

Abstract
The role of phenotypic assessment in drug discovery is discussed, along with the discovery and development of TOPAMAX (topiramate), a billion-dollar molecule for the treatment of epilepsy and migraine.
Keywords: Drug discovery, phenotype, topiramate, epilepsy, anticonvulsant
The main objective of discovering and developing new drugs is to help people with diseases and medical disorders, who are not yet being treated effectively. Unmet medical need! Do you think that sufferers really care about the means by which the drug was discovered or about a drug’s underlying mechanisms of action? So, one may wonder: Why have drug research and development (R&D) organizations made such a big deal about knowing the mechanism(s) of action for new drug candidates with great potential to improve and prolong the lives of patients? Mechanism is nice to know, and can certainly be useful, but it may not always be essential. To be sure, knowledge about mechanism of action and the molecular target can facilitate key aspects of the drug development process, while somewhat derisking late-stage clinical trials. Indeed, having surrogate biomarkers, understanding on-target vs off-target effects, using genetic tools, and applying directed imaging can be quite helpful. However, we should also avoid erecting artificial barriers to the advancement of new, nonprotein-based drug candidates that can ultimately be proven both safe and effective. Balance in thinking and approach is important. From this perspective, I am pleased to see that phenotypic assessment is making a comeback in drug discovery, mainly thanks to new methods for high-capacity screening that are serving to correct a long-standing imbalance.
Since my retirement from a 35-year career in Johnson & Johnson (J&J) pharmaceutical companies, I have consulted with several biopharma companies. One case stood out in particular (company unnamed) because it grated against my drug discovery sensibilities. An exciting oncology drug with high potential for treating difficult cancers, such as neuroblastoma, was advanced into the preclinical development pipeline, and a suitable partner was being sought to facilitate clinical development. More than 15 different, medium-to-large-size pharma companies were contacted, but the argument came back, time and again, that a clear-cut mechanism of action had to be envisioned as a prerequisite for advancing a joint-development project. Pity the cancer patients for whom this new drug might have ended up being a godsend.
During my early days at Johnson & Johnson (at the subsidiary McNeil Laboratories), in the mid-1970s, we generally hoped to discover new drug candidates through bioassays in whole animals and with isolated tissues/organs, such as the perfused heart. These tests relied on phenotypic assessment, although no one used this specific term at that time. We depended on insightful pharmacologists to develop predictive assays for identifying chemical compounds with properties that might lead to potential drugs for therapeutic areas of interest. Thus, much new drug exploration relied on low-capacity, broad screening of new chemical entities (NCEs). Drug design in that era was generally predicated on copying structural features present in existing drugs, natural hormones, or known neurotransmitters. There were few alternative approaches.
Demise of Phenotypic Assessment
The modern age of physiological receptors probably began with the report from Lefkowitz et al. in 1972 regarding the cardiac beta-adrenergic receptor.1 But big news arrived from Pert and Snyder in 1973, when they identified opiate receptors in the brain, along with a meaningful binding-vs-activity relationship for ligand molecules.2,3 Studies also revealed a surprising sodium-ion modulatory effect on ligand binding that could differentiate between opiate agonists and antagonists.4,5 These key advances, especially the first verified receptor in the central nervous system (CNS), with a very credible ligand structure–activity relationship (SAR), represented a turning-point for receptor-based pharmacology and compound screening. During the next 10 years, the field of receptology burgeoned and spread throughout the entire pharmaceutical industry. Of course, we and every other drug R&D organization jumped on the biochemical pharmacology bandwagon, seeking to discover new drugs by means of in vitro receptor-binding assays. Heady, powerful stuff!
Over the decades, more detailed molecular biology led drug researchers to prefer a drug discovery approach based on discrete molecular targets. The Human Genome Project contributed enormously to this paradigm by revealing all of those human genes. We also had the focus on combinatorial chemistry and high-throughput screening of massive chemical libraries; target identification, target validation, etc.; genomics, proteomics, metabolomics, etc. As hubris would have it, most drug discoverers rallied hard around this architecture, to the near-exclusion of phenotypic assessment, and molecular mechanism of action (MMOA) became the watchword. Although such specifically directed R&D has led to numerous clinical candidates, as well as marketed drugs, analysis has indicated an unfortunate short-fall in delivery.6−9 A wakeup call was overdue.
