

Summary
T cells play a pivotal role in controlling viral infection; however, the precise mechanisms responsible for regulating T‐cell differentiation and function during infections are incompletely understood. In this study, we demonstrated an expansion of myeloid‐derived suppressor cells (MDSCs), in particular the monocytic MDSCs (M‐MDSCs; CD14+ CD33+ CD11b+ HLA‐DR −/low), in patients with chronic hepatitis C virus (HCV) infection. Notably, HCV‐induced M‐MDSCs express high levels of phosphorylated signal transducer and activator of transcription 3 (pSTAT3) and interleukin‐10 (IL‐10) compared with healthy subjects. Blocking STAT3 signalling reduced HCV‐mediated M‐MDSC expansion and decreased IL‐10 expression. Importantly, we observed a significant increase in the numbers of CD4+ CD25+ Foxp3+ regulatory T (Treg) cells following incubation of healthy peripheral blood mononuclear cells (PBMCs) with MDSCs derived from HCV‐infected patients or treated with HCV core protein. In addition, depletion of MDSCs from PBMCs led to a significant reduction of Foxp3+ Treg cells developed during chronic HCV infection. Moreover, depletion of MDSCs from PBMCs significantly increased interferon‐γ production by CD4+ T effector (Teff) cells derived from HCV patients. These results suggest that HCV‐induced MDSCs promote Treg cell development and inhibit Teff cell function, suggesting a novel mechanism for T‐cell regulation and a new strategy for immunotherapy against human viral diseases.
Keywords: hepatitis C virus, interleukin‐10, myeloid‐derived suppressor cells, regulatory T cells, signal transducer and activator of transcription 3
Introduction
Hepatitis C virus (HCV) is a blood‐borne virus characterized by a high rate (over 80%) of chronic infection. HCV has evolved multiple strategies to evade host immunity, becoming an excellent model to study the mechanisms of persistent infections in humans.1, 2 Although the use of direct antiviral agents has resulted in a significant improvement in the outcome of HCV treatment, this therapeutic cocktail is costly and already facing new issues such as viral mutation, relapse and re‐infection following therapy.3, 4 Additionally, the lack of a vaccine against HCV is a major hurdle to control this global infection. The failure to successfully manage HCV chronic infection in specific populations, and to develop an effective vaccine, stems from our incomplete understanding of the host immune response that permits HCV persistence.
Myeloid‐derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that are generated due to aberrant myelopoiesis under pathological conditions, such as cancer, inflammatory and infectious diseases.5, 6, 7 These cells have gained special attention recently due to their potential to suppress immune responses by multiple mechanisms, including over‐expression of nitric oxide synthase, arginase 1, reactive oxygen species, interleukin‐4 receptor‐α (IL‐4Rα), programmed death‐1 ligand, IL‐10, tumour growth factor β (TGF‐β), and phosphorylated signal transducer and activator of transcription 3 (pSTAT3), all of which are important mediators of innate immune and inflammatory responses against pathogenic infections.8, 9, 10, 11, 12, 13, 14 In mice, MDSCs are defined by the co‐expression of the Gr‐1 and CD11b surface antigens. Human MDSCs are less well‐characterized because of the lack of uniform phenotypic markers.13, 14, 15 However, they usually express the common myeloid markers CD33 and CD11b, but lack the maturation marker HLA‐DR. Human MDSCs are primarily dissected into two subsets: monocytic MDSCs (M‐MDSCs; CD14+ CD11b+ CD33+ HLA‐DR−/low) and granulocytic MDSCs (G‐MDSCs; CD14− CD11b+ CD33+ HLA‐DR−/low). These two subsets of MDSCs may have different biological functions and use different mechanisms for immune suppression.6, 7
Although MDSCs may contribute to immune homeostasis after infection by limiting excessive inflammatory processes, their expansion may be at the expense of pathogen elimination and so may lead to infection persistence. Despite many studies reporting expansion of MDSCs in various disease models including viral infections,16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 the phenotypic and functional features of MDSCs remain controversial and their role in regulating T‐cell responses and viral persistence are far from clear. For example, although some studies have shown that M‐MDSC frequencies are remarkably elevated and play a role in the pathogenesis of HIV or HCV infection,20, 21, 22, 23, 24, 25 Bowers et al.26 reported an immunosuppression by neutrophils during HIV‐1 infection, whereas Nonnenmann et al.28 found no significant increases in MDSCs, which lack suppressive functions in peripheral blood of chronically HCV‐infected individuals. These discrepancies in MDSC frequency and suppressive capability might partially result from measuring different MDSC subsets, using different methodologies, investigating different patient populations, and/or focusing on different disease stages with specific cytokine milieus that may induce distinct MDSC phenotypes. Therefore, further phenotypic and functional characterization of these particular types of cells will delineate the mechanisms by which MDSCs promote immune suppression under specific disease conditions.
