

Summary
Blood–brain barrier (BBB) dysfunction complicates central nervous system lupus, an important aspect of systemic lupus erythematosus. To gain insight into the underlying mechanism, vascular corrosion casts of brain were generated from the lupus mouse model, MRL/lpr mice and the MRL/MpJ congenic controls. Scanning electron microscopy of the casts showed loss of vascular endothelial cells in lupus mice compared with controls. Immunostaining revealed a significant increase in caspase 3 expression in the brain vascular endothelial cells, which suggests that apoptosis could be an important mechanism causing cell loss, and thereby loss of BBB integrity. Complement activation occurs in lupus resulting in increased generation of circulating C5a, which caused the endothelial layer to become ‘leaky’. In this study, we show that C5a and lupus serum induced apoptosis in cultured human brain microvascular endothelial cells (HBMVECs), whereas selective C5a receptor 1 (C5aR1) antagonist reduced apoptosis in these cells, demonstrating C5a/C5aR1‐dependence. Gene expression of initiator caspases, caspase 1 and caspase 8, and pro‐apoptotic proteins death‐associated protein kinase 1, Fas‐associated protein (FADD), cell death‐inducing DNA fragmentation factor 45 000 MW subunit A‐like effector B (CIDEB) and BCL2‐associated X protein were increased in HBMVECs treated with lupus serum or C5a, indicating that both the intrinsic and extrinsic apoptotic pathways could be critical mediators of brain endothelial cell apoptosis in this setting. Overall, our findings suggest that C5a/C5aR1 signalling induces apoptosis through activation of FADD, caspase 8/3 and CIDEB in brain endothelial cells in lupus. Further elucidation of the underlying apoptotic mechanisms mediating the reduced endothelial cell number is important in establishing the potential therapeutic effectiveness of C5aR1 inhibition that could prevent and/or reduce BBB alterations and preserve the physiological function of BBB in central nervous system lupus.
Keywords: apoptosis, blood–brain barrier, complement, endothelial cells, neurodegeneration
Introduction
Systemic lupus erythematosus (SLE) is a devastating autoimmune disease that is multifactorial and leads to multiorgan failure.1, 2, 3 Between 40 and 70% of patients with SLE are affected with central nervous system (CNS) lupus, yet the exact mechanism causing the pathology remains unknown. The CNS is uniquely positioned and protected from the circulatory system by the blood–brain barrier (BBB).4 The BBB is formed by the endothelial cells surrounded and supported by the astroglial cells and the astrocytic end feet. The BBB is crucial because it maintains the brain internal milieu constant, allowing optimal neuronal function. Loss of BBB integrity could lead to influx of inflammatory cells, and molecules such as autoantibodies causing brain injury. Our earlier studies using the well‐established lupus mouse model MRL/lpr mice revealed that the BBB integrity is lost with worsening disease.5, 6
MRL/lpr mice provide a well‐established mouse model for human lupus thought to accurately reflect pathological events that occur in human SLE, including CNS lupus.7, 8, 9, 10 MRL/lpr mice differ from the congenic MRL/MpJ (MRL+/+) strain by the absence of the pro‐apoptotic membrane Fas protein, due to a retroviral insertion in the Tnfrsf6 gene.11, 12, 13 Using these mice, our studies also show that the loss of BBB integrity is complement dependent.5, 6, 14 The complement (C) cascade is as a protective mechanism in host defence. However, in autoimmune diseases such as SLE, when the complement system is excessively and chronically activated, the beneficial effects can become detrimental to the host.15, 16, 17
The complement system contains three major initiation pathways and over 40 proteins.18, 19, 20 Our studies showed for the first time that apoptosis occurred in experimental lupus brains, which was complement‐dependent.21 Complement activation results in the generation of anaphylatoxins, C3a and C5a.22, 23 Subsequent studies from our laboratory showed that C5a/C5aR signalling aggravated CNS lupus. C5a caused neuronal cells in culture to become apoptotic.24 C5a binds to two receptors, the G‐coupled, C5aR1 and the alternate receptor, C5aR2.25, 26, 27 C5a/C5aR1 signalling mediates a number of biological processes, including chemotaxis and degranulation of mast cells, basophils, neutrophils and eosinophils, increasing vascular permeability, an increased generation of reactive oxygen species, and production of cytokines from monocytes and macrophages.28 Interestingly, C5a/C5aR signalling can be either protective or neurotoxic29 depending on the setting; however, increases in circulating C5a correlate with poor outcomes in SLE.30, 31 C5a can contribute to cellular apoptosis in conditions such as lupus24 and stroke,32 or may have an anti‐apoptotic effect on neutrophils during sepsis.33, 34 C5aR1 is present predominantly on blood myeloid cells, but is also constitutively expressed on several cell types in the brain including endothelial cells.35, 36 Using cultured brain endothelial cells, this study assessed the role of C5a/C5aR signalling in rendering endothelial cells apoptotic.
Apoptosis is regulated by the sequential activation of initiator and effector caspases.37 The autocatalytic activation of an initiator caspase depends on multiprotein complexes which comprise different factors such as apoptotic protease‐activating factor 1 (APAF‐1) and cytochrome c (CytC).38, 39 One of the key adaptor proteins transmitting apoptotic signals is Fas‐associated protein (FADD).40, 41 FADD activation can follow more than one pathway: it can bind to FAS and procaspase 8 forming the death‐inducing signalling complex (DISC),42, 43, 44, 45 which then cleaves and activates caspases 3, 6 and 7. Alternatively, FADD can recruit the initiator caspase, procaspase 10 and the caspase 8/10 regulator c‐FLIP (FADD‐like interleukin‐1β‐converting enzyme‐inhibitory protein).41, 46, 47 An additional apoptotic pathway includes CIDEB, a member of the CIDE [cell death‐inducing DFF45 (DNA fragmentation factor 45)‐like effector] family of apoptosis‐inducing factors,48, 49 and was found to be up‐regulated in the spinal cord consistent with neuronal apoptosis after nerve injury. The N‐terminal region of CIDEs is homologous to the CIDE‐N domains in DFF40/CAD (caspase‐activated nuclease) and its inhibitor [DFF45/ICAD (inhibitor of CAD)], which are two subunits of the DFF complex.50, 51, 52 Cleavage of DFF45/ICAD by caspase 3 releases DFF40/CAD from the complex, which leads to DNA fragmentation and nuclear condensation.
To understand the changes that occur in the brain vasculature in lupus, this study addressed two facets. First, vascular remodelling and endothelial cell alteration in the brains of lupus mice, and second, the role of the complement protein C5a and apoptosis in human brain endothelial cells in lupus. Our studies demonstrate for the first time that vascular changes occur in lupus brain with endothelial cell loss. One of the mechanisms that could be causing the cell loss was apoptosis, which occurred when human brain microvascular endothelial cells (HBMVECs) were treated with lupus serum or C5a indicating the translational potential of these studies.
Materials and methods
Experimental protocol in lupus mice
MRL/lpr and MRL+/+ mice (n = 4 in each group) were purchased from The Jackson Laboratory (Bar Harbor, ME), and maintained in the facility with free access to food and water. They were used for experiment at 18 weeks of age. These studies were approved by the University at Buffalo Animal Care and Use Committee.
Vascular corrosion casting
Vascular corrosion casting of the brain vasculature was performed on all animals immediately after euthanasia using a Batson's No. 17 Corrosion Casting Kit (Polysciences, Inc., Warrington, PA), as described previously.53, 54 In brief, anaesthesia was induced using 2–3% isoflurane and mice were killed using 4% isoflurane. Immediately thereafter, mice were perfused with 5 ml sterile saline injected through an 18G winged needle inserted into the left ventricle. After perfusion, approximately 4 ml of Batson's No. 17 Corrosion Cast mixture was injected via the left ventricle. After injection of the casting mixture, whole specimens were kept at 4° overnight to allow for complete polymerization of the casting mixture. The brain was then extracted and placed in 20% potassium hydroxide for 2 days on a rocking platform. Once the brain tissue dissolved, the vascular corrosion cast was isolated and rinsed with distilled water.
