

Abstract

Blockade of aberrant Wnt signaling is an attractive therapeutic approach in multiple cancers. We developed and performed a cellular high-throughput screen for inhibitors of Wnt secretion and pathway activation. A lead structure (GNF-1331) was identified from the screen. Further studies identified the molecular target of GNF-1331 as Porcupine, a membrane bound O-acyl transferase. Structure–activity relationship studies led to the discovery of a novel series of potent and selective Porcupine inhibitors. Compound 19, GNF-6231, demonstrated excellent pathway inhibition and induced robust antitumor efficacy in a mouse MMTV-WNT1 xenograft tumor model.
Keywords: Wnt signaling, Porcupine inhibitor
Wnt signaling is tightly controlled during cellular proliferation, differentiation, and embryonic morphogenesis.1,2 Aberrant activation of this pathway plays a critical role in a variety of cancers, such as cutaneous squamous cell carcinoma (SCC), breast cancer, and colorectal cancer.3−6 Wnt signaling can be stimulated upon the binding of Wnt ligands to their receptors LRP5/6 and Frizzled at the plasma membrane, or be activated by downstream activating mutations.3−6 Activated Wnt signaling results in the accumulation of β-catenin in the cytoplasm through the disruption of β-catenin degradation machinery that consists of AXIN2, GSK3β, APC, and others. Increased β-catenin then translocates into the nucleus to form a complex with LEF/TCF and subsequently induces downstream gene expression. Up-regulation of Wnt signaling due to mutations in downstream components, such as APC and β-catenin, is well documented in colorectal cancer. In addition, increased pathway activity has been reported in various cancers in association with overexpression of Wnt ligands or costimulants such as R-Spondin 2/3 (RSPO2/3), or silencing of Wnt inhibitor genes.7 More recently, mutations of the RSPO coreceptors RNF43/ZNFR3 have been shown to play critical roles in several cancers, including pancreatic, colorectal, gastric cancer, and cholangiocarcinoma.8−10 Thus, inhibition of Wnt signaling is an attractive approach for anticancer therapy. Despite intensive research efforts, therapeutic intervention of activated Wnt signaling in cancers harboring downstream mutations has had limited success due to lack of tractable targets. In contrast, there have been some successes in developing therapeutics targeting the upstream components of the Wnt pathway, including LRP6 and Frizzled antibodies.11,12
As part of our effort to target this important oncogenic pathway, we are interested in searching for small molecules that specifically block Wnt ligand secretion.13 To do this, we performed a high-throughput cell-based screen of a 2.4-million compound library, using a two-cell coculture assay system that includes a Wnt ligand secreting cell, a stable L-cell line overexpressing Wnt3A, and a Wnt reporter cell, a TM3 cell line stably transfected with a Wnt-responsive luciferase reporter gene. To confirm the specificity for inhibiting Wnt secretion, the screening hits were triaged through two counter assays, a Hedgehog (Hh) coculture assay and a Wnt reporter gene assay (RGA) with the addition of Wnt3A conditioned medium. A specific Wnt secretion inhibitor should be inactive in the two counter assays. From this screening approach, we identified several chemical classes that specifically block Wnt ligand secretion. One of these is exemplified in GNF-1331 (Figure 1). Through further target elucidation efforts, we determined Porcupine (PORCN) is its molecular target.13 At the time of this discovery, an inhibitor of Wnt signaling, IWP2, was also reported to function through inhibiting PORCN by Lum and co-workers.14,15 PORCN is the enzyme responsible for post-translational palmitoylation of Wnt proteins that is required for both Wnt secretion as well as binding of Wnts to their receptors.16,17 In our recent report, we described in vitro and in vivo properties of our first-in-class PORCN inhibitor WNT974, which is currently undergoing clinical trials.13 Several other groups have also recently reported Porcupine inhibitors to inhibit Wnt signaling activity.18−21 Herein, we report the hit to lead and lead optimization efforts, which led to the discovery of WNT974 (15) and GNF-6231 (19), a molecule with further improved physicochemical properties, as a potent, selective, and orally bioavailable Porcupine inhibitor that blocks Wnt signaling in vitro and in vivo.
Figure 1.

Structure of GNF-1331, a screening hit.