Re-emergence of Phenotypic Assessment
At the beginning of the 21st century, I felt like a lumbering dinosaur in the halls of the company when arguing with hopelessly biased management that phenotypic assessment has a valid place in the drug discovery toolbox. Perhaps, my rewarding experience with the discovery and development of Topamax (topiramate) had colored my opinion, but this approach seemed well grounded in my mind from simply a logical standpoint. Moreover, I was strongly influenced by Dr. Paul Janssen, probably the greatest drug discoverer of all time, and Nobel Laureate Sir James Black, who were closely associated with J&J for many years. Their drug discovery philosophy emphasized the importance of using bioassays that predictably translate to the human disease condition of interest, which is really the foundation of phenotypic assessment. Given that drug R&D is such a complex and difficult process, it was clear to me that one should take advantage of all possible avenues to solve the problem at hand, making use of every available tool in the toolbox. To paraphrase a view put forth by psychologist Maslow, “When the only tool you have is a hammer, you tend to see every problem as a nail”.10
Molecular target-based projects are meant to produce drugs with a well-defined therapeutic application, arising from action at the specific target. However, the picture can become more complex, with therapeutic utilities that depart from the original intent. Gleevec (imatinib) might serve as one example.11,12 This drug was first advanced clinically as a selective tyrosine-kinase inhibitor for BCR-ABL to treat chronic myelogenous leukemia (CML) in patients who are Philadelphia chromosome-positive. Subsequent clinical studies found usefulness for other cancers, such as gastrointestinal stromal tumors (GIST), from targeting the tyrosine kinase domain of c-KIT and platelet-derived growth factor receptor (PDGF-R). Consequently, imatinib,11,12 as well as other “selective” kinase inhibitors,13 can exert a certain degree of polypharmacology.14,15 Other useful drugs, originally based on a particular molecular target or mode of action, have turned out to exhibit more complex pharmacology due to ancillary actions,14 which has led to a surge in drug repurposing or repositioning.13,16 Nonselective drugs with discrete profiles can have advantages in certain therapeutic areas with complex pharmacologies, such as CNS disorders.17 In fact, many diseases and disorders are not necessarily connected with a single biomolecular problem, but emanate from defects or deficiencies in a vast network of biomolecules and their complex interactions.18 Some pertinent examples include Alzheimer’s disease, asthma, migraine, arthritis, cancers, and heart disease. As a fundamental principle, physiology and pathophysiology are dependent on interconnected systems with redundant pathways and functions.18,19
Fortunately, phenotypic assessment is seeing a resurgence of interest, such that a healthy balance is being restored to drug discovery.6−8 While in vivo screening is usually available, it suffers from very limited throughput, which is especially troublesome in the early stages of drug discovery. However, newer techniques have contributed to a renaissance. There are cell-based phenotypic assays with primary or immortalized cell lines,7,8 or with various types of stem cell lines (pluripotent, induced, patient-derived).7,20−23 Reporter cell lines and single-cell analysis methodology, including molecular phenotyping, have conferred efficiency and accuracy to this arena.24−26 Human induced-pluripotent stem cells from specific patients, or patient-derived primary cells, can be used to establish high-throughput assays to screen effectively for phenotypes of interest. It is important that any phenotypic assays being developed are predictive of translation into the clinic.23 This drug discovery approach is nicely illustrated by the identification of small-molecule SMN2 splicing modulators for treating spinal muscular atrophy (SMA), which employed high-throughput screening with a motor neuron cell line expressing an SMN2 minigene reporter.27 Another facet is the application of high-capacity screening in very small animals, such as zebrafish and C. elegans.7,28−30 Phenotypic screening methods can also be used to identify new drug targets and new indications for existing drugs.7,31 The pinpointing of new molecular targets from phenotypic assays has greatly benefited from progress in chemical proteomics.32,33 In summary, technological advances in the field of phenotypic assessment offer new strategies for successful drug discovery, especially in the case of nonprotein-based medicines (i.e., small-molecule drugs).