In this study, we characterized the phenotypes and functions of MDSCs in patients with chronic HCV infection, with the primary focus on the role of MDSCs in regulating T‐cell differentiation and function, a mechanism by which HCV might modulate T‐cell responses to facilitate the establishment and maintenance of chronic infection. We provide evidence that HCV‐induced MDSCs promote regulatory T (Treg) cell development and inhibit effector T (Teff) cell function via the STAT3 pathway, representing a novel mechanism for T‐cell regulation during viral infection.
Materials and methods
Subjects
The study protocol was approved by the joint institutional review board at East Tennessee State University and James H. Quillen VA Medical Center (ETSU/VA IRB, Johnson City, TN). The study subjects were composed of two populations: 129 chronically HCV‐infected individuals and 59 healthy subjects (HS). HCV genotype (70% type 1, 30% type 2 or 3) and viral load (ranging from 12 300 to 50 000 000 IU/ml) were performed by Lexington VAMC, and all subjects were virologically and serologically positive for HCV, before antiviral treatment. Healthy subjects were serologically negative for hepatitis B virus, HCV, and HIV infection and blood buffy coats were obtained from Key Biologics, LLC, Memphis, TN. Written informed consent was obtained from all participants.
PBMC isolation and flow cytometric analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll‐Hypaque gradient centrifugation (GE Heathcare, Piscataway, NJ). For phenotypic analysis of MDSCs, PBMCs were stained with CD14‐Peridinin chlorophyll protein 710 (PerCP710), CD11b‐phycoerythrin (PE), CD33‐allophycocyanin (APC) (eBioscience, San Diego, CA), and HLA‐DR‐FITC (BD Biosciences, San Jose, CA) antibodies or isotype controls. Cytometric data were collected on an Accuri C6™ flow cytometer (BD, Franklin Lakes, NJ) and analysed using flowjo software (Tree Star, Inc., Ashland, OR). For intracellular cytokine staining, PBMCs were stimulated with 1 μg/ml lipopolysaccharide (Santa Cruz Biotechnology, Santa Cruz, CA) plus 2·5 μg/ml R848 (Santa Cruz) for 6 hr, and 1 μg/ml Brefeldin A (BioLegend, San Diego, CA) was added 5 hr before harvest. The cells were fixed and permeabilized using Inside Stain Kit (Miltenyi Biotec, Auburn, CA), and then stained with IL‐10‐PE, TGF‐β‐PE, programmed death‐1 ligand (CD274)‐PE (BD Bioscience), IL‐4Rα‐PE (R&D, Minneapolis, MN), p47phox and rat anti‐mouse IgG1‐PE. For pSTAT3‐PE (pY705, BD Bioscience) staining, PBMCs were fixed with Fix Buffer I and permeabilized with Perm Buffer III (BD) following the BD Phosflow™ protocol. The cells were also stained with surface antigens CD14‐PerCP710, CD33‐APC (clone p67.6, eBioscience) and HLA‐DR‐FITC, then analysed by flow cytometry. The fluorescence minus one strategy and isotype controls were used to adjust multicolour compensation for cell gating and to determine background levels.
MDSC induction by HCV core protein
The PBMCs from HS were incubated with recombinant HCV core or a control protein β‐gal (1 μg/ml, ViroGen, Watertoen, MA) for 72 hr in vitro, followed by analysis of MDSC induction and their IL‐10 or TGF‐β expressions by flow cytometry.