Scanning electron microscopy imaging
Scanning electron microscopy (SEM) was used to investigate the morphological features of the vascular casts, specifically imprints of endothelial cells, as described earlier.53, 55, 56, 57 Before imaging, perforating arteries were eliminated from the major vessels of the Circle of Willis. The casts were affixed to a metal stand with clay, coupled to the stand with graphite paint, and scatter coated with carbon under vacuum. Vascular casts of the mouse Circle of Willis were imaged at 50 × with a Hitachi SU‐70 SEM (Hitachi High Technologies America, Inc., Roslyn Heights, NY). Areas along major vessels of the Circle of Willis were imaged at higher magnification (300 × to 600 ×).
Quantification of endothelial cell density in the cerebral vasculature
High‐magnification SEM images of the major cerebral vessels were used to quantify endothelial cell density in lupus and control mice. For each mouse, three 50 μm by 50 μm (2500 μm2) representative interrogation windows were created on SEM images of major Circle of Willis vessels using image J software. The number of endothelial cell imprints in each window was then counted and averaged for each mouse. The average cell density of the lupus mice was compared with that of control mice using Student's t‐test (significance at P < 0·05).
Immunofluorescence staining of MRL/lpr and MRL+/+ mice brain sections
At 18 weeks of age, the MRL/lpr and MRL+/+ mice were killed and the brains were harvested. These studies were approved by the University of Buffalo Animal Care and Use Committee. Cerebral cortices were snap‐frozen in Tissue‐Tek O.C.T. (optimal cutting temperature) compound (Ted Pella, Redding, CA), placed in pre‐cooled 2‐methylbutane, and stored at −80° until use. Cryosections (7 µm) were fixed using 4% paraformaldehyde for 15 min followed by 1% Triton‐X for 5 min. Standard immunofluorescent staining procedures were followed. Briefly, sections were stained using Alexa 488‐labelled agglutinin‐1 (1 : 100; Vector Laboratories, Burlingame, CA) and rabbit anti‐mouse caspase 3 (1 : 50; Santa Cruz Biotechnology, Delaware Ave, Santa Cruz, CA) followed by Alexa 594‐labelled anti‐rabbit antibody. Sections were observed and photographed with a Zeiss microscope (Carl Zeiss, Oberkochen, Germany). The caspase 3 expression levels were quantified based on the intensity of the fluorescent signal analysed using the computer image analysis software image j (National Institutes of Health, Bethesda, MA).
Cells in culture
To determine whether endothelial cell apoptosis was a key event during loss of BBB integrity in lupus we used HBMVECs (Cat# ACBRI‐376).58, 59, 60 These primary human brain microvascular cells were obtained from Applied Cell Biology Research Institute (ACBRI, Kirkland, WA). HBMVECs were seeded on 1% gelatine‐coated 25‐cm2 tissue‐culture flasks and grown in CS‐C complete medium (ACBRI) supplemented with 10% fetal bovine serum (Gibco‐Life Technologies, Grand Island, NY), heparin (100 lg/ml), endothelial cell growth factor supplement (50 lg/ml), sodium pyruvate (2 mm), l‐glutamine (2 mm), penicillin (100 U/ml) and streptomycin (100 lg/ml) (Sigma‐Aldrich, St Louis, MO) with attachment factors (ABCRI) at 37° in a humidified 5% CO2 incubator. Cultured cells were identified as endothelial by their morphology and von Willebrand factor antibody and their viability was assessed by MTT assay. HBMVECs are obtained at passage 2 for each experiment and are used for all experimental paradigms between two and eight passages, within 6 to 27 cumulative population doublings. We observed > 98% viability for HBMVECs in culture.
Treatment of cells
HBMVEC were treated with serum isolated from control patients or lupus patients (5%), 10 nm human C5a (R & D Systems, Minneapolis, MN) or the selective C5aR1 antagonist (C5aRa, 0·1 µg/ml) PMX53.61 The concentrations of lupus serum, C5a, and C5aRa were based on the response obtained in our previous studies.62 Clinical samples were obtained from children attending the paediatric rheumatology clinics at the Women and Children's Hospital of Buffalo. Samples were obtained from two boys and six girls who ranged in age from 7 to 15 years. The serum samples were obtained from newly diagnosed patients during a clinical visit. The C3/C4 levels were below normal (C3: 80–175 mg/dl; C4: 14–40 mg/dl) indicating complement activation. Approval to acquire and use clinical materials was in accordance with the University at Buffalo Children and Youth Institutional Review Board.
Cell viability using the MTT assay
Viability of HBMVECs in culture before and after treatment with lupus serum, C5a and C5aRa was assessed using the MTT assay. The assay measures the ability of an active mitochondrial enzyme to reduce the MTT substrate (yellow to blue) in live cells. Isolated cells were plated in serum‐free conditions on 48‐well plates pre‐coated with laminin. After 24 hr of culture, 0·5 mg/ml MTT substrate (thiazolyl blue tertrazolium bromide) was added and cells were incubated for an additional 4 hr, and then solubilized with 10% SDS/HCl (0·01 M) overnight. Absorbance was measured at 595 nm.
Terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labelling assay
Terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labelling (TUNEL) assay was performed as described earlier21, 63, 64 to visualize DNA damage in cells. 1 × 104 cells were seeded into the 35‐mm culture dishes with glass‐bottom wells. Next day, the media were exchanged for fresh RPMI‐1640 medium. The cells were treated with lupus serum, C5a or lupus serum + C5aRa for 48 hr. Respective negative controls were maintained without treatment. Cells were fixed with 4% methanol‐free paraformaldehyde in PBS for 10 min at room temperature. After fixation, wells were washed with PBS, permeabilized with a 0·2% Triton X‐100 solution for 5 min, and washed twice in PBS, then 100 μl of equilibration buffer was added at room temperature and incubated for 5–10 min. Samples were washed with PBS and incubated with terminal deoxynucleotidyl transferase, recombinant (rTdT) buffer at 37° for 60 min inside the humidified chamber according to the manufacturer's protocol (Biotool.com; CAT #B31115 TUNEL Apo‐Green Detection Kit). Reaction was terminated by adding 100 μl of SSC for 15 min. The wells were washed thrice, using PBS for 5 min to remove unincorporated fluorescein‐12‐dUTP nucleotides. Fragmented DNA was examined under an inverted fluorescence microscope (Carl Zeiss). For each sample, the total number of cells and the number of TUNEL‐positive cells were quantified in 10 representative fields. The results were presented as a representation from a series of three separate experiments.
CellEvent® Caspase‐3/7 Apoptosis Detection Reagent
HBMVECs are grown to 70% confluence in a Petri dish with a glass bottom and treated with lupus serum, C5a or lupus serum + C5aRa for 48 hr. After incubation, cells are wash in PBS followed by addition of the CellEvent® Caspase‐3/7 Green Detection Reagent, which allows examination of caspase 3/7 activation in live cells. The CellEvent® Caspase‐3/7 Green Detection Reagent constitutes an intrinsically non‐fluorescent four amino acid peptide Asp‐Glu‐Val‐Asp called DEVD peptide that inhibits the ability of the dye to bind to DNA. However, after activation of caspase 3/7 in apoptotic cells, the DEVD peptide is cleaved, enabling the dye to bind to DNA and produce a bright, fluorogenic response. The fluorescence emission of the dye when bound to DNA is ~ 530 nm and can be observed in the FITC range.
RNA extraction
Cytoplasmic RNA was extracted by an acid guanidinium‐thiocyanate‐phenol‐chloroform method as described using Trizol reagent (Invitrogen‐Life Technologies, Carlsbad, CA). The amount of RNA was quantified using a Nano‐Drop ND‐1000 spectrophotometer (Nano‐Drop™, Wilmington, DE) and isolated RNA was stored at −80° until used.
