995;119:543–62. doi: 10.1016/s0002-9394(14)70212-0. [DOI] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 4.Farrar GJ, Kenna P, Redmond R, McWilliam P, Bradley DG, Humphries MM, Sharp EM, Inglehearn CF, Bashir R, Jay M. Autosomal dominant retinitis pigmentosa: absence of the rhodopsin proline—-histidine substitution (codon 23) in pedigrees from Europe. Am J Hum Genet. 1990;47:941–5. [PMC free article] [PubMed] [Google Scholar]
- 5.Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–3. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 6.Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;38:1972–82. [PubMed] [Google Scholar]
- 7.Bowne SJ, Daiger SP, Hims MM, Sohocki MM, Malone KA, McKie AB, Heckenlively JR, Birch DG, Inglehearn CF, Bhattacharya SS, Bird A, Sullivan LS. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–8. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bessant DA, Payne AM, Mitton KP, Wang QL, Swain PK, Plant C, Bird AC, Zack DJ, Swaroop A, Bhattacharya SS. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet. 1999;21:355–6. doi: 10.1038/7678. [DOI] [PubMed] [Google Scholar]
- 9.Payne AM, Downes SM, Bessant DA, Plant C, Moore T, Bird AC, Bhattacharya SS. Genetic analysis of the guanylate cyclase activator 1B (GUCA1B) gene in patients with autosomal dominant retinal dystrophies. J Med Genet. 1999;36:691–3. [PMC free article] [PubMed] [Google Scholar]
- 10.Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell. 2001;8:375–81. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 11.McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, Mackey DA, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet. 2001;10:1555–62. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 12.Wada Y, Abe T, Takeshita T, Sato H, Yanashima K, Tamai M. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2001;42:2395–400. [PubMed] [Google Scholar]
- 13.Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, Rosenberg T, Webster AR, Bird AC, Gal A, Hunt D, Vithana EN, Bhattacharya SS. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 14.Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–68. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maita H, Kitaura H, Keen TJ, Inglehearn CF, Ariga H, Iguchi-Ariga SM. PAP-1, the mutated gene underlying the RP9 form of dominant retinitis pigmentosa, is a splicing factor. Exp Cell Res. 2004;300:283–96. doi: 10.1016/j.yexcr.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 16.Rebello G, Ramesar R, Vorster A, Roberts L, Ehrenreich L, Oppon E, Gama D, Bardien S, Greenberg J, Bonapace G, Waheed A, Shah GN, Sly WS. Apoptosis-inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci USA. 2004;101:6617–22. doi: 10.1073/pnas.0401529101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abid A, Ismail M, Mehdi SQ, Khaliq S. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J Med Genet. 2006;43:378–81. doi: 10.1136/jmg.2005.035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coppieters F, Leroy BP, Beysen D, Hellemans J, De BK, Haegeman G, Robberecht K, Wuyts W, Coucke PJ, De BE. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007;81:147–57. doi: 10.1086/518426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, Theis T, Friedman J, Maubaret C, Bujakowska K, Veraitch B, Abd El-Aziz MM. Prescott dQ, Parapuram SK, Bickmore WA, Munro PM, Gal A, Hamel CP, Marigo V, Ponting CP, Wissinger B, Zrenner E, Matter K, Swaroop A, Koenekoop RK, Bhattacharya SS. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet. 2007;81:1098–103. doi: 10.1086/521953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fingert JH, Oh K, Chung M, Scheetz TE, Andorf JL, Johnson RM, Sheffield VC, Stone EM. Association of a novel mutation in the retinol dehydrogenase 12 (RDH12) gene with autosomal dominant retinitis pigmentosa. Arch Ophthalmol. 2008;126:1301–7. doi: 10.1001/archopht.126.9.1301. [DOI] [PubMed] [Google Scholar]