Treg cell induction
(i) CD33+ myeloid cells isolated from HCV PBMCs, using CD33 microbeads (Miltenyi Biotec., cell purity > 95%), were co‐cultured with healthy PBMCs in a ratio of 1 : 5 in the presence of 20 U/ml IL‐2 (eBioscience) for 5 days. (ii) The CD33+ myeloid cells or CD33− non‐myeloid cells, purified from healthy PBMCs treated with HCV core or β‐gal control protein (1 μg/ml, ViroGen) in vitro for 5 days, were also co‐cultured with autologous naive CD4+ T cells in a ratio of 1 : 3 in the presence of 1 μg/ml anti‐CD3 and 2 μg/ml anti‐CD28 (BD Bioscience) for 3 days. (iii) The CD33+ myeloid cells isolated from HCV PBMCs were also co‐cultured with autologous naive CD4+ T cells in a ratio of 1 : 3 in the presence of 1 μg/ml anti‐CD3 and 2 μg/ml anti‐CD28 (BD Bioscience) for 3 days. (iv) For depletion experiments, HCV PBMCs and CD33+‐depleted PBMCs were incubated ex vivo for 5 days in the presence of 20 U/ml IL‐2 (eBioscience), respectively. (v) To determine whether the conversion is a cytokine‐dependent event, we also incubated the CD33+/− cells with CD4+ T cells in the presence of 10 μg/ml anti‐IL‐10 blocking antibody or IgG control (Biolegend, San Diego, CA) for 3 days. (vi) To determine whether cell–cell direct contact is required for Treg cell induction, a transwell plate containing 0·4‐μm pores (Corning, Corning, NY) was employed to culture the isolated cells in the same condition. To analyse in vitro‐generated Treg cells, the cells were stained with CD4‐PE, CD25‐Alexa488, Foxp3‐PE‐Cy5 (eBioscience) or isotype controls using Foxp3 Staining Buffer Kit (Miltenyi Biotec). The stained cells were collected by flow cytometry and analysed using flowjo software as described above.
Teff cell interferon‐γ assays
The PBMCs derived from HCV‐infected subjects, with or without depletion of CD33+ myeloid cells, were stimulated with 1 μg/ml anti‐CD3 and 2 μg/ml anti‐CD28 (BD Bioscience) for 3 days. CD4+ Teff intracellular cytokine expression was assessed by interferon‐γ (IFN‐γ) ‐PE (BD Bioscience) or isotype control staining, followed by flow cytometric analysis as described above.
STAT3 inhibition
The PBMCs from patients with chronic HCV infection were cultured in the presence of the specific STAT3 inhibitor STA‐21 (Santa Cruz, 20 μm) or DMSO control for 48 hr. The cells were stimulated by lipopolysaccharide/R848 for 6 hr and Brefeldin A was added for 5 hr before harvest, then subjected to flow analysis for the MDSC phenotype as well as IL‐10 and TGF‐β expression in M‐MDSCs as described above.
Statistical analysis
The data were summarized as mean ± standard error of the mean (SEM) or median with interquartile, depending on the characteristics of the data distribution. An independent t‐test or paired t‐test was used to compare the difference of mean between two groups. A Mann–Whitney test or Wilcoxon signed rank test was used to compare the difference of median between two groups. All the data were analysed using graphpad prism 5·0 (GraphPad Prism, San Diego, CA). *P < 0·05, **P < 0·01, and ***P < 0·001 were considered significant or very significant.