Quantitative PCR analysis of apoptosis responsive genes
Apoptosis‐related gene expression was analysed with quantitative RT‐PCR (Applied Biosystems 7500 Fast, Foster City, CA) using a real‐time SYBR Green/ROX gene expression assay kit (Qiagen, Valencia, CA). The cDNA was directly prepared from cultured cells using a Fastlane® Cell cDNA kit (Qiagen); and mRNA levels of apoptotic genes such as, Bax, FADD and caspase 1, 4, 8 and 10 as well as the reference gene, β‐actin, were analysed using gene‐specific SYBR Green‐based QuantiTect® Primer assays (Qiagen). Quantitative PCR was performed in a reaction volume of 25 μl according to the manufacturer's instructions. Briefly, 12·5 μl of master mix, 2·5 μl of assay primers (10 ×) and 10 μl of template cDNA (100 ng) were added to each well. After a brief centrifugation, the PCR plate was subjected to 35 cycles under the following conditions: (i) PCR activation at 95° for 5 min, (ii) denaturation at 95° for 5 seconds and (iii) annealing/extension at 60° for 10 seconds. All samples and controls were run in triplicate on a Stratagene MX3000P Real‐Time PCR system. To provide precise quantification of the initial target in each PCR, the amplification plot was examined and the data were calculated as described. Relative expression of mRNA species was calculated using the comparative threshold cycle number (CT) method.65 Briefly, for each sample, a difference in CT values (∆CT) is calculated for each mRNA by taking the mean CT of duplicate tubes and subtracting the mean CT of the duplicate tubes for the reference RNA (β‐actin) measured on an aliquot from the same RT reaction. The ∆CT for the treated sample is then subtracted from the ∆CT for the untreated control sample to generate a ∆∆CT. The mean of these ∆∆CT measurements is then used to calculate the levels in the targeted cytoplasmic RNA relative to the reference gene and normalized to the control as follows: Relative levels or Transcript Accumulation Index = 2–∆∆CT. This calculation assumes that all PCRs are working with 100% efficiency. All PCR efficiencies were found to be > 95%; therefore, this assumption introduces minimal error into the calculations. All data were controlled for quantity of RNA input and by performing measurements on an endogenous reference gene, β‐actin.
Statistical analysis
Data are expressed as means ± SE and were analysed using minitab 12 statistical software (Minitab, State College, PA). For comparisons between two groups, Student's t‐test was used for parametric data and Mann–Whitney U‐test for nonparametric data. Statistical comparisons between more than two groups were performed using an analysis of variance. A post‐hoc analysis using Bonferroni's test was carried out. A statistically significant difference was accepted when the P value was < 0·05.
Results
Vascular pathological changes in lupus brain
We measured endothelial cell density in the cerebral vasculature on vascular corrosion casts of the Circle of Willis of lupus and control mice. In both groups, endothelial cells aligned with the direction of blood flow through the vessels. Quantification of endothelial cell number on high magnification SEM images of the casts demonstrated that lupus brain vessels had significantly lower cell density (3·23 × 103 cells/mm2) than vessels from congenic controls (5·07 × 103 cells/mm2, P = 0·016) (Fig. 1).
Figure 1.
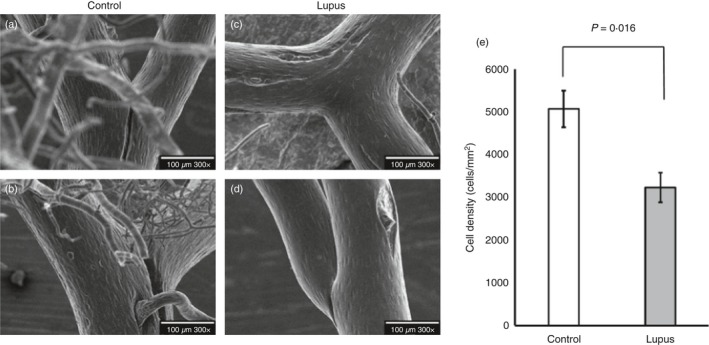
Vascular damage occurs in experimental lupus brain. Representative scanning electron microscopy images of brain vasculature casts show endothelial cell irregularities in MRL/lpr mice (c,d) compared with congenic MRL +/+ controls (a,b). The endothelial cell number was quantified using image J (e). Endothelial cell number was significantly reduced in lupus mice as shown in (e). BA, basilar artery; ACA, anterior cerebral artery; MCA, middle cerebral artery; PCA, posterior cerebral artery. Statistical analysis showed significant change, P < 0·05.
Apoptosis leads to loss of endothelial cells in brain vasculature
To determine whether the effect of reduced number of endothelial cells observed in the casts from MRL/lpr mice could be due to apoptosis, brain sections were stained for caspase 3 using Alexa 488‐labelled isolectin and rabbit anti‐mouse caspase 3 followed by Alexa 594‐labelled anti‐rabbit antibody. Sections were observed and photographed with a Zeiss microscope (Carl Zeiss). Isolectin staining overlaid with caspase 3 staining demonstrates that the walls of the microvessels in MRL/lpr mouse underwent caspase‐dependent apoptosis, which could alter the integrity of the brain microvasculature. (Fig. 2).
Figure 2.
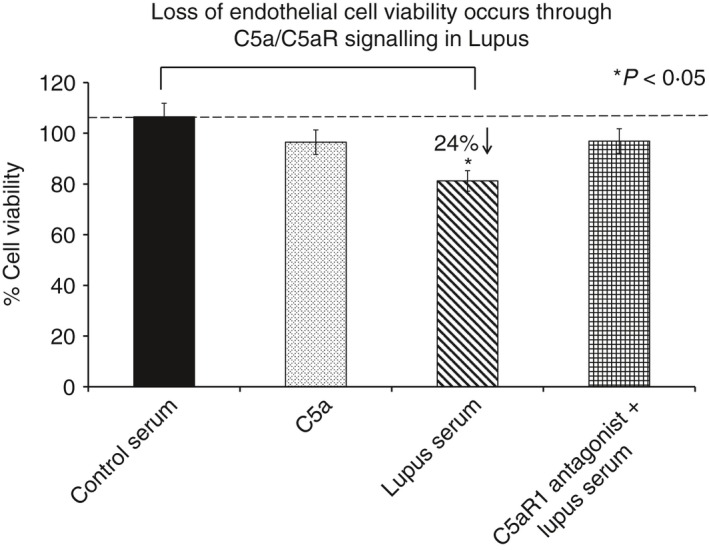
Endothelial cell viability is reduced in experimental lupus. human brain microvascular endothelial cells treated with lupus serum, showed a significant decrease in cell viability as measured by an MTT assay. Treatment of lupus‐treated cells with C5aRa reversed the lupus‐induced effect, indicating that the lupus‐induced effects are mediated via C5a/C5aR1 signalling. Results are expressed as the mean ± SD from n = 3 separate experiments. P < 0·05 is considered statistically significant.
C5a reduces cell viability in brain endothelial cells in lupus
Viability of HBMVECs in culture before and after treatment with lupus serum, C5a and a C5aR1 antagonist (C5aRa) was assessed using the MTT assay. A significant decrease in cell viability was observed in HBMVECs after treatment with lupus serum compared with the cells treated with normal serum (Fig. 3). To determine the mechanism/s by which the cell number is reduced, apoptosis was assessed in HBMVECs treated with lupus serum and C5a. Apoptosis was determined by two independent methods: anti‐active caspase 3 assay and TUNEL.
Figure 3.