- 21.Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O’Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85:581–92. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, Sullivan LS, Ponjavic V, Granse L, Khanna R, Trager EH, Gieser LM, Hughbanks-Wheaton D, Cojocaru RI, Ghiasvand NM, Chakarova CF, Abrahamson M, Goring HH, Webster AR, Birch DG, Abecasis GR, Fann Y, Bhattacharya SS, Daiger SP, Heckenlively JR, Andreasson S, Swaroop A. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009;84:792–800. doi: 10.1016/j.ajhg.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, Bowne SJ, Sullivan LS, Daiger SP, Chen LJ, Pang CP, Zhao K, Staley JP, Larsson C. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. 2009;85:617–27. doi: 10.1016/j.ajhg.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menotti-Raymond M, Deckman KH, David V, Myrkalo J, O’Brien SJ, Narfstrom K. Mutation discovered in a feline model of human congenital retinal blinding disease. Invest Ophthalmol Vis Sci. 2010;51:2852–9. doi: 10.1167/iovs.09-4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL, Rivolta C. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2011;88:643–9. doi: 10.1016/j.ajhg.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowne SJ, Humphries MM, Sullivan LS, Kenna PF, Tam LC, Kiang AS, Campbell M, Weinstock GM, Koboldt DC, Ding L, Fulton RS, Sodergren EJ, Allman D, Millington-Ward S, Palfi A, McKee A, Blanton SH, Slifer S, Konidari I, Farrar GJ, Daiger SP, Humphries P. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19:1074–81. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Liu Y, Sheng X, Tam PO, Zhao K, Chen X, Rong W, Liu Y, Liu X, Pan X, Chen LJ, Zhao Q, Vollrath D, Pang CP, Zhao C. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2014;23:2926–39. doi: 10.1093/hmg/ddu005. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan LS, Koboldt DC, Bowne SJ, Lang S, Blanton SH, Cadena E, Avery CE, Lewis RA, Webb-Jones K, Wheaton DH, Birch DG, Coussa R, Ren H, Lopez I, Chakarova C, Koenekoop RK, Garcia CA, Fulton RS, Wilson RK, Weinstock GM, Daiger SP. A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55:7147–58. doi: 10.1167/iovs.14-15419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Chen X, Xu Q, Gao X, Tam PO, Zhao K, Zhang X, Chen LJ, Jia W, Zhao Q, Vollrath D, Pang CP, Zhao C. SPP2 Mutations Cause Autosomal Dominant Retinitis Pigmentosa. Sci Rep. 2015;5:14867. doi: 10.1038/srep14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X, Guan L, Wu W, Zhang Y, Zheng W, Gao YT, Long J, Wu N, Wu L, Xiang Y, Xu B, Shen M, Chen Y, Wang Y, Yin Y, Li Y, Xu H, Xu X, Li Y. Whole-exome sequencing identifies OR2W3 mutation as a cause of autosomal dominant retinitis pigmentosa. Sci Rep. 2015;5:9236. doi: 10.1038/srep09236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1:209–13. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- 32.Bayes M, Giordano M, Balcells S, Grinberg D, Vilageliu L, Martinez I, Ayuso C, Benitez J, Ramos-Arroyo MA, Chivelet P. Homozygous tandem duplication within the gene encoding the beta-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum Mutat. 1995;5:228–34. doi: 10.1002/humu.1380050307. [DOI] [PubMed] [Google Scholar]
- 33.Huang SH, Pittler SJ, Huang X, Oliveira L, Berson EL, Dryja TP. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet. 1995;11:468–71. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 34.Dryja TP, Finn JT, Peng YW, McGee TL, Berson EL, Yau KW. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:10177–81. doi: 10.1073/pnas.92.22.10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–7. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 36.Maw MA, Kennedy B, Knight A, Bridges R, Roth KE, Mani EJ, Mukkadan JK, Nancarrow D, Crabb JW, Denton MJ. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet. 1997;17:198–200. doi: 10.1038/ng1097-198. [DOI] [PubMed] [Google Scholar]