Genesis of Topiramate
Certain classes of therapeutic
agents are more readily discovered by phenotypic assessment, and anticonvulsant
drugs are legendary in this regard. Pharmacologist Joe Gardocki and
I discovered topiramate (McN-4853, 1) by using standard
in vivo models that are highly predictive of clinical efficacy in
humans.34 In fact, compounds with demonstrable
activity in the Maximal Electroshock Seizure (MES) test in mice and
rats have been considered to present a 95% chance of effectiveness
in treating at least a sizable subgroup of epileptic patients. Since
epilepsy is a multifactorial disorder with limited understanding about
its etiology (i.e., the origin of seizures is idiopathic), drug discovery
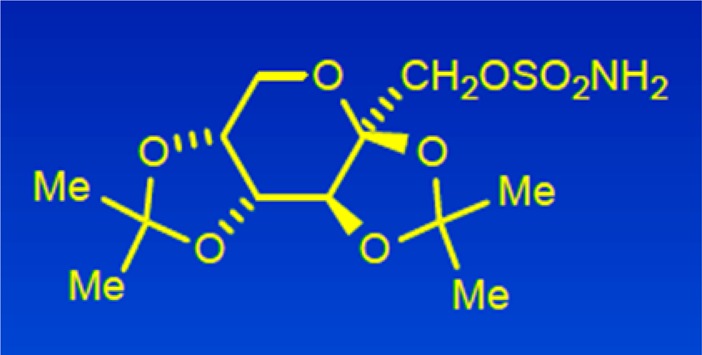
in this field has often taken a phenotypic route.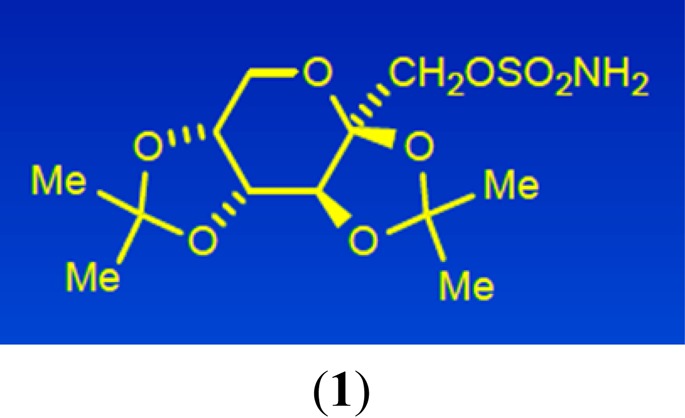
Topiramate (1) was first synthesized as a chemical intermediate for another purpose, in 1979. Several grams were submitted to the corporate compound library, and it was assayed in whole animal tests for different potential therapies. The MES test was not part of the standard stable of animal assays because the company had little commercial interest in developing a medicine for epilepsy. However, after reviewing the compound submission sheet, Joe became intrigued with the chemical structure, especially the SO2NH2 moiety, and selected 1 for testing in the MES test in mice. The anticonvulsant activity became apparent right away.
In the world of anticonvulsant chemotypes, topiramate is a rather unusual structure, a sulfamate derivative of the monosaccharide fructose. Of course, the polar hydroxyl groups of fructose are tied up, so to speak, as ketals of acetone, which facilitates its penetration into the CNS. Given this novel structure for an anticonvulsant agent, we thought that there might be a novel mode of action with the ability to treat epileptic patients unresponsive to the current therapies, which constituted a large percentage of the afflicted population.
After assembling a large package of supporting data, we presented the compound to management for advancement into preclinical development, in 1981. The reception was less than enthusiastic because epilepsy therapy was not a company priority, given the lack of a commercial franchise in the neurology space. We enlisted the assistance of Dr. Harvey Kupferberg at the National Institute of Neurological Disorders and Stroke (NINDS), a division of the National Institutes of Health (NIH), for support. His drug discovery unit was involved in pharmacological testing to identify and characterize new anticonvulsant agents for treating epilepsy. Their workup not only corroborated our results but also ranked topiramate among the top-ten antiseizure compounds that they had found over many years of study.