Results
Expansion of MDSCs in patients with chronic HCV infection
As an initial approach to characterize the role of MDSCs in the chronicity of HCV infection, we first compared the phenotypic frequencies of MDSCs in the peripheral blood of 56 chronically HCV‐infected individuals and 22 HS. As shown in Fig. 1, the representative dot plots and summary data of flow cytometry, PBMCs were first gated on CD11b+ and CD33+ myeloid cells, and then analysed for the immature HLA‐DR−/low populations within the monocytic (CD14+) and granulocytic (CD14−) subsets (Fig. 1a). Although the overall numbers of myeloid cells (CD33+ CD11b+) in chronic HCV patients were significantly lower than those in HS (Fig. 1b), the numbers of MDSCs (CD33+ CD11b+ HLA‐DR−/low, Fig. 1c), in particular M‐MDSCs (CD14+ CD33+ CD11b+ HLA‐DR−/low, Fig. 1d), but not G‐MDSCs (CD14− CD33+ CD11b+ HLA‐DR−/low, Fig. 1e), were significantly higher in patients with chronic HCV infection. Of note, we did not find any direct relationship between MDSC frequencies and HCV genotype, viral load, and alanine aminotransferase or aspartate aminotransferase levels in this study.
Figure 1.
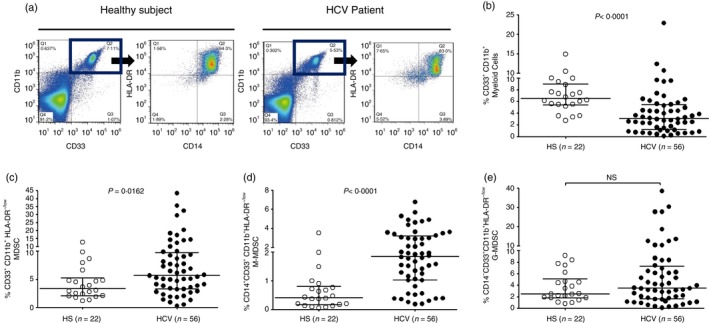
Phenotypic analysis of myeloid cell frequencies in chronic hepatitis C virus (HCV) patients versus healthy subjects (HS). (a) Representative dot plots analysis of myeloid‐derived suppressor cells (MDSCs) by flow cytometry. CD33+ CD11b+ myeloid cells were first gated in peripheral blood mononuclear cells (PBMCs) of HS and HCV patients; monocytic MDSCs (M‐MDSC, CD33+ CD11b+ CD14+ HLA‐DR −/low) and granulocytic MDSCs (G‐MDSC, CD33+ CD11b+ CD14− HLA‐DR −/low) were further characterized by the expression levels of CD14 and HLA‐DR, respectively. The values in the quadrants indicate the percentages of the related subsets of cells. (b) Summary data of myeloid cell (CD33+ CD11b+) frequencies in HS (hollow circle, n = 22) versus chronically HCV‐infected patients (filled circle, n = 56). (c) Summary data of immature myeloid cell (CD33+ CD11b+ HLA‐DR −/low) frequencies in HS versus HCV. The frequencies of circulating MDSCs in the pooled data were calculated by the frequencies of CD33+ CD11b+ cells in PBMCs multiplied by the frequencies of CD14+/− HLA‐DR −/low cells. (d) Summary data of M‐MDSC (CD14+ CD33+ CD11b+ HLA‐DR −/low) frequencies in HS versus HCV. (e) Summary data of G‐MDSC (CD14− CD33+ CD11b+ HLA‐DR −/low) frequencies in HS versus HCV. Each symbol represents an individual subject, and the horizontal bars represent median with interquartile. P‐value with significant difference is shown in each panel; NS, no significance.
Elevation of regulatory proteins in M‐MDSCs from patients with chronic HCV infection
MDSCs are more accurately identified by their immunosuppressive functions rather than phenotypic surface markers. MDSCs exert their suppressive activities by producing copious amounts of immunosuppressive mediators.5, 6, 7, 8, 9, 10, 11, 12, 13, 14 To functionally characterize MDSCs in patients with chronic HCV infection, we measured the most common inhibitory cytokines or proteins, including pSTAT3, IL‐10, TGF‐β, p47phox, IL‐4Rα and programmed death‐1 ligand, which have been suggested to play a role in MDSC development and/or suppressive functions.6, 7 As shown in Fig. 2, the representative dot plots and summary data of flow cytometry, we first gated the monocytic myeloid cell populations (CD14+ CD33+) in PBMCs, and then measured the expressions of these regulatory proteins in the immature (HLA‐DR−/low) myeloid cells. Although M‐MDSCs express these regulator proteins, only pSTAT3+ and IL‐10+ M‐MDSCs (Fig. 2a,b) were found to be significantly elevated in PBMCs in patients with chronic HCV infection.