- 37.Banerjee P, Kleyn PW, Knowles JA, Lewis CA, Ross BM, Parano E, Kovats SG, Lee JJ, Penchaszadeh GK, Ott J, Jacobson SG, Gilliam TC. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet. 1998;18:177–9. doi: 10.1038/ng0298-177. [DOI] [PubMed] [Google Scholar]
- 38.Cremers FP, van De Pol DJ. van DM, den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 39.Nakazawa M, Wada Y, Tamai M. Arrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch Ophthalmol. 1998;116:498–501. doi: 10.1001/archopht.116.4.498. [DOI] [PubMed] [Google Scholar]
- 40.Morimura H, Saindelle-Ribeaudeau F, Berson EL, Dryja TP. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat Genet. 1999;23:393–4. doi: 10.1038/70496. [DOI] [PubMed] [Google Scholar]
- 41.den Hollander AI, ten Brink JB, de Kok YJ. van SS, van den Born LI, van Driel MA, van De Pol DJ, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FP, Bergen AA. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23:217–21. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 42.Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG. pfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–1. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 43.Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66:1975–8. doi: 10.1086/302926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bareil C, Hamel CP, Delague V, Arnaud B, Demaille J, Claustres M. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet. 2001;108:328–34. doi: 10.1007/s004390100496. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz A, Kuehn MH, Andorf JL, Stone E, Hageman GS, Bok D. Genomic organization and mutation analysis of the gene encoding lecithin retinol acyltransferase in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:31–7. [PubMed] [Google Scholar]
- 46.Nishiguchi KM, Friedman JS, Sandberg MA, Swaroop A, Berson EL, Dryja TP. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc Natl Acad Sci USA. 2004;101:17819–24. doi: 10.1073/pnas.0408183101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuson M, Marfany G, Gonzalez-Duarte R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am J Hum Genet. 2004;74:128–38. doi: 10.1086/381055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riazuddin SA, Zulfiqar F, Zhang Q, Sergeev YV, Qazi ZA, Husnain T, Caruso R, Riazuddin S, Sieving PA, Hejtmancik JF. Autosomal recessive retinitis pigmentosa is associated with mutations in RP1 in three consanguineous Pakistani families. Invest Ophthalmol Vis Sci. 2005;46:2264–70. doi: 10.1167/iovs.04-1280. [DOI] [PubMed] [Google Scholar]
- 49.Zangerl B, Goldstein O, Philp AR, Lindauer SJ, Pearce-Kelling SE, Mullins RF, Graphodatsky AS, Ripoll D, Felix JS, Stone EM, Acland GM, Aguirre GD. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics. 2006;88:551–63. doi: 10.1016/j.ygeno.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abd El-Aziz MM, El-Ashry MF, Chan WM, Chong KL, Barragan I, Antinolo G, Pang CP, Bhattacharya SS. A novel genetic study of Chinese families with autosomal recessive retinitis pigmentosa. Ann Hum Genet. 2007;71:281–94. doi: 10.1111/j.1469-1809.2006.00333.x. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet. 2007;122:293–9. doi: 10.1007/s00439-007-0395-2. [DOI] [PubMed] [Google Scholar]