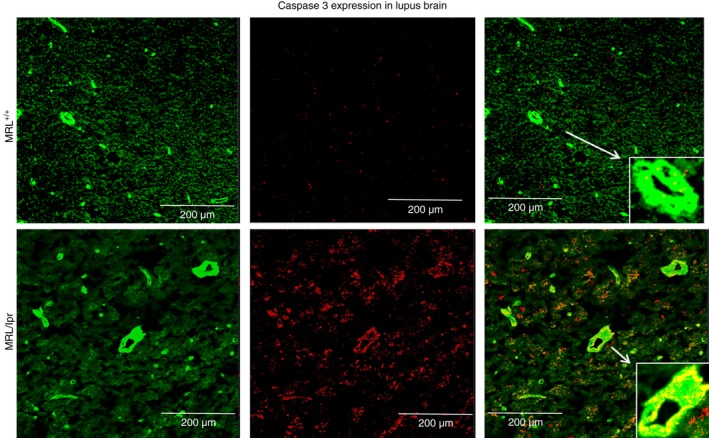
Endothelial cells in brain vasculature undergo apoptosis in experimental lupus. MRL +/+ mice and MRL/lpr mice were killed at 18 weeks of age. Brains were harvested, cryosectioned, immunostained and observed using a Zeiss confocal microscope. Representative sections were stained with Alexa 594 labelled anti‐caspase‐3 and observed at × 60 under oil. Lining of the vasculature was stained using Alexa 488‐labelled agglutinin1. Overlay (c) shows increased caspase 3 activity in the brain vasculature of MRL/lpr mouse brain (b) compared with controls (a).
C5a induces caspase activation in lupus‐serum‐treated endothelial cells
Caspase‐3/7 activation was significantly increased in C5a‐ and lupus‐serum‐treated cells compared with the normal‐serum‐treated HBMVEC controls (Fig. 4). image J quantification of fluorescence signal intensity in pixel units indicates a significant increase in the fluorescence signal in C5a‐treated (46% increase, P < 0·05) and lupus‐serum‐treated (65% increase, P < 0·01) HBMVECs compared with the normal controls. Treatment of HBMVECs with the C5aRa and subsequently with lupus serum resulted in a significant decrease in activation of caspase 3/7, as indicated by a lower fluorescence signal intensity compared with HBMVECs treated with lupus serum alone (57% decrease P < 0·05) indicative of the involvement of C5a/C5aR1 signalling in lupus.
Figure 4.
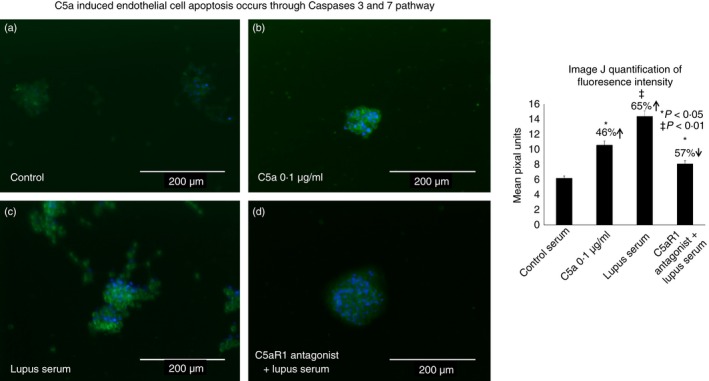
C5a‐induced endothelial cell apoptosis occurs through caspases 3 and 7 pathway. Brain endothelial cells were treated with (a) control serum, (b) C5a, (c) lupus serum and (d) C5aR1 antagonist and lupus serum. Cells were stained with CellEvent caspase 3/7 Green Detection Reagent with absorption/emission maxima of 502/530nm. The representative histogram shows the ImageJ quantification (e) Cell Imaging System.
DNA damage induced by lupus serum is C5a dependent
DNA damage was analysed using TUNEL assay, which confirmed the presence of terminal DNA damage in lupus‐serum‐treated cells compared with HBMVECs treated with normal serum (Fig. 5). Increased APO‐green florescence intensity indicates the degree of DNA damage induced by lupus serum or C5a. Significant increase in APO‐green fluorescence intensity in C5a‐ and lupus‐serum‐treated cell was observed as compared to the normal‐serum‐treated HBMVEC controls. image J quantification of fluorescence signal intensity in pixel units indicates an increase in the fluorescence signal in C5a‐treated (31% increase, P < 0·05) and lupus‐serum‐treated (61% increase, P < 0·01) HBMVECs compared with the normal controls. Treatment of HBMVECs with the C5aRa and subsequently with lupus serum, resulted in a significant decrease in APO‐green fluorescence intensity compared with HBMVECs treated with lupus serum alone (45% decrease, P < 0·05). The results confirm that lupus‐serum‐treated cells undergo cell death by apoptosis and that complement involvement via C5a/C5aR1 signalling is likely to be a mechanism through which endothelial cell death and BBB breakdown occur in lupus‐associated neurodegeneration.
Figure 5.
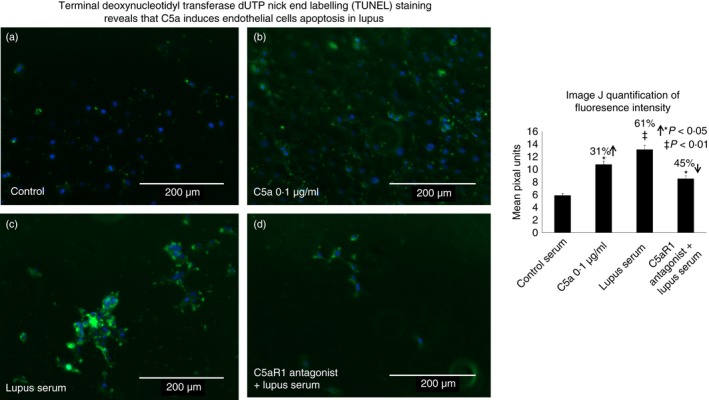
TUNEL staining reveals that C5a induces endothelial cell apoptosis in lupus brain. DNA fragmentation was detected using a TUNEL Apo‐Green Detection Kit. The number of TUNEL‐positive cells was quantified in 10 representative fields. The results are presented as a representation of three separate experiments and the histogram shows the image J quantification of fluorescence signal intensity in pixel units. Increased TUNEL staining is observed in human brain microvascular endothelial cells treated with lupus serum or C5a compared with controls, which was reduced by treatment with C5aRa. Imaging was performed with the EVOS ® FL Cell Imaging System.
Apoptotic gene expression profiling using quantitative PCR
Changes in apoptotic gene expression levels were analysed in endothelial cells treated with lupus serum or C5a by real‐time PCR. An array of apoptotic genes were evaluated, selected based on their role in the intrinsic and extrinsic apoptotic pathways. Therefore, the effect of C5a/C5aR1 signalling on the gene expression of BCL2‐L11 BAX, BIK, APAF‐1, CASP1, CASP4, CASP8, CASP10, CIDEB, FADD, DAPK1 and DFFA genes were examined in HBMVECs treated with lupus serum for 24 hr, C5a or the C5aRa. mRNA was extracted and subjected to quantitative RT‐PCR using specific primers. Table 1 provides the list of primers used as well as the PCR conditions. We observed a significant increase in gene expression levels of pro‐apoptotic genes such as BAX (123%, P < 0·001); BCL2‐L11 (82%, P < 0·01); BIK (41%, P < 0·05) and APAF‐1 (34%, P < 0·05) in HBMVECs treated with lupus serum (Fig. 6). Although treatment with C5a showed an increase in gene expression levels of BAX, BCL2‐L11, APAF‐1 and BIK, significant increase was observed in BAX (82%, P < 0·01) and BCL2‐L11 (80%, P < 0·05) gene expression. HBMVECs treated with normal serum were used as controls. However, when HBMVECs were pre‐treated with the C5aRa and subsequently with lupus serum, BAX, BCL2‐L11, APAF‐1 and BIK, gene expression was significantly decreased compared with HBMVECs treated with lupus serum alone [72% decrease, P < 0·01 (BAX); 55% decrease P < 0·01 (BCL2‐L11); 27% decrease (NS) (APAF‐1) and 62% decrease P < 0·01 (BIK)] indicating that C5a/C5aR1 signalling is an important aspect of BBB integrity in lupus.
Table 1.
