

Abstract

ROMK, the renal outer medullary potassium channel, is involved in potassium recycling at the thick ascending loop of Henle and potassium secretion at the cortical collecting duct in the kidney nephron. Because of this dual site of action, selective inhibitors of ROMK are expected to represent a new class of diuretics/natriuretics with superior efficacy and reduced urinary loss of potassium compared to standard-of-care loop and thiazide diuretics. Following our earlier work, this communication will detail subsequent medicinal chemistry endeavors to further improve lead selectivity against the hERG channel and preclinical pharmacokinetic properties. Pharmacological assessment of highlighted inhibitors will be described, including pharmacodynamic studies in both an acute rat diuresis/natriuresis model and a subchronic blood pressure model in spontaneous hypertensive rats. These proof-of-biology studies established for the first time that the human and rodent genetics accurately predict the in vivo pharmacology of ROMK inhibitors and supported identification of the first small molecule ROMK inhibitor clinical candidate, MK-7145.
Keywords: ROMK, diuretics, natriuresis, hypertension, heart failure, MK-7145
The renal outer medullary potassium channel (ROMK, encoded by the KCNJ1 gene) is an inward rectifying potassium channel (Kir1.1) that plays a key role in renal salt recycling and potassium homeostasis.1,2 In the kidney nephron, ROMK is expressed at both the thick ascending loop of Henle (TALH) and the cortical collecting duct (CCD).3 At the TALH, ROMK facilitates potassium recycling across the luminal membrane, which is critical for the proper function of the furosemide-sensitive Na+/K+/2Cl– cotransporter. At the CCD, ROMK is involved in potassium secretion that is tightly coupled to sodium uptake through the amiloride-sensitive epithelial sodium channel (ENaC).4,5 This dual site of function suggests that ROMK inhibitors may provide superior diuretic/natriuretic efficacy with reduced urinary loss of potassium compared to standard-of-care loop and thiazide diuretics. This hypothesis is supported by a growing body of evidence in both human6,7 and rodent8,9 genetics. However, the human pharmacology of ROMK is unexplored largely due to lack of selective ROMK inhibitors. Thus, suitable inhibitors are needed for further understanding of ROMK pharmacology, which may ultimately lead to development of a new class of diuretics for the treatment of hypertension and edematous states such as heart failure.10
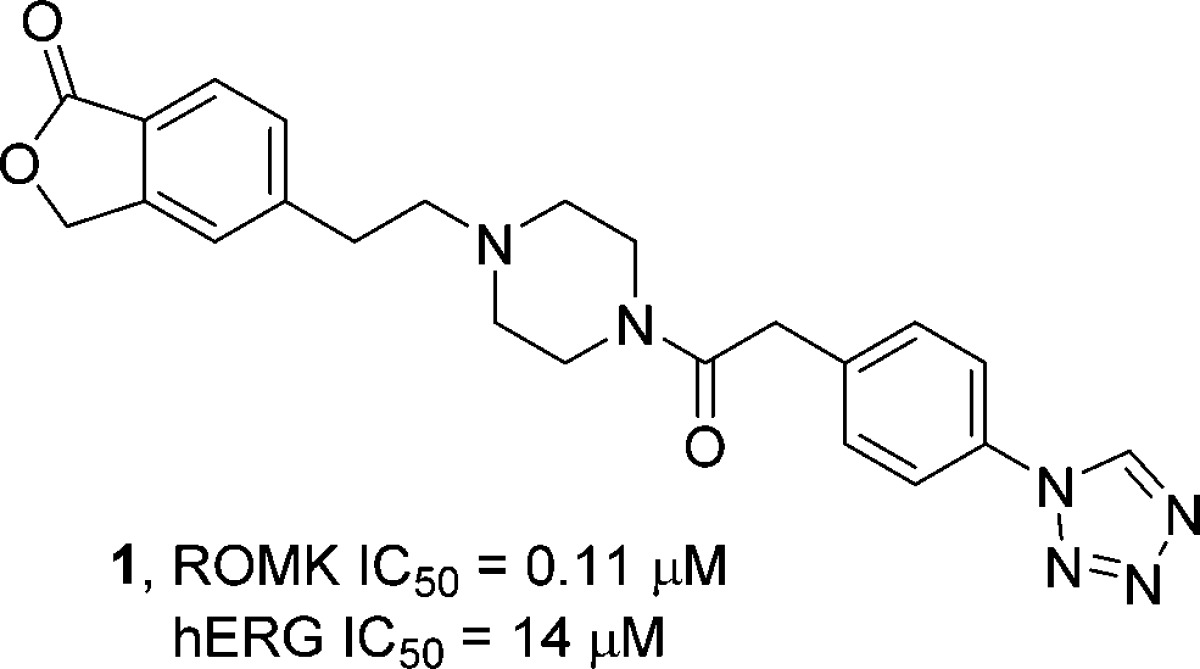
Recently, we have disclosed the development of a new class of small molecule ROMK inhibitors exemplified by compound 1.11 Although compound 1 was shown to display diuretic and natriuretic activity, when dosed orally in animals, it features somewhat high clearance rates across three preclinical species with a relatively low exposure and short half-life (Table 1). The major metabolite observed from a bile duct cannulated rat study (>parent in urine and primary component in bile) and from in vitro incubation in human and rat microsomes and liver S9 fractions was the hydrolyzed lactone, with comparable stability seen from rat and human microsomes and liver S9 fractions. Based on the short half-lives in preclinical species and comparable metabolism profile in human microsomes and liver S9 fractions, it is unlikely that compound 1 would have a human half-life consistent with once daily dosing. In addition, despite its 100-fold selectivity against hERG, QTc prolongation (i.e., >5% increase over vehicle control) was observed when 1 was evaluated in an anesthetized and vagotomized cardiovascular dog model.12 Thus, significant improvement in hERG selectivity, with an improved PK profile, would be required in order to identify a small molecule clinical candidate ROMK inhibitor.