Our initial focus was to develop a tool compound that is suitable for in vivo target validation work. GNF-1331 shows good potency in the Wnt secretion coculture assay with an IC50 of 12 nM. It binds to PORCN in a radioligand binding assay with an IC50 of 8 nM. However, it exhibits poor pharmacokinetic properties with rapid clearance and low systemic exposure after oral administration in mice (dose normalized Cmax and AUC are 0.03 μM·h and 0.014 μM, respectively), which prevent it from being used in vivo. From the initial structure–activity relationship (SAR) analysis of existing analogues in our compound collection, the pyridine nitrogen appears to be important for activity. We envisioned the thioether moiety can be one of the key metabolically labile spots that result in poor pharmacokinetic properties. And we further hypothesized that the thioether moiety together with the N-propyl triazole functions as a linker between the two key binding motifs on both sides of the molecule. To test this hypothesis, we replaced the entire moiety with a simple phenyl group as shown in 1 (Figure 2) in our first attempt. This modification led to not only drastically improved in vivo pharmacokinetic properties but also much improved in vitro potency. In mice, compound 1 exhibits low clearance (2.49 mL/min/kg) and nearly complete oral bioavailability with dose normalized AUC and Cmax of 26 μM·h and 4.1 μM, respectively. It shows a 6-fold improved potency in the coculture reporter gene assay with an IC50 of 2 nM.
Figure 2.

Structure–Activity Relationship of 1.
While 1 is a suitable in vivo validation tool, it bears some liabilities, such as strong inhibition of CYPs (e.g., IC50 of 0.8 μM on CYP3A4). Our initial chemistry effort was thus directed toward understanding the general SAR of this scaffold in order to further optimize the drug-like properties. The selected results are summarized in Figure 2. The Wnt coculture RGA is used to guide the SAR studies.
It is well-known that pyridine nitrogen with no adjacent substitution, as in 1, can cause strong inhibition of CYP enzymes.22 One of the tactics to address this issue is to introduce a substituent at the adjacent carbon next to the nitrogen. Indeed, introduction of a methyl group at the 2-position as shown in 2 ameliorated the CYP inhibitory activity, up to 10 μM testing concentration. More encouragingly, this modification led to a 4-fold increase in the in vitro potency. However, significant loss of activity was observed when moving the 4-pyridine nitrogen to the 3-position as shown in 3. Methylation of the amide nitrogen in 4 or reversal of the amide configuration in 5 was also less tolerated. Interestingly, the replacement of the fused benzothiazole ring with phenyl thiazole in 6 led to only a slight decrease in potency. This loss of activity can be completely compensated by the further replacement of thiazole with either a pyridine or a phenyl as shown in 7 or 8, respectively. The amide linkage appears essential, since either urea 9 or carbamate 10 result in complete loss of activity, up to 10 μM.
Based on these initial SAR and pharmacokinetic data, we chose 7 as a new lead for further optimization with the emphasis on aqueous solubility and other drug-like properties. Given the highly lipophilic nature of 7, which has a cLogP of 4.8, one of our strategies to improve solubility was focused on reducing lipophilicity. The selected SAR is summarized in Table 1.
Table 1. IC50 Values for Select Compounds in the Wnt3a Co-culture RGA.
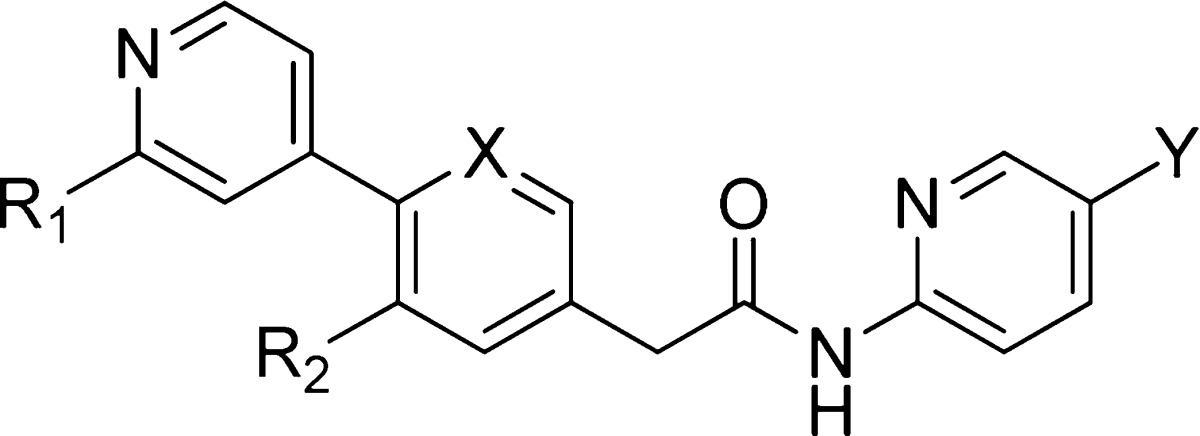

See Supporting Information for detailed assay descriptions.