Armed with this information, Joe, Harvey, and I revisited the subject in 1983 with R&D management, and they were sufficiently impressed to place topiramate into preclinical development, at 25th position on a list of 25 development projects. Topiramate was easily synthesized, had an excellent pharmacological profile, exhibited low toxicity, and had drug-like properties. The attrition of other development projects over the years caused topiramate to rise in priority, such that it entered into the top ten. An Investigational New Drug (IND) application was filed with the U.S. Food and Drug Administration (FDA) in 1986 and marketing approval from the FDA was received at the end of 1996. This new drug was effective in epileptic patients with generalized tonic–clonic and/or complex partial seizures. Topamax (topiramate) was first introduced to the marketplace as an antiepileptic drug for adjunctive therapy with other agents, and its use in monotherapy was approved later. Clinical trials for other potential indications were performed, which led to the finding that topiramate is very useful for migraine prophylaxis. Peak annual sales of Topamax (topiramate) reached 2.7 billion U.S. dollars.35
The U.S. Patent for topiramate expired in 2009, and annual sales at J&J dropped off sharply, as was expected. Interestingly, there had been numerous postmarketing reports of useful anti-obesity activity, which led VIVUS, Inc., to create a combination product containing topiramate and phentermine, in an extended-release formulation. In 2012, the pharmaceutical world was shocked when the FDA conferred marketing approval to this drug combination, known as Qsymia, for chronic weight-loss management in adults. That was the first prescription anti-obesity drug to enter the U.S. market in decades.
In the realm of mechanism of action, topiramate can modulate voltage-gated sodium channels, enhance γ-aminobutyric acid (GABA) at GABA-A receptors, affect the AMPA/kainate subtype of glutamate receptors, and attenuate voltage-gated R-type calcium channels.36−38 Topiramate also inhibits some isozymes of carbonic anhydrase (CA), such as CA-II and CA-IV.37,39 Although topiramate blocked voltage-sensitive sodium channels at therapeutically relevant concentrations, it was less effective than the anticonvulsants phenytoin and carbamazepine, eliminating Na-channel blockade as a primary mechanism. Topiramate potentiated chloride-ion currents evoked by GABA-A receptors at clinically relevant concentrations, similar to the effect of diazepam, but topiramate’s effect was not blocked by the benzodiazepine ligand flumazenil. Unlike barbiturates, topiramate does not affect channel-open time. These observations support direct modulation of GABA’s action on GABA-A receptors, in a nonbenzodiazepine or nonbarbiturate manner. Topiramate was found to be a negative modulator of kainate-evoked currents on AMPA receptors, but it did not affect N-methyl-d-aspartate (NMDA)-evoked currents. The body of evidence indicates that topiramate has a constellation of neuronal actions that contribute to its antiseizure and antimigraine activity. By virtue of attenuating the excitability of brain neuronal pathways, topiramate can be classified as a “neurostabilizer.”
Innovation with topiramate relates to two aspects: its novel chemical structure in the realm of broad-spectrum anticonvulsants and, importantly, its action as a neurostabilizer, which is a class of drugs that was essentially unrecognized prior to 1990. In addition to their therapeutic utility for treating epilepsy, anticonvulsants of this type often turn out to be effective medications for various neurological disorders, such as migraine (topiramate; valproic acid), bipolar disorder (valproic acid; lamotrigine), neuropathic pain and diabetic neuropathy (gabapentin; pregabalin), postherpetic neuraglia (gabapentin; pregabalin), and fibromyalgia (pregabalin).40 These drugs have benefited many patients, and each one achieved peak annual sales of more than one billion U.S. dollars. It is important to note that their nonepilepsy medical uses were mainly determined in human clinical trials, that is, through human phenotypic assessment.
Acknowledgments
I am very grateful to many excellent colleagues who helped make Topamax (topiramate) a great success, and to Johnson & Johnson for support and encouragement.