Table 1. Pharmacokinetic Properties of Compound 1 (dose, 1 mg/kg IV, 2 mg/kg PO; n = 3).
| species | rat | dog | Rhesus |
|---|---|---|---|
| Cl (mL/min/kg) | 40 | 36 | 34 |
| Vdss (L/kg) | 0.66 | 1.9 | 1.4 |
| AUCNpo (μM·h·kg/mg) | 0.34 | 0.88 | 0.12 |
| half-life (h) | 0.62 | 1.4 | 1.1 |
| F% | 33 | 80 | 11 |
Unless otherwise noted, the ROMK inhibitory activity of compounds was initially assessed in a 86Rb+ efflux functional assay,13 whereas hERG inhibition was determined in a 35S-MK499 radioligand binding assay.14 Initial structure–activity relationship (SAR) work on the phthalide phenyl ring revealed that substitution at the 4-position of the phthalide is tolerated. For instance, the 4-methyl analogue (2)11 showed comparable ROMK potency and hERG selectivity to 1 (Figure 1).15 Interestingly, a further improvement in hERG selectivity was observed when a benzylic methyl was introduced to 2 (see compound 3). Given these data, we decided to synthesize the α-F analogue hoping for an improvement in hERG selectivity while retaining ROMK activity. This expectation is based on the idea that fluoride would reduce basicity of the piperizine nitrogen and help to diminish the extent of hERG inhibition.16,17 Fluoride 5 was accessed via alcohol 4. Notably, although some loss of ROMK potency was observed for 4, we did observe weaker hERG inhibition. Encouragingly, the fluoro analogue 5 retained similar ROMK potency and demonstrated about 1000-fold selectivity over hERG. Thus, 5 was assessed in a rat pharmacokinetic study and shown to still display high clearance and a short half-life.
Figure 1.

α-Substitution in the piperazine carboxamide series.
In a parallel SAR effort, the phthalide pharmacophore was introduced into our previously described piperazine diamine series (Figure 2).18 Compared to the original lead compound 6, compound 7(18) retained similar ROMK inhibitory potency. While compound 6 is 7-fold selective for hERG, compound 7 displays 21-fold selectivity for ROMK. Further incorporation of 4-methyl phthalide led to compound 8, with similar ROMK potency and marginally improved hERG selectivity. Next, as described in the piperazine carboxamide series, one α-hydroxyl group was added to furnish compound 9. Interestingly, racemate 9 showed a small improvement in ROMK activity and a decrease in hERG potency, resulting in a significant overall improvement in hERG selectivity. Importantly, hERG selectivity could be improved an additional 4-fold when the symmetric diol 10 was synthesized. When the isomer mixture 10 was evaluated in the ROMK and hERG electrophysiology (EP) assays,13 potency and selectivity were even more remarkable (ROMK EP IC50 0.015 μM, hERG EP IC50 28 μM; hERG EP/ROMK EP = 1870). The diol 10 was further converted into the corresponding difluoro analogue 11, which showed reduced ROMK potency compared to compound 10 in both the 86Rb+ efflux and EP assays, and reduced potency in 35S-MK499 binding and hERG EP assays.19 To further differentiate 10 and 11, the isomeric mixtures of both compounds were evaluated in an acute Sprague–Dawley (SD) rat diuresis model.20 Diol 10 demonstrated remarkable diuresis and natriuresis at either 3 or 10 mg/kg oral dosing. In comparison, 11 showed significant but modest efficacy only at 10 mg/kg (Table 2), probably due to its weaker ROMK inhibitory activity, as well as lower compound levels in urine as compared to 10.21 Thus, further efforts were devoted to prepare and profile stereoisomers of diol 10.