HT: high throughput.
ND: not determined.
Realizing that multiple phenyl rings in the structure of compound 7 contribute to the high lipophilicity, we focused our initial attempts on introducing nitrogen into the rings to lower cLogP and subsequently to improve aqueous solubility. As seen from the results in the table, noticeable increases in solubility were achieved using this approach, as shown in 11–13, and the resulting compounds retained excellent in vitro activity. Interestingly, the substitution of a methyl group at the R2 position (14 and 15) led to further increases in solubility, especially for the latter. This is likely due to the increased dihedral angle between the two pyridines, which leads to reduced π stacking. The methyl group at the R1-position can be replaced by an electron withdrawing fluorine (16) without losing potency. However, a hydroxymethyl group (17) is less tolerated at this position. In addition to reducing lipophilicity, we also tried to increase the sp3 fraction in the molecules to improve the physicochemical properties. Replacement of the terminal aromatic ring by piperizine was tolerated (18), but resulted in a 30-fold decrease in potency. This loss of activity, however, can be completely restored by the formation of either acetamides (19–20) or methyl carbamate (21). As expected, these modifications resulted in further improvement of aqueous solubility as measured in the high-throughput solubility assay. To further confirm this improved property, the aqueous solubility was determined in a neutral pH buffer using crystalline materials. The free base of 19 showed a good solubility of 357 μM in contrast to 5 μM observed for the free base of 15.
Based on favorable potency, specificity, and other drug-like properties, such as physicochemical properties, in vitro ADMET, and in vivo pharmacokinetics, compound 19, GNF-6231, was chosen as one of the advanced compounds for further in vitro and in vivo testing. Compound 19 exhibits exquisite specificity for PORCN. In our broad off-target panel profiling, there are no appreciable activities observed, at least up to 10 μM testing concentrations, for more than 200 off-targets, which include GPCRs, kinases, proteases, transporters, ion channels, and nuclear receptors. Like the other analogues, compound 19 shows IC50s of greater than 10 μM on all CYP isoforms tested (2C9, 2D6, 3A4). Compound 19 is moderately bound to mouse, rat, dog, monkey, and human plasma proteins (88.0, 83.1, 90.9, 71.2, and 95%, respectively) as determined by rapid equilibrium dialysis. It shows high permeability in a Caco-2 human cell permeability assay (PappA-B of 7.5 × 10–6 cm/s) with a possible efflux (efflux ratio of 3.2). In vitro metabolic stability studies indicate that the microsomal clearance of 19 is low across preclinical species and humans. It shows good oral bioavailability, ranging from 72 to 96% in preclinical species (mouse, rat, and dog) when dosed in solution formulations (Table 2). Consistent with the in vitro microsomal clearance data, the systemic clearance (CL) is low in all species, relative to respective hepatic blood flow (Table 2). The terminal half-lives are 2.4, 2.8, and 8.9 h in mouse, rat, and dog, respectively. Compound 19 is expected to have minimal to marginal distribution to tissues compared to total body water following intravenous administration to mouse (Vss 0.57 L/kg), rat (Vss 0.70 L/kg), and dog (Vss 0.25 L/kg).