Glossary
ABBREVIATIONS
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- CNS
central nervous system
- GABA
γ-aminobutyric acid
- J&J
Johnson & Johnson
- MES
maximal electroshock seizure
- R&D
research and development
The author declares no competing financial interest.
References
- Lefkowitz R. J.; Haber E.; O’Hara D. Identification of the cardiac beta-adrenergic receptor protein: solubilization and purification by affinity chromatography. Proc. Natl. Acad. Sci. U. S. A. 1972, 69, 2828–2832. 10.1073/pnas.69.10.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert C. B.; Snyder S. H. Opiate receptor: demonstration in nervous tissue. Science 1973, 179 (4077), 1011–1014. 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- Pert C. B.; Snyder S. H. Properties of opiate-receptor binding in rat brain. Proc. Natl. Acad. Sci. U. S. A. 1973, 70, 2243–2247. 10.1073/pnas.70.8.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert C. B.; Snyder S. H. Opiate receptor binding of agonists and antagonists affected differentially by sodium. Mol. Pharmacol. 1974, 10, 868–879. [Google Scholar]
- Simon E. J.; Hiller J. M.; Groth J.; Edelman I. Further properties of stereospecific opiate binding sites in rat brain: on the nature of the sodium effect. J. Pharmacol. Exp. Ther. 1975, 192, 531–537. [PubMed] [Google Scholar]
- Priest B. T.; Erdemli G. Phenotypic screening in the 21st century. Front. Pharmacol. 2014, 5, 1–2. 10.3389/fphar.2014.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Thorne N.; McKew J. C. Phenotypic screens as a renewed approach for drug discovery. Drug Discovery Today 2013, 18, 1067–1073. 10.1016/j.drudis.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior M.; Chiruta C.; Currais A.; Goldberg J.; Ramsey J.; Dargusch R.; Maher P. A.; Schubert D. Back to the future with phenotypic screening. ACS Chem. Neurosci. 2014, 5, 503–513. 10.1021/cn500051h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney D. C.; Anthony J. How were new medicines discovered?. Nat. Rev. Drug Discovery 2011, 10, 507–519. 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- Maslow A. H.The Psychology of Science; Harper & Row: New York, 1966; p 15 [qv. https://en.wikipedia.org/wiki/Abraham_Maslow#Maslow.27s_hammer; accessed in March 2016]. [Google Scholar]
- Capdeville R.; Buchdunger E.; Zimmermann J.; Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discovery 2002, 1, 493–502. 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Druker B. J.; Lydon N. B. Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Invest. 2000, 105, 3–7. 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J.-U. Polypharmacology–foe or friend?. J. Med. Chem. 2013, 56, 8955–8971. 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Poly-pharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- The inhibitors target the ATP-binding site of protein kinases, 518 of which exist in the human kinome (see: https://en.wikipedia.org/wiki/kinome; accessed in March 2016).
- Ashburn T. T.; Thor K. B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discovery 2004, 3, 673–683. 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discovery 2004, 3, 353–359. 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Jalencas X.; Mestres J. On the origins of drug polypharmacology. MedChemComm 2013, 4, 80–87. 10.1039/C2MD20242E. [DOI] [Google Scholar]
- Lukaszewicz A. I.; McMillan M. K.; Kahn M. Small molecules and stem cells. Potency and lineage commitment: the new quest for the fountain of youth. J. Med. Chem. 2010, 53, 3439–3453. 10.1021/jm901361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan P.; Gaskell T.; Moens N.; Culley O. J.; Hansen D.; Gervasio M. K. R.; Yeap Y. J.; Danovi D. Human pluripotent stem cells on artificial microenviroments: a high content perspective. Front. Pharmacol. 2014, 5 (150), 1–14. 10.3389/fphar.2014.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Munoz A. L.; Minter R. R.; Rust S. J. Phenotypic screening: the future of antibody discovery. Drug Discovery Today 2016, 21, 150–156. 10.1016/j.drudis.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Vincent F.; Loria P.; Pregel M.; Stanton R.; Kitching L.; Nocka K.; Doyannas R.; Steppan C.; Gilbert A.; Schroeter T.; Peakman M.-C. Developing predictive assays: the phenotypic screening ″rule of 3″. Sci. Transl. Med. 2015, 7 (293), 1–5. 10.1126/scitranslmed.aab1201. [DOI] [PubMed] [Google Scholar]