Figure 2.

α-Substitution in the piperazine diamine series (ROMK and hERG IC50 values determined in 86Rb+ efflux and 35S-MK499 binding assays, respectively, unless otherwise indicated; EP, electrophysiology).
Table 2. Acute (4 h) Diuresis/Natriuresis of 10 and 11 in SD Rata after Oral Dosing.
| compd | dose | diuresis fold increaseb | natriuresis fold increaseb |
|---|---|---|---|
| 10 | 3 mg/kg | 6.7c | 9.4c |
| 10 | 10 mg/kg | 6.8c | 9.5c |
| 11 | 3 mg/kg | 1.6 | 1.7 |
| 11 | 10 mg/kg | 3.0c | 3.0c |
n = 5 per group.
Compared to vehicle.
p < 0.05 vs. vehicle.
Although it is possible to resolve the isomers of 10 by chiral HPLC, it is much more efficient to prepare them via the chiral epoxides. Heating of piperazine with either the R or S epoxide gave rise to the R,R or S,S enantiomer, 12 and 13 (Figure 3).22 The meso-isomer can be prepared by stepwise epoxide opening reactions. The R-epoxide was treated with 1-Boc piperazine to furnish 14, which was followed by deprotection of Boc under acidic conditions. Subsequent reaction of the revealed amine (15) with the S-epoxide gave rise to the meso-isomer 16.
Figure 3.

Synthesis of stereoisomers of 10.
In the ROMK 86Rb+ efflux and EP assays, the three isomers showed very similar potency, although their potency is significantly higher in the EP as compared to the 86Rb+ efflux assay. All three isomers are weak inhibitors of the hERG channel in both the 35S-MK499 binding and hERG EP assays. In fact, all three isomers are at least 1800-fold selective for ROMK in the EP assays. In a third type of ROMK functional assay based on Tl+ influx,13 where compounds’ potencies display a good correlation with the EP assay, the S,S-isomer 13 seemed to be 10-fold less potent than the R,R-isomer 12, while the meso-isomer 16 showed similar potency to the R,R-isomer (Table 3).
Table 3. ROMK and hERG Activity of Stereoisomers of 10 (in μM).
| compd | 12 | 13 | 16 |
|---|---|---|---|
| ROMK 86Rb+ efflux | 0.068 | 0.134 | 0.112 |
| ROMK Tl+ flux | 0.006 | 0.059 | 0.010 |
| ROMK EP | 0.009 | 0.012 | 0.009 |
| hERG binding | 23 | 47 | 100 |
| hERG EP | 22 | 22 | >30 |
| hERG EP/ROMK EP | 2440 | 1800 | >3000 |
All three isomers were tested in rat pharmacokinetic studies, showing similar oral exposure and half-lives (Table 4). Surprisingly, quite different efficacies were observed when dosed in the acute rat diuresis assay (Table 5). The R,R-isomer 12 evoked remarkable diuresis and natriuresis at doses as low as 0.3 mg/kg PO, while the S,S-isomer 13 did not display significant activity at any of the tested doses.23 In comparison, moderate efficacy was observed when the meso-isomer 16 was tested at 1 and 3 mg/kg PO. Potassium excretion was increased approximately 2-fold in animals that received HCTZ and 1.4- to 2.1-fold for those that received compound 12, but the latter effect was not dose-dependent and was not significantly different relative to vehicle treated animals for the 1, 3, and 10 mg/kg treatment groups. Moreover, the ratio of potassium to sodium excretion was lower for all three ROMK inhibitors compared to HCTZ. Based on these results, the R,R-isomer 12 was selected for further profiling.24
Table 4. Rat PK Properties of Compounds 12, 13, and 16.