Primer sequences
| Gene | Accession # | Forward | Reverse | Amplicon length bp | Tm |
|---|---|---|---|---|---|
| β‐actin | NM_001101.3 | 5′‐TTCTACAATGAGCTGCGTGTG‐3′ | 5′‐GGGGTGTTGAAGGTCTCAAA‐3′ | 114 | 58 |
| BCL2 L11 | NM_001204106.1 | 5′‐CTGCTGGACACACACATACA ‐3′ | 5′‐GGGCTGAGGAAACAGAGTAAA ‐3′ | 135 | 58 |
| BAX‐ BCL2 associated X protein | NM_000633.2 | 5′‐TGG AGCTGCAGAGGATGATTG‐3′ | 5′‐GAAGTTGCCGTCAGAAAACATG‐3′ | 121 | 60 |
| BIK‐ BCL2 interacting killer (apoptosis‐inducing) | NM_001197.4 | 5′‐CTGGAACACTGCTGAGGTTT‐3′ | 5′‐GACAATTGCAGAGCCATAGGA ‐3′ | 100 | 62 |
| DFFA‐DNA fragmentation factor 45 000 MW subunit A | NM_004401.2 | 5′‐CACCCTCAGCCTCATCATTT ‐3′ | 5′‐GCGTATGTTGAGACCTGGAATA ‐3′ | 94 | 58 |
| CIDEB cell death‐inducing DFFA‐like effector b | XM_005267540.3 | 5′‐CATCTCTGTCACGTCCACTAATC‐3′ | 5′‐CTCACTGCTCTGGCTTCATT‐3′ | 104 | 62 |
| FADD Fas associated via death domain | NM_003824.3 | 5′‐TCTCCTCTCTGAGACTGCTAAG ‐3′ | 5′‐AGAGAGTGCTGTGTGTCAATC‐3′ | 102 | 58 |
| DAPK1 death‐associated protein kinase 0 | NM_001288729.0 | 5′‐GTGGATGGTCATTGCAGTTTAAG‐3′ | 5′‐TACTGGAGGATGAGAGATGGAG‐3′ | 109 | 58 |
| APAF‐1 Apototic protease activating factor | NM_001160.2 | 5′‐GGACGACAGCCATTTCCTAATA‐3′ | 5′‐GCAGCTTAGCTTGCTGATAAAC‐3′ | 82 | 58 |
| CASP1 ‐caspase 1 | NM_001223.4 | 5′‐ACACAAGAAGGGAGGAGAGA‐3′ | 5′‐CTTCACCCATGGAACGGATAA‐3′ | 84 | 60 |
| CASP4‐ caspase 4 | NM_001225.3 | 5′‐GAATCTGACAGCCAGGGATATG‐3′ | 5′‐CCATGAGACATGAGTACCAAGAA ‐3′ | 102 | 60 |
| CASP8‐ caspase 8 | NM_001080124.1 | 5′‐GGAGCTGCTCTTCCGAATTA ‐3′ | 5′‐CATGACCCTGTAGGCAGAAA‐3′ | 124 | 62 |
| CASP10‐ caspase 10 | NM_001206524.1 | 5′‐ACAACTCTCCCTCCCAGATAA‐3′ | 5′‐CTTAGTCTGTAAGTGAGTGGGC‐3′ | 112 | 58 |
Figure 6.
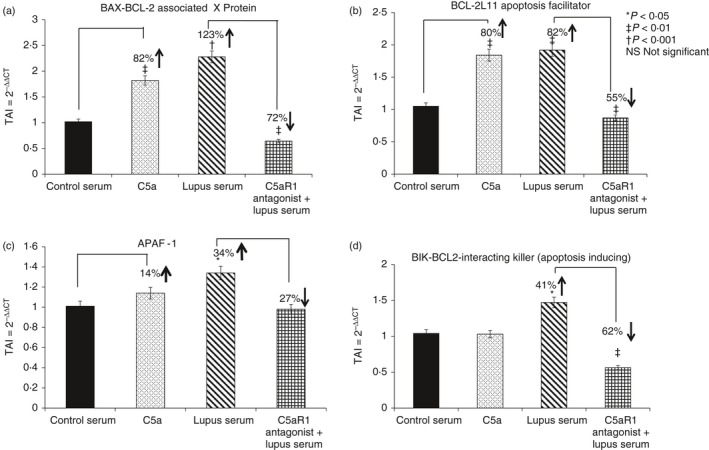
Gene expression of apoptotic regulatory proteins is C5a dependent in lupus. Effects of C5a, C5aRa and/or lupus serum on gene expression of pro‐apoptotic regulatory proteins (a) BAX, (b) BCL‐2L11, (c) APAF‐1 and (d) BIK were assessed. RNA was extracted from human brain microvascular endothelial cells and subjected to real‐time PCR. Expression levels of BAX, BCL‐2L11, APAF‐1 and BIK genes were significantly increased in lupus‐serum‐treated cells compared with control, whereas C5aRa treatment alleviated the change. Results are expressed as the mean ± SD, n = 3 separate experiments. P < 0·05 is considered statistically significant.
Caspase 1 and caspase 8 gene expression was significantly higher in cells treated with lupus serum compared with those treated with control serum (40% increase in caspase 1 versus control, P < 0·05) and 89% increase in caspase 8 versus control P < 0·001, respectively) (Fig. 7). No significant changes were observed in caspase 4 and 10 gene expression, when treated with C5a or lupus serum. In HBMVECs pre‐treated with the C5aRa and subsequently with lupus serum, caspase 8 gene expression was also significantly decreased compared with HBMVECs treated with lupus serum alone (29% decrease P < 0·05) indicative of the involvement of C5a/C5aR1 signalling.
Figure 7.
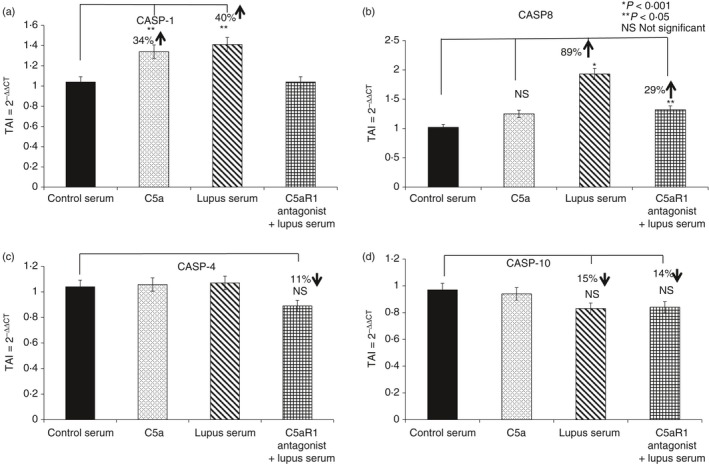
C5a/C5aR1 signalling causes endothelial cell apoptosis through the caspase 1/8 pathway. Expression of (a) caspase 1 (Casp‐1), (b) Casp‐8, (c) Casp‐4 and (d) Casp‐10 were assessed in human brain microvascular endothelial cells (HBMVECs) treated with C5a, lupus serum alone and in combination with C5aRa. Treatment with lupus serum significantly increased Casp‐1 and Casp‐8 gene expression levels in HBMVEC cells, whereas no significant differences were observed in Casp‐4 and Casp‐10 gene expression levels. All statistical comparisons were made to the untreated controls. Results are expressed as the mean ± SD from n = 3 separate experiments. P < 0·05 is considered statistically significant.
The HBMVECs treated with lupus serum showed a significant increase in the gene expression levels of several cell death‐inducing effector proteins such as FADD (48% increase, P < 0·01); CIDEB (314% increase, P < 0·001); DFFA (38% increase, P < 0·05) and DAPK1 (60%, increase P < 0·01) (Fig. 8). Treatment with C5a also showed increases in FADD, CIDEB, DFFA and DAPK1 gene expression levels with significant increases in FADD (51%, P < 0·01), CIDEB (94%, P < 0·01) DFFA (58% increase, P < 0·01) and DAPK1 (243%, increase P < 0·001). Statistical control comparators were HBMVECs treated with normal serum. In HBMVECs pre‐treated with the C5aRa and subsequently with lupus serum, FADD and CIDEB gene expression was significantly decreased compared with HBMVEC treated with lupus serum alone [22% decrease, P < 0·05 (FADD) and 80% decrease, P < 0·001 (CIDEB)] indicative of the role of C5a/C5aR1 signalling in SLE.