- Kang J.; Hsu C.-H.; Wu Q.; Liu S.; Coster A. D.; Posner B. A.; Altschuler S. J.; Wu L. F. Improving drug discovery with high-content phenotypic screens by systematic selection of reported cell lines. Nat. Biotechnol. 2016, 34, 70–77. 10.1038/nbt.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath J. R.; Ribas A.; Mischel P. S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discovery 2016, 15, 204–216. 10.1038/nrd.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. D.; Kung E.; Boess F.; Certa U.; Ebeling M. Pathway reporter genes define molecular phenotypes of human cells. BMC Genomics 2015, 16, 1–10. 10.1186/s12864-015-1532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino J.; Swalley S. E.; Song C.; Cheung A. K.; Shu L.; Zhang X.; Van Hoosear M.; Shin Y.; Chin D. N.; Keller C. G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- Rihel J.; Prober D. A.; Arvanites A.; Lam K.; Zimmerman S.; Jang S.; Haggarty S. J.; Kokel D.; Rubin L. L.; Peterson R. T.; Schier A. F. Zebrafish behavioral profiling links to biological targets and rest/wake regulation. Science 2010, 327, 348–351. 10.1126/science.1183090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni G.; Lakhani P.; Kokel D. Discovering novel neuroactive drugs through high-throughput behavior-based chemical screening in the zebrafish. Front. Pharmacol. 2014, 5 (153), 1–7. 10.3389/fphar.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll M. N.; Gendelev L.; Keiser M. J.; Kokel D. Leveraging large-scale behavioral profiling in zebrafish to explore neuroactive pharmacology. ACS Chem. Biol. 2016, 11, 842–849. 10.1021/acschembio.5b00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle M.; Jenkins J. L. Identifying compound efficacy targets in phenotypic drug discovery. Drug Discovery Today 2016, 21, 82–89. 10.1016/j.drudis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Rix U.; Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009, 5, 616–624. 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- Kotz J. Phenotypic screening, take two. SciBX: Science-Business eXchange 2012, 5 (15), 1–3. 10.1038/scibx.2012.380. [DOI] [Google Scholar]
- Maryanoff B. E. Pharmaceutical “gold” from neurostabilizing agents: topiramate and successor molecules. J. Med. Chem. 2009, 52, 3431–3440. 10.1021/jm900141j. [DOI] [PubMed] [Google Scholar]
- The 2008 Johnson & Johnson Annual Report (http://www.investor.jnj.com/annual-reports.cfm, p 23; accessed in April 2016).
- Shank R. P.; Gardocki J. F.; Streeter A. J.; Maryanoff B. E. An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia 2000, 41, S3–S9. 10.1111/j.1528-1157.2000.tb02163.x. [DOI] [PubMed] [Google Scholar]
- Porter R. J.; Dhir A.; Macdonald R. L.; Rogawski M. A. Mechanisms of action of antiseizure drugs. Handbook of Clinical Neurology, Epilepsy Part II 2012, 108, 663–681. 10.1016/B978-0-444-52899-5.00021-6. [DOI] [PubMed] [Google Scholar]
- Zamponi G. W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discovery 2016, 15, 19–34. 10.1038/nrd.2015.5. [DOI] [PubMed] [Google Scholar]
- Maryanoff B. E.; McComsey D. F.; Costanzo M. J.; Hochman C.; Smith-Swintosky V.; Shank R. P. Comparison of sulfamate and sulfamide groups for the inhibition of carbonic anhydrase-II by using topiramate as a structural platform. J. Med. Chem. 2005, 48, 1941–1947. 10.1021/jm040124c. [DOI] [PubMed] [Google Scholar]
- Landmark C. J. Antiepileptic drugs in non-epilepsy disorders: relations between mechanisms of action and clinical efficacy. CNS Drugs 2008, 22, 27–47. 10.2165/00023210-200822010-00003. [DOI] [PubMed] [Google Scholar]