| compd | 12 | 13 | 16 |
|---|---|---|---|
| dose IV/PO (mg/kg) | 1/2 | 0.5/1 | 0.5/1 |
| Cl (mL/min/kg) | 56 | 82 | 54 |
| Vdss (L/kg) | 3.8 | 5.8 | 4.5 |
| AUCNpo (μM·h·kg/mg) | 0.14 | 0.13 | 0.30 |
| half-life (h) | 1.1 | 0.86 | 1.4 |
| F% | 21 | 45 | 44 |
Table 5. Acute (4 h) Diuresis/Natriuresis of Isomers of 10 in SD Rata after Oral Dosing.
| compd | dose | diuresis fold increaseb | natriuresis fold increaseb | kaliuresis fold increaseb |
|---|---|---|---|---|
| 12 | 0.3 mg/kg | 6.4d | 6.2d | 2.1c |
| 12 | 1 mg/kg | 8.6d | 8.6d | 1.8 |
| 12 | 3 mg/kg | 9.0d | 8.0d | 1.4 |
| 12 | 10 mg/kg | 9.4d | 8.5d | 1.7 |
| 13 | 0.3 mg/kg | 1.3 | 1.4 | 1.1 |
| 13 | 1 mg/kg | 1.6 | 1.5 | 1.0 |
| 13 | 3 mg/kg | 1.9 | 2.2 | 0.9 |
| 16 | 1 mg/kg | 3.5d | 3.7d | 1.3 |
| 16 | 3 mg/kg | 4.5d | 4.1d | 1.1 |
n = 5 per group.
Compared to vehicle.
p < 0.01 vs. vehicle.
p < 0.05 vs. vehicle.
Compound 12 was further dosed in dog and rhesus to assess higher species pharmacokinetic properties (Table 6). Dog PK of 12 indicated a high clearance rate, which led to relatively short half-life and low exposure. Compared to 1 (Table 1), compound 12 in rhesus did show significant improvement in half-life, benefiting mainly from a larger volume of distribution. In a bile duct cannulated rat study with 3H-12, the major clearance mechanisms were oxidative metabolism (by CYP3A4) and urinary excretion of unchanged parent; minor metabolism pathways included glucuronidation and lactone hydrolysis. The major circulating component was parent. Incubation of 3H-12 (1 μM) in rat, dog, monkey, and human hepatocytes resulted in 15–37% turnover at 120 min with oxidation products observed. In addition to the Phase 1 metabolites, the lactone hydrolysis product and the parent glucuronide were also detected at very low levels (each <1%). There were no human-specific metabolites. The projected human half-life of compound 12 was estimated to be approximately 5 h based upon a combination of in vitro to in vivo correlation and allometric scaling, averaged from preclinical species. Overall, compound 12 has moderately improved pharmacokinetic properties and significantly improved pharmacodynamic properties at lower doses and has better hERG selectivity relative to compound 1.
Table 6. Dog and Rhesus PK Properties of Compound 12 (1 mpk IV, 2 mpk PO).
| species | dog | Rhesus |
|---|---|---|
| Cl (mL/min/kg) | 56 | 29 |
| Vdss (L/kg) | 7.5 | 6.7 |
| AUCNpo (μM·h·kg/mg) | 0.32 | 0.30 |
| half-life (h) | 1.7 | 4.0 |
| F% | 51 | 25 |
Compound 12 was screened against other members of the Kir family of channels. It did not show any significant activity on Kir2.1, Kir2.3, Kir4.1, or Kir7.1 channels when tested at concentrations up to 30 μM. Compound 12 is also selective against other cardiac ion channels such as Cav1.2 and Nav1.5 (IC50 > 30 μM). In addition, 12 was not a potent reversible inhibitor of human CYP3A4, CYP2C9, or CYP2D6 (IC50 > 50 μM) and was not a time-dependent inhibitor of CYP3A4 at 10 and 50 μM. In a broad counterscreen panel conducted at Panlabs and including over 150 receptors, enzymes, and ion channels, 12 only exhibited three activities at <10 μM: acetyl cholinesterase, ACES, IC50 = 9.94 μM, somatostatin subtype 1, sst1 IC50 = 2.63 μM, and human serotonin transporter, SERT, IC50 = 0.12 μM. The sst1 activity of 12 was followed up with functional assays, and no functional activity was observed. In a functional assay using HEK293 cells stably transfected with human SERT, uptake of 3H-serotonin was inhibited by 12 with an IC50 value of 2.40 ± 0.32 μM (n = 5). The superior ROMK potency and in vivo efficacy of 12, coupled with the fact that the compound is a substrate of human Pgp (human Mdr1 BAAB ratio = 12), should be able to impart a significant safety window with respect to the SERT off-target activity.25
Finally, the cardiovascular safety of 12 was assessed in the Merck cardiovascular dog model. The compound was administered intravenously during three sequential 30 min periods at 1, 2, and 7 mg/kg (10 mg/kg cumulative dose) to determine its effects on cardiovascular function in anesthetized and vagotomized dogs. Heart rate, mean arterial pressure, and electrocardiographic parameters (PR, QRS, and QT/QTc intervals) were monitored predose and during each infusion period. No treatment-related effects were observed during the course of the experiment at plasma concentrations of up to 11.6 μM, illustrating the fact that cardiovascular risks of ROMK inhibitors, such as 1, can be mitigated by enhancing their selectivity against the hERG channel.