Figure 8.
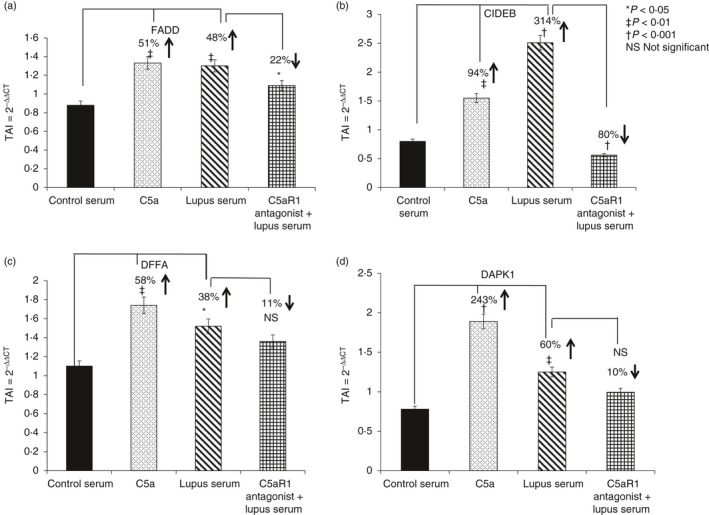
Apoptotic biomarker gene expression is C5a/C5aR1 dependent in central nervous system lupus. Gene expression of apoptotic biomarkers (a) FADD, (b) CIDEB, (c) DFFA and (d) DAPK1 was determined in human brain microvascular endothelial cells treated with C5a or lupus serum. The gene expression levels of FADD, CIDEB, DFFA and DAPK1 were measured using real‐time PCR. C5a and lupus serum treatment significantly increased FADD, CIDEB, DFFA and DAPK1 gene expression levels. Treatment with C5aRa resulted in a reversal of the lupus‐mediated effects. All statistical comparisons were made to the untreated controls. Results are expressed as the mean ± SD from n = 3 separate experiments. P < 0·05 is considered statistically significant.
Discussion
Our earlier results demonstrate complement‐dependent loss of BBB integrity in mouse brain in vivo and in brain endothelial cells in vitro.5, 6 Results of the present study suggest that pathological vascular remodelling occurs in lupus, resulting in the loss of BBB integrity. At 18 weeks of age there was a significant reduction in the number of brain vascular endothelial cells. Significant apoptosis was observed in the vascular endothelium, which could lead to the reduced cell number. In addition, our data show that these results are translatable, because HBMVECs in culture, when exposed to lupus serum from patients, underwent apoptosis.
The pathology in CNS lupus includes both vascular and parenchymal damage. The parenchymal injury occurs only after the BBB is disrupted, allowing the entry of antibodies, infiltrating cells and inflammatory mediators.66 In addition, breach of the BBB results in altered metabolic profiles.67 Our results are the first to show that pathological remodelling occurred in brains of MRL/lpr mice, indicating loss of endothelial cells that results in increased intercellular and transcellular transport across the BBB. This is in line with our earlier results where monolayer of bEnd3 cells (mouse brain endothelial cells) in culture was rendered leaky when treated with lupus serum.
Several studies have shown that apoptosis occurs in lupus brains. Entry of a number of toxic effectors such as autoantibodies and other inflammatory mediators could cause cell apoptosis. One of the circulating proteins in lupus serum that correlate with CNS lupus is the complement protein C5a.30, 68, 69, 70 C5a was shown to cause apoptosis in different settings, such as sepsis.71 Our studies have clearly demonstrated that C5a induces apoptosis in lupus brain, which was alleviated by inhibition of C5a/C5aR1 signalling.24 In addition, C5a induced apoptosis in bEnd3 cells.5, 35 Brain endothelial cells are susceptible to complement‐mediated effects, due to their exposure to both systemic and CNS‐synthesized complement proteins. Complement‐mediated endothelial damage promotes BBB breakdown, resulting in neuro‐inflammation due to an influx of active complement fragments and cytokines.
Our earlier studies using the HBMVECs showed reduced transendothelial electrical resistance when treated with lupus serum, indicating reduced resistance to transport across the layer.62 In this study our results show that both lupus serum and C5a caused endothelial cell apoptosis in HBMVECs, which could be reduced by inhibition of C5a/C5aR1 signalling, indicating complement dependence.
In non‐immune cells such as endothelial cells, apoptosis signals may be initiated by both the intrinsic and extrinsic pathways. The intrinsic pathway, or the BCL2 pathway, is induced by activation of caspase 9 via APAF‐1 and CytC causing rupture of the mitochondrial membrane, through the release of Bcl‐2 family proteins into the cytoplasm.72 Our results show a significant increase in APAF‐1 gene expression in C5a and lupus‐treated HBMVECs. We also observed a significant increase in gene expression levels of BAX; (a BCL2‐associated pro‐apoptotic); BIK (an apoptosis‐inducing protein) and BCL2‐L11 (an apoptosis facilitator) in HBMVECs treated with lupus serum. These three BCL2 family members act as anti‐ or pro‐apoptotic regulators that are involved in a wide variety of cellular activities. BAX forms a heterodimer with BCL2, and functions as an apoptotic activator and interacts with the mitochondrial voltage‐dependent anion channel, leading to the loss in membrane potential and the release of CytC. BCL2‐L11 induces apoptosis via the cysteine‐aspartic acid protease (caspase) dependent pathway.
The extrinsic pathway, on the other hand, is caspase 8/10 dependent. Sequential activation of caspases plays a central role in the execution‐phase of cell apoptosis.73 In our study, treatment with lupus serum resulted in increased gene expression of caspases 1 and 8. Caspase 10 is suggested to be modulated by caspase 8 and activates caspases 3 and 7, resulting in cell apoptosis. We did not observe a significant change in caspase 10 gene expression but observed activation of caspases 3 and 7. Increased caspase 1 in lupus‐treated HBMVECs can lead to activation of effector caspases resulting in endothelial cell apoptosis and a decrease in BBB function. Caspases 1 and 10 are initiator caspases that cleave inactive pro‐forms of effector caspases, thereby activating them, the effector caspases in turn cleave other protein substrates within the cell, to trigger the apoptotic process.
In addition, our results show that treatment of HBMVECs with C5a and lupus serum resulted in a significant increase in gene expression of several cell death‐inducing effector family proteins that include FADD, CIDEB, DAPK1, DFFA and RIPK1, which was C5a/C5aR1 signalling dependent. FADD is an adaptor molecule that is recruited by TNFRSF6/Fas‐receptor, tumour necrosis factor receptor, TNFRSF25 and TNFSF10/TRAIL‐receptor, to participate in the death signalling initiated by these receptors.74 DNA fragmentation factor (DFF), is the substrate for caspase‐3 and triggers DNA fragmentation during apoptosis, whereas cell death‐inducing DFFA‐like effector (CIDE) proteins contribute to the chromatin condensation and DNA fragmentation events of apoptosis. The CIDE_N domain is believed to regulate the activity of ICAD/DFF45, and the CAD/DFF40 and CIDE nucleases during apoptosis.75 The extrinsic apoptotic pathway is activated by several extracellular ligands binding to cell‐surface death receptors, which leads to the formation of the death‐inducing signalling complex (DISC). The increased expression, observed in this study, of FADD and the initiator caspases, caspases 1 and 8, suggest that apoptosis may occur through DISC and not through caspases 4 and 10, the expression of which remained unchanged in lupus.
In conclusion, our studies demonstrate for the first time that vascular remodelling with reduction in number of endothelial cells occurs in experimental lupus brain. Apoptosis plays a key role in this setting. Another important feature of this study is the demonstration that the results obtained in the mouse cells could be replicated in human vascular endothelial cells, suggesting that the results could be translated to human settings. Lastly, our results show that the observed changes in brain vasculature are C5a/C5aR1 dependent, and therefore continue to strengthen the possibility that interrupting C5a/C5aR1 signalling could be an effective therapeutic target for CNS lupus and other neurodegenerative diseases.