Table 2. In Vivo Pharmacokinetics in Preclinical Speciesa.
| CL (mL/min/kg) | Vss (L/kg) | t1/2 (h) | F (%) | |
|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |
| Mouse | 5.33 ± 0.81 | 0.57 ± 0.10 | 2.44 ± 0.13 | 96 ± 19 |
| Rat | 5.35 ± 0.65 | 0.70 ± 0.05 | 2.77 ± 0.14 | 84 ± 3 |
| Dog | 0.67 ± 0.14 | 0.25 ± 0.06 | 8.86 ± 1.76 | 72 ± 22 |
The compound was administered as a solution in 20% PEG300/3% ETPGS for mouse and rat or 25% PEG300/5% Solutol for dog.
The mouse mammary tumor virus (MMTV)-Wnt1 transgenic mice spontaneously develop murine breast tumors. The xenograft model derived from these tumors has been effectively used to evaluate the in vivo pharmacodynamics and antitumor efficacy of Wnt inhibitors.12 Given that 19 shows favorable potency and a PK profile across preclinical species upon oral administration, we elected to assess its in vivo activity in the subcutaneous MMTV-Wnt1 xenograft mouse model.
To determine the inhibition of Wnt signaling by 19 in the MMTV-Wnt1 tumors, Wnt target gene Axin2 expression (Axin2 mRNA) was measured by quantitative RT-PCR through TaqMan assays. In the single dose PK/PD relationship study, compound 19 was dosed at 3 mg/kg orally in MMTV-Wnt1 tumor bearing mice, and plasma and tumor samples were collected at various time points (1, 5, 7, 10, and 24 h post-dosing). The plasma was analyzed for PK measurements, and the tumor was analyzed for both PK and PD measurements. As we can see from the results shown in Figure 3, compound 19 inhibited the signaling activity effectively, as shown by the reduction of Axin2 mRNA levels with the maximal inhibition of about 90% observed at 5 h post-dosing, a delayed PD response in relation to the maximal drug concentrations (Cmax). The Axin2 expression was back to baseline level 24 h after treatment accompanying decreasing drug concentrations.
Figure 3.
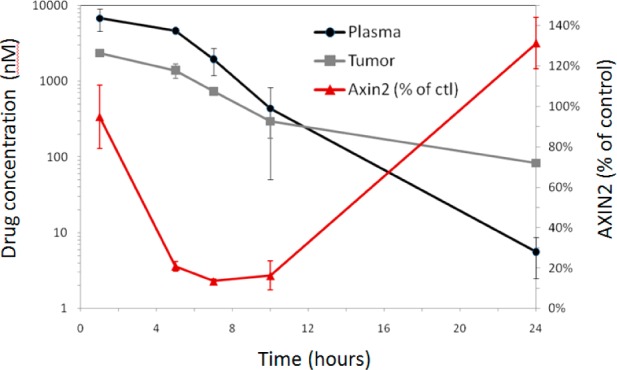
PK–PD relationship of 19 in the mouse MMTV-Wnt1 tumor model (n = 3 per time point).
To test its in vivo antitumor efficacy, the MMTV-Wnt1 tumor bearing mice were treated with 19 by daily oral administration for 2 weeks at three different dose levels. Changes in tumor volume for each of the treated (T) and control (C) groups were measured, and the T/C ratio was calculated to express the level of efficacy. As shown in Figure 4, compound 19 showed very robust dose-related antitumor efficacy. Significant tumor growth inhibition (T/C 15%, p < 0.0001) was observed at a dose of 0.3 mg/kg, while tumor regression was achieved at 1 and 3 mg/kg (T/C −74% and −84%, respectively, p < 0.0001). The compound was well tolerated with no significant body weight loss in all treated groups (data not shown).
Figure 4.
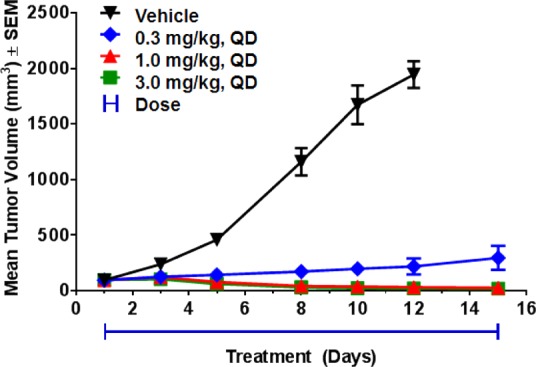
In vivo antitumor efficacy of 19 in the mouse MMTV-Wnt1 tumor model (n = 8 per group).