With a safe and efficacious compound in hand, 12 was tested in a subchronic blood pressure study in spontaneously hypertensive rats (SHR). The compound was dosed once daily (QD) via oral gavage for 4 days, and changes in blood pressure were monitored by implanted C40 telemetry devices. As shown in Table 7, compound 12 evoked dose-dependent decreases in systolic blood pressure (SBP) at day 4.26 At 3 mg/kg/day of 12, the magnitude of SBP lowering (∼12 mmHg) was similar to that achieved with hydrochlorothiazide (HCTZ) (25 mg/kg/day).27 At 10 mg/kg/day, the SBP lowering efficacy (∼20 mmHg) exceeded that of HCTZ at 25 mg/kg/day. To the best of our knowledge, this is the first time that a small molecule ROMK inhibitor has been used in an animal model to show a blood pressure lowering effect. Based on its potency, selectivity, safety, and superior in vivo efficacy, compound 12 was selected as a clinical development compound and designated as MK-7145.
Table 7. Changes in SBP at Day 4 in SHR Dosed with Compound 12.
| compd/dose (mg/kg/day) | baseline (mmHg) | SBP @ day 4 (mmHg) | ΔSBP @ day 4 (mmHg) |
|---|---|---|---|
| vehicle | 194 ± 6.1 | 191 ± 6.9 | –2.5 ± 2.2 |
| 12/1 | 191 ± 5.3 | 182 ± 5.0 | –9.4 ± 1.8 |
| 12/3 | 194 ± 4.5 | 182 ± 4.9 | –12.4 ± 0.7 |
| 12/10 | 193 ± 5.3 | 173 ± 3.6 | –19.9 ± 2.9 |
| HCTZ/25 | 194 ± 4.5 | 179 ± 4.6 | –15.0 ± 2.1 |
In summary, a new subseries of novel ROMK inhibitors were developed by combining SAR information from the piperazine carboxamide series and piperazine diamine series.28 Compared to compound 1, diol 12 demonstrated much improved hERG selectivity and did not display cardiac liabilities in a subsequent assessment in a cardiovascular dog model. In addition, compound 12 has moderately improved PK properties in preclinical species. In the acute rat diuresis model, 12 evoked remarkable diuresis and natriuresis at doses as low as 0.3 mg/kg. For the first time with a small molecule ROMK inhibitor, we were also able to show that compound 12 caused dose-dependent lowering of blood pressure in a subchronic SHR model. Following extensive profiling, 12 was selected as MK-7145, the first small molecule ROMK inhibitor to enter clinical development. Human data from clinical trials conducted with MK-7145 will be reported in future publications.
Acknowledgments
We thank Junling Gao and Dr. Li-Kang Zhang for help with HR–MS analysis of key compounds.
Glossary
ABBREVIATIONS
- ROMK
renal outer medullary potassium channel
- hERG
the Human Ether-a-go-go-Related Gene
- PK
pharmacokinetics
- PD
pharmacodynamics
- EP
electrophysiology
- SHR
spontaneous hypertensive rat
- HCTZ
hydrochlorothiazide
- QD
once daily
- PO
oral dosing
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00122.