Funding
The authors would like to acknowledge the research funding received from the Dr Louis Sklarow Memorial Trust 2015–2016. This work was also supported by NIH grant R01‐AR‐060605 (JNJ).
Competing interest
The authors have declared that no competing interests exist.
Disclosure
No financial conflicts.
References
- 1. Huizinga TW, Diamond B. Lupus and the central nervous system. Lupus 2008; 17:376–9. [DOI] [PubMed] [Google Scholar]
- 2. van Dam AP. Diagnosis and pathogenesis of CNS lupus. Rheumatol Int 1991; 11:1–11. [DOI] [PubMed] [Google Scholar]
- 3. Zvaifler NJ, Bluestein HG. The pathogenesis of central nervous system manifestations of systemic lupus erythematosus. Arthritis Rheum 1982; 25:862–6. [DOI] [PubMed] [Google Scholar]
- 4. Abbott NJ, Mendonca LL, Dolman DE. The blood–brain barrier in systemic lupus erythematosus. Lupus 2003; 12:908–15. [DOI] [PubMed] [Google Scholar]
- 5. Jacob A, Hack B, Chen P, Quigg RJ, Alexander JJ. C5a/CD88 signaling alters blood–brain barrier integrity in lupus through nuclear factor‐κB. J Neurochem 2011; 119:1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, Alexander JJ. C5a alters blood–brain barrier integrity in experimental lupus. FASEB J 2010; 24:1682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gulinello M, Putterman C. The MRL/lpr mouse strain as a model for neuropsychiatric systemic lupus erythematosus. J Biomed Biotechnol 2011; 2011:207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brey RL, Sakic B, Szechtman H, Denburg JA. Animal models for nervous system disease in systemic lupus erythematosus. Ann N Y Acad Sci 1997; 823:97–106. [DOI] [PubMed] [Google Scholar]
- 9. Stock AD, Wen J, Doerner J, Herlitz LC, Gulinello M, Putterman C. Neuropsychiatric systemic lupus erythematosus persists despite attenuation of systemic disease in MRL/lpr mice. J Neuroinflammation 2015; 12:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stock AD, Wen J, Putterman C. Neuropsychiatric lupus, the blood‐brain barrier, and the TWEAK/Fn14 pathway. Front Immunol 2013; 4:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dixon FJ. Murine SLE models and autoimmune disease. Hosp Pract (Hosp Ed) 1982; 17:63–73. [DOI] [PubMed] [Google Scholar]
- 12. Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol 1985; 37:269–390. [DOI] [PubMed] [Google Scholar]
- 13. Theofilopoulos AN, Kofler R, Singer PA, Dixon FJ. Molecular genetics of murine lupus models. Adv Immunol 1989; 46:61–109. [DOI] [PubMed] [Google Scholar]
- 14. Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry‐Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim Biophys Acta 2003; 1639:169–76. [DOI] [PubMed] [Google Scholar]
- 15. Atkinson JP. Complement system on the attack in autoimmunity. J Clin Invest 2003; 112:1639–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barrington R, Zhang M, Fischer M, Carroll MC. The role of complement in inflammation and adaptive immunity. Immunol Rev 2001; 180:5–15. [DOI] [PubMed] [Google Scholar]
- 17. Carroll MV, Sim RB. Complement in health and disease. Adv Drug Deliv Rev 2011; 63:965–75. [DOI] [PubMed] [Google Scholar]
- 18. Muller‐Eberhard HJ. Complement. Annu Rev Biochem 1969; 38:389–414. [DOI] [PubMed] [Google Scholar]
- 19. Muller‐Eberhard HJ. The membrane attack complex of complement. Annu Rev Immunol 1986; 4:503–28. [DOI] [PubMed] [Google Scholar]
- 20. Pangburn MK, Morrison DC, Schreiber RD, Muller‐Eberhard HJ. Activation of the alternative complement pathway: recognition of surface structures on activators by bound C3b. J Immunol 1980; 124:977–82. [PubMed] [Google Scholar]
- 21. Alexander JJ, Jacob A, Bao L, Macdonald RL, Quigg RJ. Complement‐dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J Immunol 2005; 175:8312–9. [DOI] [PubMed] [Google Scholar]
- 22. Hugli TE, Muller‐Eberhard HJ. Anaphylatoxins: C3a and C5a. Adv Immunol 1978; 26:1–53. [DOI] [PubMed] [Google Scholar]
- 23. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re‐evaluating the evidence and the myth. J Immunol 2015; 194:3542–8. [DOI] [PubMed] [Google Scholar]
- 24. Jacob A, Hack B, Bai T, Brorson JR, Quigg RJ, Alexander JJ. Inhibition of C5a receptor alleviates experimental CNS lupus. J Neuroimmunol 2010; 221:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barnum SR. Complement in central nervous system inflammation. Immunol Res 2002; 26:7–13. [DOI] [PubMed] [Google Scholar]
- 26. Nataf S, Stahel PF, Davoust N, Barnum SR. Complement anaphylatoxin receptors on neurons: new tricks for old receptors? Trends Neurosci 1999; 22:397–402. [DOI] [PubMed] [Google Scholar]
- 27. Li R, Coulthard LG, Wu MC, Taylor SM, Woodruff TM. C5L2: a controversial receptor of complement anaphylatoxin, C5a. FASEB J 2013; 27:855–64. [DOI] [PubMed] [Google Scholar]
- 28. Manthey HD, Woodruff TM, Taylor SM, Monk PN. Complement component 5a (C5a). Int J Biochem Cell Biol 2009; 41:2114–7. [DOI] [PubMed] [Google Scholar]
- 29. Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber‐Lang M, Mackay CR, et al Functional roles for C5a receptors in sepsis. Nat Med 2008; 14:551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hopkins P, Belmont HM, Buyon J, Philips M, Weissmann G, Abramson SB. Increased levels of plasma anaphylatoxins in systemic lupus erythematosus predict flares of the disease and may elicit vascular injury in lupus cerebritis. Arthritis Rheum 1988; 31:632–41. [DOI] [PubMed] [Google Scholar]
- 31. Buyon JP, Tamerius J, Ordorica S, Young B, Abramson SB. Activation of the alternative complement pathway accompanies disease flares in systemic lupus erythematosus during pregnancy. Arthritis Rheum 1992; 35:55–61. [DOI] [PubMed] [Google Scholar]
- 32. Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J 2012; 26:3680–90. [DOI] [PubMed] [Google Scholar]
- 33. Ward PA. Sepsis, apoptosis and complement. Biochem Pharmacol 2008; 76:1383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ward PA. Role of the complement in experimental sepsis. J Leukoc Biol 2008; 83:467–70. [DOI] [PubMed] [Google Scholar]
- 35. Eadon MT, Jacob A, Cunningham PN, Quigg RJ, Garcia JG, Alexander JJ. Transcriptional profiling reveals that C5a alters microRNA in brain endothelial cells. Immunology 2014; 143:363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krych M, Atkinson JP, Holers VM. Complement receptors. Curr Opin Immunol 1992; 4:8–13. [DOI] [PubMed] [Google Scholar]
- 37. Los M, Wesselborg S, Schulze‐Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity 1999; 10:629–39. [DOI] [PubMed] [Google Scholar]
- 38. Cecconi F, Alvarez‐Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED‐4 homolog) regulates programmed cell death in mammalian development. Cell 1998; 94:727–37. [DOI] [PubMed] [Google Scholar]
- 39. Yuan J, Yankner BA. Apoptosis in the nervous system. Nature 2000; 407:802–9. [DOI] [PubMed] [Google Scholar]
- 40. Hartmann A, Mouatt‐Prigent A, Faucheux BA, Agid Y, Hirsch EC. FADD: a link between TNF family receptors and caspases in Parkinson's disease. Neurology 2002; 58:308–10. [DOI] [PubMed] [Google Scholar]