- 52.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008;40:1230–4. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.den Hollander AI, McGee TL, Ziviello C, Banfi S, Dryja TP, Gonzalez-Fernandez F, Ghosh D, Berson EL. A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2009;50:1864–72. doi: 10.1167/iovs.08-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, den Hollander AI, Moayedi Y, Abulimiti A, Li Y, Collin RW, Hoyng CB, Lopez I, Abboud EB, Al-Rajhi AA, Bray M, Lewis RA, Lupski JR, Mardon G, Koenekoop RK, Chen R. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet. 2009;84:380–7. doi: 10.1016/j.ajhg.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Escher P, Gouras P, Roduit R, Tiab L, Bolay S, Delarive T, Chen S, Tsai CC, Hayashi M, Zernant J, Merriam JE, Mermod N, Allikmets R, Munier FL, Schorderet DF. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum Mutat. 2009;30:342–51. doi: 10.1002/humu.20858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riazuddin SA, Iqbal M, Wang Y, Masuda T, Chen Y, Bowne S, Sullivan LS, Waseem NH, Bhattacharya S, Daiger SP, Zhang K, Khan SN, Riazuddin S, Hejtmancik JF, Sieving PA, Zack DJ, Katsanis N. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am J Hum Genet. 2010;86:805–12. doi: 10.1016/j.ajhg.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bandah-Rozenfeld D, Collin RW, Banin E, van den Born LI, Coene KL, Siemiatkowska AM, Zelinger L, Khan MI, Lefeber DJ, Erdinest I, Testa F, Simonelli F, Voesenek K, Blokland EA, Strom TM, Klaver CC, Qamar R, Banfi S, Cremers FP, Sharon D, den Hollander AI. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:199–208. doi: 10.1016/j.ajhg.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collin RW, Safieh C, Littink KW, Shalev SA, Garzozi HJ, Rizel L, Abbasi AH, Cremers FP, den Hollander AI, Klevering BJ, Ben-Yosef T. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;86:783–8. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dvir L, Srour G, Abu-Ras R, Miller B, Shalev SA, Ben-Yosef T. Autosomal-recessive early-onset retinitis pigmentosa caused by a mutation in PDE6G, the gene encoding the gamma subunit of rod cGMP phosphodiesterase. Am J Hum Genet. 2010;87:258–64. doi: 10.1016/j.ajhg.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmann T, Di Gioia SA, Rau I, Stohr H, Maksimovic NS, Corbo JC, Renner AB, Zrenner E, Kumaramanickavel G, Karlstetter M, Arsenijevic Y, Weber BH, Gal A, Rivolta C. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:376–81. doi: 10.1016/j.ajhg.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li L, Nakaya N, Chavali VR, Ma Z, Jiao X, Sieving PA, Riazuddin S, Tomarev SI, Ayyagari R, Riazuddin SA, Hejtmancik JF. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:400–9. doi: 10.1016/j.ajhg.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan MI, Kersten FF, Azam M, Collin RW, Hussain A, Shah ST, Keunen JE, Kremer H, Cremers FP, Qamar R, den Hollander AI. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2011;118:1444–8. doi: 10.1016/j.ophtha.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 63.Stone EM, Luo X, Heon E, Lam BL, Weleber RG, Halder JA, Affatigato LM, Goldberg JB, Sumaroka A, Schwartz SB, Cideciyan AV, Jacobson SG. Autosomal recessive retinitis pigmentosa caused by mutations in the MAK gene. Invest Ophthalmol Vis Sci. 2011;52:9665–73. doi: 10.1167/iovs.11-8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, Cideciyan AV, Jacobson SG, Sharon D. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011;88:207–15. doi: 10.1016/j.ajhg.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Estrada-Cuzcano A, Neveling K, Kohl S, Banin E, Rotenstreich Y, Sharon D, Falik-Zaccai TC, Hipp S, Roepman R, Wissinger B, Letteboer SJ, Mans DA, Blokland EA, Kwint MP, Gijsen SJ, van Huet RA, Collin RW, Scheffer H, Veltman JA, Zrenner E, den Hollander AI, Klevering BJ, Cremers FP. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet. 2012;90:102–9. doi: 10.1016/j.ajhg.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abu-Safieh L, Alrashed M, Anazi S, Alkuraya H, Khan AO, Al-Owain M, Al-Zahrani J, Al-Abdi L, Hashem M, Al-Tarimi S, Sebai MA, Shamia A, Ray-Zack MD, Nassan M, Al-Hassnan ZN, Rahbeeni Z, Waheeb S, Alkharashi A, Abboud E, Al-Hazzaa SA, Alkuraya FS. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013;23:236–47. doi: 10.1101/gr.144105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davidson AE, Schwarz N, Zelinger L, Stern-Schneider G, Shoemark A, Spitzbarth B, Gross M, Laxer U, Sosna J, Sergouniotis PI, Waseem NH, Wilson R, Kahn RA, Plagnol V, Wolfrum U, Banin E, Hardcastle AJ, Cheetham ME, Sharon D, Webster AR. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2013;93:321–9. doi: 10.1016/j.ajhg.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M, Holder GE, Robson AG, Moore AT, Plagnol V, Webster AR. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–14. doi: 10.1002/humu.22264. [DOI] [PubMed] [Google Scholar]
- 69.Nishiguchi KM, Tearle RG, Liu YP, Oh EC, Miyake N, Benaglio P, Harper S, Koskiniemi-Kuendig H, Venturini G, Sharon D, Koenekoop RK, Nakamura M, Kondo M, Ueno S, Yasuma TR, Beckmann JS, Ikegawa S, Matsumoto N, Terasaki H, Berson EL, Katsanis N, Rivolta C. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci USA. 2013;110:16139–44. doi: 10.1073/pnas.1308243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Siemiatkowska AM, van den Born LI, van Hagen PM, Stoffels M, Neveling K, Henkes A, Kipping-Geertsema M, Hoefsloot LH, Hoyng CB, Simon A, den Hollander AI, Cremers FP, Collin RW. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2013;120:2697–705. doi: 10.1016/j.ophtha.2013.07.052. [DOI] [PubMed] [Google Scholar]
- 71.Ajmal M, Khan MI, Neveling K, Khan YM, Azam M, Waheed NK, Hamel CP, Ben-Yosef T, De BE, Koenekoop RK, Collin RW, Qamar R, Cremers FP. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J Med Genet. 2014;51:444–8. doi: 10.1136/jmedgenet-2014-102316. [DOI] [PubMed] [Google Scholar]
- 72.El SS, Neuille M, Terray A, Orhan E, Condroyer C, Demontant V, Michiels C, Antonio A, Boyard F, Lancelot ME, Letexier M, Saraiva JP, Leveillard T, Mohand-Said S, Goureau O, Sahel JA, Zeitz C, Audo I. Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am J Hum Genet. 2014;94:625–33. doi: 10.1016/j.ajhg.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin ZB, Huang XF, Lv JN, Xiang L, Li DQ, Chen J, Huang C, Wu J, Lu F, Qu J. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat Commun. 2014;5:3517. doi: 10.1038/ncomms4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ali S, Khan SY, Naeem MA, Khan SN, Husnain T, Riazuddin S, Ayyagari R, Riazuddin S, Hejtmancik JF, Riazuddin SA. Phenotypic variability associated with the D226N allele of IMPDH1. Ophthalmology. 2015;122:429–31. doi: 10.1016/j.ophtha.2014.07.057. [DOI] [PubMed] [Google Scholar]
- 75.Wang F, Li H, Xu M, Li H, Zhao L, Yang L, Zaneveld JE, Wang K, Li Y, Sui R, Chen R. A homozygous missense mutation in NEUROD1 is associated with nonsyndromic autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2015;56:150–5. doi: 10.1167/iovs.14-15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haer-Wigman L, Newman H, Leibu R, Bax NM, Baris HN, Rizel L, Banin E, Massarweh A, Roosing S, Lefeber DJ, Zonneveld-Vrieling MN, Isakov O, Shomron N, Sharon D, den Hollander AI, Hoyng CB, Cremers FP, Ben-Yosef T. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum Mol Genet. 2015;24:3742–51. doi: 10.1093/hmg/ddv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Avila-Fernandez A, Perez-Carro R, Corton M, Lopez-Molina MI, Campello L, Garanto A, Fernandez-Sanchez L, Duijkers L, Lopez-Martinez MA, Riveiro-Alvarez R, Da Silva LR, Sanchez-Alcudia R, Martin-Garrido E, Reyes N, Garcia-Garcia F, Dopazo J, Garcia-Sandoval B, Collin RW, Cuenca N, Ayuso C. Whole-exome sequencing reveals ZNF408 as a new gene associated with autosomal recessive retinitis pigmentosa with vitreal alterations. Hum Mol Genet. 2015;24:4037–48. doi: 10.1093/hmg/ddv140. [DOI] [PubMed] [Google Scholar]