Synthesis of key compounds and characterization, protocols of animal models, and discussions on compound hERG activity, ROMK PD efficacy in acute rat diuresis model, and derisking off-target activity for compound 12 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ho K.; Nichols C. G.; Lederer W. J.; Lytton J.; Vassilev P. M.; Kanazirska M. V.; Hebert S. C. Cloning and Expression of an Inwardly Rectifying ATP-regulated Potassium Channel. Nature 1993, 362 (6415), 31–8. 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- Shuck M. E.; Bock J. H.; Benjamin C. W.; Tsai T. D.; Lee K. S.; Slightom J. L.; Bienkowski M. J. Cloning and Characterization of Multiple Forms of the Human Kidney ROM-K Potassium Channel. J. Biol. Chem. 1994, 269 (39), 24261–70. [PubMed] [Google Scholar]
- Hebert S. C.; Desir G.; Giebisch G.; Wang W. Molecular Diversity and Regulation of Renal Potassium Channels. Physiol. Rev. 2005, 85 (1), 319–371. 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinalter S. C.; Jeck N.; Peters M.; Seyberth H. W. Pharmacotyping of Hypokalaemic Salt-losing Tubular Disorders. Acta Physiol. Scand. 2004, 181 (4), 513–521. 10.1111/j.1365-201X.2004.01325.x. [DOI] [PubMed] [Google Scholar]
- ROMK inhibition in the CCD is anticipated to limit sodium reabsorption by the ENaC channel, however there is conflicting evidence on this issue, and it remains to be resolved. Please see:Kharade S. V.; Flores D.; Lindsley C. W.; Satlin L. M.; Denton J. S. Am. J. Physiol. Renal Physiol. 2015, 310, 732–737. 10.1152/ajprenal.00423.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W.; Foo J. N.; O’Roak B. J.; Zhao H.; Larson M. G.; Simon D. B.; Newton-Cheh C.; State M. W.; Levy D.; Lifton R. P. Rare Independent Mutations in Renal Salt Handling Genes Contribute to Blood Pressure Variation. Nat. Genet. 2008, 40 (5), 592–599. 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin M. D.; Timpson N. J.; Wain L. V.; Ring S.; Jones L. R.; Emmett P. M.; Palmer T. M.; Ness A. R.; Samani N. J.; Smith G. D.; Burton P. R. Common Variation in the WNK1 Gene and Blood Pressure in Childhood: the Avon Longitudinal Study of Parents and Children. Hypertension 2008, 52 (5), 974–979. 10.1161/HYPERTENSIONAHA.108.118414. [DOI] [PubMed] [Google Scholar]
- Lorenz J. N.; Baird N. R.; Judd L. M.; Noonan W. T.; Andringa A.; Doetschman T.; Manning P. A.; Liu L. H.; Miller M. L.; Shull G. E. Impaired Renal NaCl Absorption in Mice Lacking the ROMK Potassium Channel, a Model for Type II Bartter’s Syndrome. J. Biol. Chem. 2002, 277 (40), 37871–37880. 10.1074/jbc.M205627200. [DOI] [PubMed] [Google Scholar]
- Lu M.; Wang T.; Yan Q.; Yang X.; Dong K.; Knepper M. A.; Wang W.; Giebisch G.; Shull G. E.; Hebert S. C. Absence of Small Conductance K+ Channel (SK) Activity in Apical Membranes of Thick Ascending Limb and Cortical Collecting Duck in ROMK (Bartter’s) Knockout Mice. J. Biol. Chem. 2002, 277 (40), 37881–37887. 10.1074/jbc.M206644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifton R. P.; Gharavi A. G.; Geller D. S. Molecular Mechanisms of Human Hypertension. Cell 2001, 104 (4), 545–556. 10.1016/S0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- Tang H.; de Jesus R. K.; Walsh S. P.; Zhu Y.; Yan Y.; Priest B. T.; Swensen A. M.; Alonso-Galicia M.; Felix J. P.; Brochu R. M.; Bailey T.; Thomas-Fowlkes B.; Zhou X.; Pai L.-Y.; Hampton C.; Hernandez M.; Owens K.; Roy S.; Kaczorowski G. J.; Yang L.; Garcia M. L.; Pasternak A. Discovery of a novel sub-class of ROMK channel inhibitors typified by 5-(2-(4-(2-(4-(1H-Tetrazol-1-yl)phenyl)acetyl)piperazin-1-yl)ethyl)-isobenzofuran-1(3H)-one. Bioorg. Med. Chem. Lett. 2013, 23 (21), 5829–5832. 10.1016/j.bmcl.2013.08.104. [DOI] [PubMed] [Google Scholar]
- The CV dog model was a Merck internal model used to assess cardiovascular safety of research compounds.