- 41. Huang P, Rani MR, Ahluwalia MS, Bae E, Prayson RA, Weil RJ, et al Endothelial expression of TNF receptor‐1 generates a proapoptotic signal inhibited by integrin α6β1 in glioblastoma. Cancer Res 2012; 72:1428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tatton WG, Chalmers‐Redman R, Brown D, Tatton N. Apoptosis in Parkinson's disease: signals for neuronal degradation. Ann Neurol 2003; 53 (Suppl. 3):S61–70 [DOI] [PubMed] [Google Scholar]
- 43. Thomas S, Mayer L, Sperber K. Mitochondria influence Fas expression in gp120‐induced apoptosis of neuronal cells. Int J Neurosci 2009; 119:157–65. [DOI] [PubMed] [Google Scholar]
- 44. Thompson SJ, Loftus LT, Ashley MD, Meller R. Ubiquitin‐proteasome system as a modulator of cell fate. Curr Opin Pharmacol 2008; 8:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wosik K, Becher B, Ezman A, Nalbantoglu J, Antel JP. Caspase 8 expression and signaling in Fas injury‐resistant human fetal astrocytes. Glia 2001; 33:217–24. [DOI] [PubMed] [Google Scholar]
- 46. Dzietko M, Boos V, Sifringer M, Polley O, Gerstner B, Genz K, et al A critical role for Fas/CD‐95 dependent signaling pathways in the pathogenesis of hyperoxia‐induced brain injury. Ann Neurol 2008; 64:664–73. [DOI] [PubMed] [Google Scholar]
- 47. Hainsworth AH, Bermpohl D, Webb TE, Darwish R, Fiskum G, Qiu J, et al Expression of cellular FLICE inhibitory proteins (cFLIP) in normal and traumatic murine and human cerebral cortex. J Cereb Blood Flow Metab 2005; 25:1030–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Inohara N, Ding L, Chen S, Nunez G. Harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival‐promoting proteins Bcl‐2 and Bcl‐X(L). EMBO J 1997; 16:1686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Inohara N, Nunez G. Genes with homology to mammalian apoptosis regulators identified in zebrafish. Cell Death Differ 2000; 7:509–10. [DOI] [PubMed] [Google Scholar]
- 50. Konishi S, Ishiguro H, Shibata Y, Kudo J, Terashita Y, Sugiura H, et al Decreased expression of DFF45/ICAD is correlated with a poor prognosis in patients with esophageal carcinoma. Cancer 2002; 95:2473–8. [DOI] [PubMed] [Google Scholar]
- 51. Sabol SL, Li R, Lee TY, Abdul‐Khalek R. Inhibition of apoptosis‐associated DNA fragmentation activity in nonapoptotic cells: the role of DNA fragmentation factor‐45 (DFF45/ICAD). Biochem Biophys Res Commun 1998; 253:151–8. [DOI] [PubMed] [Google Scholar]
- 52. Takahashi M, Ozaki T, Takahashi A, Miyauchi M, Ono S, Takada N, et al DFF45/ICAD restores cisplatin‐induced nuclear fragmentation but not DNA cleavage in DFF45‐deficient neuroblastoma cells. Oncogene 2007; 26:5669–73. [DOI] [PubMed] [Google Scholar]
- 53. Tutino VM, Mandelbaum M, Choi H, Pope LC, Siddiqui A, Kolega J, et al Aneurysmal remodeling in the circle of Willis after carotid occlusion in an experimental model. J Cereb Blood Flow Metab 2014; 34:415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jamous MA, Nagahiro S, Kitazato KT, Satoh K, Satomi J. Vascular corrosion casts mirroring early morphological changes that lead to the formation of saccular cerebral aneurysm: an experimental study in rats. J Neurosurg 2005; 102:532–5. [DOI] [PubMed] [Google Scholar]
- 55. Meng H, Tutino VM, Xiang J, Siddiqui A. High WSS or low WSS? Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. AJNR Am J Neuroradiol 2014; 35:1254–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tutino VM, Liaw N, Spernyak JA, Ionita CN, Siddiqui AH, Kolega J, et al Assessment of vascular geometry for bilateral carotid artery ligation to induce early basilar terminus aneurysmal remodeling in rats. Curr Neurovasc Res 2016; 13:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tutino VM, Liaw N, Spernyak JA, Ionita CN, Siddiqui AH, Kolega J, et al Assessment of vascular geometry for bilateral carotid artery ligation to induce early basilar terminus aneurysmal remodeling in rats. Curr Neurovasc Res 2016; 13:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, et al Methamphetamine alters blood–brain barrier permeability via the modulation of tight junction expression: implication for HIV‐1 neuropathogenesis in the context of drug abuse. Brain Res 2008; 1203:133–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Fernandez SF, et al Tight junction regulation by morphine and HIV‐1 tat modulates blood–brain barrier permeability. J Clin Immunol 2008; 28:528–41. [DOI] [PubMed] [Google Scholar]
- 60. Parikh NU, Aalinkeel R, Reynolds JL, Nair BB, Sykes DE, Mammen MJ, et al Galectin‐1 suppresses methamphetamine induced neuroinflammation in human brain microvascular endothelial cells: neuroprotective role in maintaining blood–brain barrier integrity. Brain Res 2015; 1624:175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benson MJ, Thomas NK, Talwar S, Hodson MP, Lynch JW, Woodruff TM, et al A novel anticonvulsant mechanism via inhibition of complement receptor C5ar1 in murine epilepsy models. Neurobiol Dis 2015; 76:87–97. [DOI] [PubMed] [Google Scholar]
- 62. Mahajan SD, Parikh NU, Woodruff TM, Jarvis JN, Lopez M, Hennon T, et al C5a alters blood–brain barrier integrity in a human in vitro model of systemic lupus erythematosus. Immunology 2015; 146:130–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor‐1. Neurochem Int 2008; 52:447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol 2007; 37:1691–701. [DOI] [PubMed] [Google Scholar]
- 65. Bustin SA. Quantification of mRNA using real‐time reverse transcription PCR (RT‐PCR): trends and problems. J Mol Endocrinol 2002; 29:23–39. [DOI] [PubMed] [Google Scholar]
- 66. Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011; 365:2110–21. [DOI] [PubMed] [Google Scholar]
- 67. Vo A, Volpe BT, Tang CC, Schiffer WK, Kowal C, Huerta PT, et al Regional brain metabolism in a murine systemic lupus erythematosus model. J Cereb Blood Flow Metab 2014; 34:1315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abramson S, Belmont HM, Hopkins P, Buyon J, Winchester R, Weissmann G. Complement activation and vascular injury in systemic lupus erythematosus. J Rheumatol Suppl 1987; 14(Suppl. 13):43–6. [PubMed] [Google Scholar]
- 69. Belmont HM, Hopkins P, Edelson HS, Kaplan HB, Ludewig R, Weissmann G, et al Complement activation during systemic lupus erythematosus. C3a and C5a anaphylatoxins circulate during exacerbations of disease. Arthritis Rheum 1986; 29:1085–9. [DOI] [PubMed] [Google Scholar]
- 70. Crookston KP, Sibbitt WL Jr, Chandler WL, Qualls CR, Roldan CA. Circulating microparticles in neuropsychiatric systemic lupus erythematosus. Int J Rheum Dis 2013; 16:72–80. [DOI] [PubMed] [Google Scholar]
- 71. Flierl MA, Rittirsch D, Chen AJ, Nadeau BA, Day DE, Sarma JV, et al The complement anaphylatoxin C5a induces apoptosis in adrenomedullary cells during experimental sepsis. PLoS One 2008; 3:e2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cunningham RE, Abbondanzo SL, Chu WS, Emory TS, Sobin LH, O'Leary TJ. Apoptosis, bcl‐2 expression, and p53 expression in gastrointestinal stromal/smooth muscle tumors. Appl Immunohistochem Mol Morphol 2001; 9:19–23. [PubMed] [Google Scholar]
- 73. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ 2012; 19:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chan SC, Lin SC, Li P. Regulation of Cidea protein stability by the ubiquitin‐mediated proteasomal degradation pathway. Biochem J 2007; 408:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]