- 78.Buraczynska M, Wu W, Fujita R, Buraczynska K, Phelps E, Andreasson S, Bennett J, Birch DG, Fishman GA, Hoffman DR, Inana G, Jacobson SG, Musarella MA, Sieving PA, Swaroop A. Spectrum of mutations in the RPGR gene that are identified in 20% of families with X-linked retinitis pigmentosa. Am J Hum Genet. 1997;61:1287–92. doi: 10.1086/301646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mears AJ, Gieser L, Yan D, Chen C, Fahrner S, Hiriyanna S, Fujita R, Jacobson SG, Sieving PA, Swaroop A. Protein-truncation mutations in the RP2 gene in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 1999;64:897–900. doi: 10.1086/302298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, Ngu LH, Budny B. van WE, Gorden NT, Azhimi M, Thauvin-Robinet C, Veltman JA, Boink M, Kleefstra T, Cremers FP, van BH, de Brouwer AP. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–81. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.den Hollander AI, van Lith-Verhoeven JJ, Arends ML, Strom TM, Cremers FP, Hoyng CB. Novel compound heterozygous TULP1 mutations in a family with severe early-onset retinitis pigmentosa. Arch Ophthalmol. 2007;125:932–5. doi: 10.1001/archopht.125.7.932. [DOI] [PubMed] [Google Scholar]
- 82.North MA, Naggert JK, Yan Y, Noben-Trauth K, Nishina PM. Molecular characterization of TUB, TULP1, and TULP2, members of the novel tubby gene family and their possible relation to ocular diseases. Proc Natl Acad Sci USA. 1997;94:3128–33. doi: 10.1073/pnas.94.7.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iqbal M, Naeem MA, Riazuddin SA, Ali S, Farooq T, Qazi ZA, Khan SN, Husnain T, Riazuddin S, Sieving PA, Hejtmancik JF, Riazuddin S. Association of pathogenic mutations in TULP1 with retinitis pigmentosa in consanguineous Pakistani families. Arch Ophthalmol. 2011;129:1351–7. doi: 10.1001/archophthalmol.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU, Khan SN, Husnain T, Akram J, Hejtmancik JF, Riazuddin S. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis. 2010;16:511–7. [PMC free article] [PubMed] [Google Scholar]
- 85.Lathrop GM, Lalouel JM. Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–5. [PMC free article] [PubMed] [Google Scholar]
- 86.Schaffer AA, Gupta SK, Shriram K, Cottingham RW., Jr Avoiding recomputation in linkage analysis. Hum Hered. 1994;44:225–37. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 87.Katagiri S, Akahori M, Sergeev Y, Yoshitake K, Ikeo K, Furuno M, Hayashi T, Kondo M, Ueno S, Tsunoda K, Shinoda K, Kuniyoshi K, Tsurusaki Y, Matsumoto N, Tsuneoka H, Iwata T. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS One. 2014;9:e108721. doi: 10.1371/journal.pone.0108721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet. 1998;18:174–6. doi: 10.1038/ng0298-174. [DOI] [PubMed] [Google Scholar]
- 89.Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier JL, Munnich A, Rozet JM, Kaplan J. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23:306–17. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 90.Beryozkin A, Zelinger L, Bandah-Rozenfeld D, Shevach E, Harel A, Storm T, Sagi M, Eli D, Merin S, Banin E, Sharon D. Identification of mutations causing inherited retinal degenerations in the israeli and palestinian populations using homozygosity mapping. Invest Ophthalmol Vis Sci. 2014;55:1149–60. doi: 10.1167/iovs.13-13625. [DOI] [PubMed] [Google Scholar]
- 91.Paloma E, Hjelmqvist L, Bayes M, Garcia-Sandoval B, Ayuso C, Balcells S, Gonzalez-Duarte R. Novel mutations in the TULP1 gene causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:656–9. [PubMed] [Google Scholar]
- 92.Gu S, Lennon A, Li Y, Lorenz B, Fossarello M, North M, Gal A, Wright A. Tubby-like protein-1 mutations in autosomal recessive retinitis pigmentosa. Lancet. 1998;351:1103–4. doi: 10.1016/S0140-6736(05)79384-3. [DOI] [PubMed] [Google Scholar]