In summary, a novel chemical series was developed from GNF-1331, a screening hit identified by a Wnt coculture reporter gene assay, as Porcupine inhibitor to block Wnt ligand secretion and subsequently Wnt signaling activity. Compounds with exquisite potency and specificity, excellent pharmacokinetics, and desirable drug-like properties were identified. Treatment with 19, GNF-6231, in MMTV-Wnt1 subcutaneously implanted tumor bearing mice led to pronounced inhibition of the Wnt signaling activity as measured by the reduction of Wnt target gene Axin2 expression. This compound exhibited robust antitumor efficacy in the same model upon repeat dosing treatment. Porcupine inhibitors may represent a new therapeutic approach for diseases with aberrant Wnt signaling activities.
Glossary
Abbreviations
- ADMET
absorption, distribution, metabolism, excretion, toxicity
- CYP
cytochromes P450
- PK/PD
pharmacokinetic/pharmacodynamics
- GPCR
G-protein coupled receptor
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00038.
Synthetic schemes, procedure, and analytical data for 15 and 19; analytical data of 1–14, 16–18, and 20–21; general animal study protocols; high-throughput solubility assay protocols (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Barker N.; Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discovery 2006, 5, 997–1014. 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- Moon R. T.; Kohn A. D.; De Ferrari G. V.; Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701. 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Turashvili G.; Bouchal J.; Burkadze G.; Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology 2006, 73, 213–223. 10.1159/000098207. [DOI] [PubMed] [Google Scholar]
- Braun K. M. Cutaneous cancer stem cells: beta-catenin strikes again. Cell Stem Cell 2008, 2, 406–408. 10.1016/j.stem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Polakis P.Wnt signaling in cancer. Cold Spring Harbor Perspect. Biol. 2012, 4 ( (5), ), pii:a008052. 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R.; Varmus H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. 10.1038/emboj.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshagiri S.; Stawiski E. W.; Durinck S.; Modrusan Z.; Storm E. E.; Conboy C. B.; Chaudhuri S.; Guan Y.; Janakiraman V.; Jaiswal B. S.; Guillory J.; Ha C.; Dijkgraaf G. J.; Stinson J.; Gnad F.; Huntley M. A.; Degenhardt J. D.; Haverty P. M.; Bourgon R.; Wang W.; Koeppen H.; Gentleman R.; Starr T. K.; Zhang Z.; Largaespada D. A.; Wu T. D.; de Sauvage F. J. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H. X.; Xie Y.; Zhang Y.; Charlat O.; Oster E.; Avello M.; Lei H.; Mickanin C.; Liu D.; Ruffner H.; Mao X.; Ma Q.; Zamponi R.; Bouwmeester T.; Finan P. M.; Kirschner M. W.; Porter J. A.; Serluca F. C.; Cong F. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. 10.1038/nature11019. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Hao H. X.; Growney J. D.; Woolfenden S.; Bottiglio C.; Ng N.; Lu B.; Hsieh M. H.; Bagdasarian L.; Meyer R.; Smith T. R.; Avello M.; Charlat O.; Xie Y.; Porter J. A.; Pan S.; Liu J.; McLaughlin M. E.; Cong F. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12649–54. 10.1073/pnas.1307218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M.; Hodis E.; Jasmine M. X.; Yamauchi M.; Rosenbluh J.; Cibulskis K.; Saksena G.; Lawrence M. S.; Qian Z. R.; Nishihara R.; Van Allen E. M.; Hahn W. C.; Gabriel S. B.; Lander E. S.; Getz G.; Ogino S.; Fuchs C. S.; Garraway L. A. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–6. 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney A.; Axelrod F.; Bond C. J.; Cain J.; Chartier C.; Donigan L.; Fischer M.; Chaudhari A.; Ji M.; Kapoun A. M.; Lam A.; Lazetic S.; Ma S.; Mitra S.; Park I. K.; Pickell K.; Sato A.; Satyal S.; Stroud M.; Tran H.; Yen W. C.; Lewicki J.; Hoey T. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11717–11722. 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettenberg S. A.; Charlat O.; Daley M. P.; Liu S.; Vincent K. J.; Stuart D. D.; Schuller A. G.; Yuan J.; Ospina B.; Green J.; Yu Q.; Walsh R.; Li S.; Schmitz R.; Heine H.; Bilic S.; Ostrom L.; Mosher R.; Hartlepp K. F.; Zhu Z.; Fawell S.; Yao Y. M.; Stover D.; Finan P. M.; Porter J. A.; Sellers W. R.; Klagge I. M.; Cong F. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand-binding regions by LRP6 antibodies. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15473–15478. 10.1073/pnas.1007428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Pan S.; Hsieh M. H.; Ng N.; Sun F.; Wang T.; Kasibhatla S.; Schuller A. G.; Li A. G.; Cheng D.; Li J.; Tompkins C.; Pferdekamper A.; Steffy A.; Cheng J.; Kowal C.; Phung V.; Guo G.; Wang Y.; Graham M. P.; Flynn S.; Brenner J. C.; Li C.; Villarroel M. C.; Schultz P. G.; Wu X.; McNamara P.; Sellers W. R.; Petruzzelli L.; Boral A. L.; Seidel H. M.; McLaughlin M. E.; Che J.; Carey T. E.; Vanasse G.; Harris J. L. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20224–9. 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C.-W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth M. G.; Amatruda J. F.; Chen C.; Lum L. Small moledule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge M. E.; Moon J.; Tuladhar R.; Lu J.; Jacob L. S.; Zhang L.-S.; Shi H.; Wang X.; Moro E.; Mongera A.; Argenton F.; Karner C. M.; Carroll T. J.; Chen C.; Amatruda J. F.; Lum L. Diverse chemical scaffolds support direct inhibition of the membrane-bound O-acyltransferase porcupine. J. Biol. Chem. 2012, 287, 23246–23254. 10.1074/jbc.M112.372029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nile A. H.; Hannoush R. N. Fatty acylation of Wnt proteins. Nat. Chem. Biol. 2016, 12, 60–69. 10.1038/nchembio.2005. [DOI] [PubMed] [Google Scholar]
- Tuladhar R.; Lum L. Fatty acyl donor selectivity in membrane bound O-acyltransferases and communal cell fate decision-making. Biochem. Soc. Trans. 2015, 43, 235–239. 10.1042/BST20140282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraiswamy A. J.; Lee M. A.; Madan B.; Ang S. H.; Tan E. S.; Cheong W. W.; Ke Z.; Pendharkar V.; Ding L. J.; Chew Y. S.; Manoharan V.; Sangthongpitag K.; Alam J.; Poulsen A.; Ho S. Y.; Virshup D. M.; Keller T. H. Discovery and optimization of a porcupine inhibitor. J. Med. Chem. 2015, 58, 5889–99. 10.1021/acs.jmedchem.5b00507. [DOI] [PubMed] [Google Scholar]
- Wang X.; Moon J.; Dodge M. E.; Pan X.; Zhang L.; Hanson J. M.; Tuladhar R.; Ma Z.; Shi H.; Williams N. S.; Amatruda J. F.; Carroll T. J.; Lum L.; Chen C. The development of highly potent inhibitors for porcupine. J. Med. Chem. 2013, 56, 2700–4. 10.1021/jm400159c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y.; Li K.; Xu Z.; Ma H.; Zheng J.; Hu Z.; He S.; Wu Y.; Sun Z.; Luo L.; Li J.; Zhang H.; Zhang X. Exploration of the linkage elements of porcupine antagonists led to potent Wnt signaling pathway inhibitors. Bioorg. Med. Chem. 2015, 23, 6855–6868. 10.1016/j.bmc.2015.09.048. [DOI] [PubMed] [Google Scholar]
- Madan B.; Ke Z.; Harmston N.; Ho S. Y.; Frois A. O.; Alam J.; Jeyaraj D. A.; Pendharkar V.; Ghosh K.; Virshup I. H.; Manobaran V.; Ong E. H. Q.; Sangthongpitag K.; Hill J.; Petretoo E.; Keller T. H.; Lee M. A.; Matter A.; Virshup D. M. Wnt addition of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 1–11. 10.1038/onc.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokarnik G.; Grootenhuis D. J.; Watson J. B. High throughput P450 inhibition screens in early drug discovery. Drug Discovery Today 2005, 10, 1443–1450. 10.1016/S1359-6446(05)03580-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.