- Felix J. P.; Priest B. T.; Solly S.; Bailey T.; Brochu R. M.; Liu C. J.; Kohler M. G.; Kiss L.; Alonso-Galicia M.; Tang H.; Pasternak A.; Kaczorowski G. J.; Garcia M. L. The inwardly rectifying potassium channel Kir1.1: development of functional assays to identify and characterize channel inhibitors. Assay Drug Dev. Technol. 2012, 10, 417. 10.1089/adt.2012.462. [DOI] [PubMed] [Google Scholar]
- Wang J.; Della Penna K.; Wang H.; Karczewski J.; Connolly T. M.; Koblan K. S.; Bennett P. B.; Salata J. J. Functional and pharmacological properties of canine ERG potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2003, 284 (1), H256–267. 10.1152/ajpheart.00220.2002. [DOI] [PubMed] [Google Scholar]
- The substitution SAR on the phthalide ring was thoroughly explored, and only small substituents at the C-4 position afforded compounds with similar ROMK potency and selectivity over hERG.
- Bell I. M.; Bilodeau M. T. The Impact of Ikr Blockade on Medicinal Chemistry Programs. Curr. Top. Med. Chem. 2008, 8 (13), 1128–1139. 10.2174/156802608785700034. [DOI] [PubMed] [Google Scholar]
- For a recent review on the role of fluorine in medicinal chemistry, please see:Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58 (21), 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Tang H.; Walsh S. P.; Yan Y.; de Jesus R. K.; Shahripour A.; Teumelsan N.; Zhu Y.; Ha S.; Owens K. A.; Thomas-Fowlkes B. S.; Felix J. P.; Liu J.; Kohler M.; Priest B. T.; Bailey T.; Brochu R.; Alonso-Galicia M.; Kaczorowski G. J.; Roy S.; Yang L.; Mills S. G.; Garcia M. L.; Pasternak A. Discovery of Selective Small Molecule ROMK Inhibitors as Potential New Mechanism Diuretics. ACS Med. Chem. Lett. 2012, 3 (5), 367–372. 10.1021/ml3000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The improvement of hERG off-target activity can be rationalized by lowering of pKa and compound lipophilicity. Please see the Supporting Information for correlation of hERG selectivity with calculated pKa and LogD.
- Compounds 10 and 11 showed similar rat PK properties as mixture of isomers.
- Please see the Supporting Information for further discussion on ROMK PD efficacy of compounds 10 and 11.
- Please see the Supporting Information for preparation of both chiral epoxides.
- Please see the Supporting Information for further discussion on ROMK PD efficacy of compounds 12, 13, and 16.
- Despite of good efficacy in the acute rat diuresis model, meso-isomer 16 showed quite low aqueous solubility based on qualitative assessment. In comparison, R,R-isomer 12 has aqueous solubility of at least 1 mg/mL.
- Please see Supporting Information for detailed discussion on derisking of hSERT off-target activity.
- Only changes in SBP at day 4 are disclosed in this communication. For a more thorough description of the in vivo pharmacology of compound 12, a manuscript by Caryn Hampton et al. will be separately submitted for publication.
- Based on internal dose titration, HCTZ achieves approximately the maximum blood pressure lowering efficacy in SHR at 25 mg/kg oral dosing QD.
- For more SAR work on this end, please see:Walsh S. P.; Shahripour A.; Tang H.; Teumelsan N.; Frie J.; Zhu Y.; Priest B. T.; Swensen A. M.; Liu J.; Margulis M.; Visconti R.; Weinglass A.; Felix J. P.; Brochu R. M.; Bailey T.; Thomas-Fowlkes B.; Alonso-Galicia M.; Zhou X.; Pai L.-Y.; Corona A.; Hampton C.; Hernandez M.; Bentley R.; Chen J.; Shah K.; Metzger J.; Forrest M.; Owens K.; Tong V.; Ha S.; Roy S.; Kaczorowski G. J.; Yang L.; Parmee E.; Garcia M. L.; Sullivan K.; Pasternak A. Discovery of Selective Small Molecule ROMK Inhibitors as Potential New Mechanism Diuretics. ACS Med. Chem. Lett. 2015, 6 (7), 747–752. It is worth noting that most of the SAR work in the mentioned publication was done after the discovery of MK-7145. 10.1021/ml500440u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.