- 93.Gonzalez-del PM, Borrego S, Barragan I, Pieras JI, Santoyo J, Matamala N, Naranjo B, Dopazo J, Antinolo G. Mutation screening of multiple genes in Spanish patients with autosomal recessive retinitis pigmentosa by targeted resequencing. PLoS One. 2011;6:e27894. doi: 10.1371/journal.pone.0027894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang H, Wang X, Zou X, Xu S, Li H, Soens ZT, Wang K, Li Y, Dong F, Chen R, Sui R. Comprehensive Molecular Diagnosis of a Large Chinese Leber Congenital Amaurosis Cohort. Invest Ophthalmol Vis Sci. 2015;56:3642–55. doi: 10.1167/iovs.14-15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Glockle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, Weisschuh N, Bernd A, Rudolph G, Schubach M, Poloschek C, Zrenner E, Biskup S, Berger W, Wissinger B, Neidhardt J. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet. 2014;22:99–104. doi: 10.1038/ejhg.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.den Hollander AI, Lopez I, Yzer S, Zonneveld MN, Janssen IM, Strom TM, Hehir-Kwa JY, Veltman JA, Arends ML, Meitinger T, Musarella MA, van den Born LI, Fishman GA, Maumenee IH, Rohrschneider K, Cremers FP, Koenekoop RK. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci. 2007;48:5690–8. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 97.Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, Bowles S, Khan A, Salvo J, Jacobson SG, Iannaccone A, Wang F, Birch D, Heckenlively JR, Fishman GA, Traboulsi EI, Li Y, Wheaton D, Koenekoop RK, Chen R. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50:674–88. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Y, Wang H, Peng J, Gibbs RA, Lewis RA, Lupski JR, Mardon G, Chen R. Mutation survey of known LCA genes and loci in the Saudi Arabian population. Invest Ophthalmol Vis Sci. 2009;50:1336–43. doi: 10.1167/iovs.08-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khan AO, Bergmann C, Eisenberger T, Bolz HJA. TULP1 founder mutation, p.Gln301*, underlies a recognisable congenital rod-cone dystrophy phenotype on the Arabian Peninsula. Br J Ophthalmol. 2015;99:488–92. doi: 10.1136/bjophthalmol-2014-305836. [DOI] [PubMed] [Google Scholar]
- 100.Hebrard M, Manes G, Bocquet B, Meunier I, Coustes-Chazalette D, Herald E, Senechal A, Bolland-Auge A, Zelenika D, Hamel CP. Combining gene mapping and phenotype assessment for fast mutation finding in non-consanguineous autosomal recessive retinitis pigmentosa families. Eur J Hum Genet. 2011;19:1256–63. doi: 10.1038/ejhg.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, Wang DY, Au ED, Sims KB, Sweetser DA, Fulton AB, Liu Q, Wiggs JL, Gai X, Pierce EA. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2015;17:253–61. doi: 10.1038/gim.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kannabiran C, Singh H, Sahini N, Jalali S, Mohan G. Mutations in TULP1, NR2E3, and MFRP genes in Indian families with autosomal recessive retinitis pigmentosa. Mol Vis. 2012;18:1165–74. [PMC free article] [PubMed] [Google Scholar]
- 103.Boulanger-Scemama E, El SS, Demontant V, Condroyer C, Antonio A, Michiels C, Boyard F, Saraiva JP, Letexier M, Souied E, Mohand-Said S, Sahel JA, Zeitz C, Audo I. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J Rare Dis. 2015;10:85. doi: 10.1186/s13023-015-0300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guo Y, Prokudin I, Yu C, Liang J, Xie Y, Flaherty M, Tian L, Crofts S, Wang F, Snyder J, Donaldson C, Abdel-Magid N, Vazquez L, Keating B, Hakonarson H, Wang J, Jamieson RV. Advantage of Whole Exome Sequencing over Allele-specific and Targeted Segment Sequencing, in Detection of Novel TULP1 Mutation in Leber Congenital Amaurosis. Ophthalmic Genet. 2015 doi: 10.3109/13816810.2014.886269. [DOI] [PubMed] [Google Scholar]
- 105.Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, Bowne SJ, Sullivan LS, Luo H, Zhao L, Wang X, Zaneveld JE, Salvo JS, Siddiqui S, Mao L, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Wen C, Flagg K, Ferreyra H, Pei J, Khan A, Ren H, Wang K, Lopez I, Qamar R, Zenteno JC. yala-Ramirez R, Buentello-Volante B, Fu Q, Simpson DA, Li Y, Sui R, Silvestri G, Daiger SP, Koenekoop RK, Zhang K, Chen R. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133:331–45. doi: 10.1007/s00439-013-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McKibbin M, Ali M, Mohamed MD, Booth AP, Bishop F, Pal B, Springell K, Raashid Y, Jafri H, Inglehearn CF. Genotype-phenotype correlation for leber congenital amaurosis in Northern Pakistan. Arch Ophthalmol. 2010;128:107–13. doi: 10.1001/archophthalmol.2010.309. [DOI] [PubMed] [Google Scholar]
- 107.Ajmal M, Khan MI, Micheal S, Ahmed W, Shah A, Venselaar H, Bokhari H, Azam A, Waheed NK, Collin RW, den Hollander AI, Qamar R, Cremers FP. Identification of recurrent and novel mutations in TULP1 in Pakistani families with early-onset retinitis pigmentosa. Mol Vis. 2012;18:1226–37. [PMC free article] [PubMed] [Google Scholar]
- 108.Kondo H, Qin M, Mizota A, Kondo M, Hayashi H, Hayashi K, Oshima K, Tahira T, Hayashi K. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Invest Ophthalmol Vis Sci. 2004;45:4433–9. doi: 10.1167/iovs.04-0544. [DOI] [PubMed] [Google Scholar]
- 109.Chen Y, Zhang Q, Shen T, Xiao X, Li S, Guan L, Zhang J, Zhu Z, Yin Y, Wang P, Guo X, Wang J, Zhang Q. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2013;54:4351–7. doi: 10.1167/iovs.13-11606. [DOI] [PubMed] [Google Scholar]
- 110.Jacobson SG, Cideciyan AV, Huang WC, Sumaroka A, Roman AJ, Schwartz SB, Luo X, Sheplock R, Dauber JM, Swider M, Stone EM. TULP1 mutations causing early-onset retinal degeneration: preserved but insensitive macular cones. Invest Ophthalmol Vis Sci. 2014;55:5354–64. doi: 10.1167/iovs.14-14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Singh HP, Jalali S, Narayanan R, Kannabiran C. Genetic analysis of Indian families with autosomal recessive retinitis pigmentosa by homozygosity screening. Invest Ophthalmol Vis Sci. 2009;50:4065–71. doi: 10.1167/iovs.09-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roosing S, van den Born LI, Hoyng CB, Thiadens AA, De BE, Collin RW, Koenekoop RK, Leroy BP, van Moll-Ramirez N, Venselaar H, Riemslag FC, Cremers FP, Klaver CC, den Hollander AI. Maternal uniparental isodisomy of chromosome 6 reveals a TULP1 mutation as a novel cause of cone dysfunction. Ophthalmology. 2013;120:1239–46. doi: 10.1016/j.ophtha.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 113.Maria M, Ajmal M, Azam M, Waheed NK, Siddiqui SN, Mustafa B, Ayub H, Ali L, Ahmad S, Micheal S, Hussain A, Shah ST, Ali SH, Ahmed W, Khan YM, den Hollander AI, Haer-Wigman L, Collin RW, Khan MI, Qamar R, Cremers FP. Homozygosity mapping and targeted sanger sequencing reveal genetic defects underlying inherited retinal disease in families from pakistan. PLoS One. 2015;10:e0119806. doi: 10.1371/journal.pone.0119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Abbasi AH, Garzozi HJ, Ben-Yosef T. A novel splice-site mutation of TULP1 underlies severe early-onset retinitis pigmentosa in a consanguineous Israeli Muslim Arab family. Mol Vis. 2008;14:675–82. [PMC free article] [PubMed] [Google Scholar]
- 115.Mataftsi A, Schorderet DF, Chachoua L, Boussalah M, Nouri MT, Barthelmes D, Borruat FX, Munier FL. Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:5160–7. doi: 10.1167/iovs.06-1013. [DOI] [PubMed] [Google Scholar]
- 116.Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, Bieg A, Gliem M, Charbel IP, Holz FG, Baig SM, Hellenbroich Y, Galvez A, Platzer K, Wollnik B, Laddach N, Ghaffari SR, Rafati M, Botzenhart E, Tinschert S, Borger D, Bohring A, Schreml J, Kortge-Jung S, Schell-Apacik C, Bakur K, Al-Aama JY, Neuhann T, Herkenrath P, Nurnberg G, Nurnberg P, Davis JS, Gal A, Bergmann C, Lorenz B, Bolz HJ. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013;8:e78496. doi: 10.1371/journal.pone.0078496. [DOI] [PMC free article] [PubMed] [Google Scholar]