Figure 2.
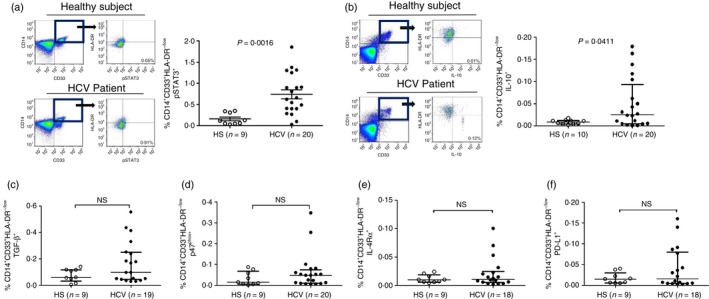
Analysis of regulatory protein expression in monocytic myeloid‐derived suppressor cells (M‐MDSCs) from hepatitis C virus (HCV) patients versus healthy subjects (HS). Peripheral blood mononuclear cells (PBMCs) isolated from HS and HCV patients were stimulated with Toll‐like receptor (TLR) ligand lipopolysaccharide (LPS)/R848 for 6 hr, immune stained for phosphorylated signal transducer and activator of transcription 3 (pSTAT3), interleukin‐10 (IL‐10), transforming growth factor‐β (TGF‐β), p47phox, IL‐4Rα, and programmed death‐1 ligand (PD‐L1) in M‐MDSC, followed by flow cytometric analysis. (a), and (b) left panel: representative dot plots for pSTAT3 and IL‐10 expression in M‐MDSCs from HS and HCV patients; right panel: summary data for the percentage of pSTAT3+ or IL‐10+ M‐MDSCs in PBMCs from HS (hollow circle) and HCV patients (filled circle). The percentages of pSTAT3+ (or IL‐10+) M‐MDSCs in the pooled data were calculated by the frequencies of CD14+ CD33+ cells in PBMCs multiplied by the frequencies of HLA‐DR −/low pSTAT3+ (or IL‐10+) cells. (c), (d), (e), and (f) Summary data of TGF‐β +, p47phox +, IL‐4Rα + and PD‐L1+ M‐MDSCs in PBMCs from HS and HCV patients. Each symbol represents an individual subject, n = the number of subjects to be studied, and the horizontal bars represent mean ± SEM (pSTAT3) or median with interquartile. P‐value with significant difference is shown in each panel; NS, no significance.
Induction of MDSCs and suppressive proteins by HCV (core) via the STAT3 pathway
Despite several reports showing increases in the numbers of MDSCs in HCV‐infected patients, the factors responsible for MDSC expansion during HCV infection are unclear. As patients with chronic HCV infection often suffer from confounding factors that may drive MDSC expansion, we determined whether the increases of M‐MDSCs in patients with HCV infection were directly caused by HCV or other factors. To this end, purified PBMCs from HS were incubated with recombinant HCV core protein, an important HCV structural protein that has been found to circulate in the peripheral blood of HCV patients and exerts immunosuppressive activity,29, 30 or a control protein β‐gal (1 μg/ml, ViroGen) for 3 days, followed by the analysis of immature myeloid cell differentiation and maturation by flow cytometry. As shown in Fig. 3(a), healthy PBMCs treated with HCV core protein in vitro generated an increased number of M‐MDSCs (CD14+ CD33+ HLA‐DR−/low cells) when compared with those exposed to the control protein. In addition, healthy PBMCs treated with HCV core protein generated higher numbers of pSTAT3+ M‐MDSCs (Fig. 3b), IL‐10+ M‐MDSCs (Fig. 3c), and TGF‐β + M‐MDSCs (no significance, Fig. 3d) compared with those exposed to the β‐gal control protein. Moreover, healthy PBMCs co‐cultured with Huh‐7R hepatocytes expressing HCV also resulted in an increase in MDSC frequency, though the increase is not statistically significant compared with those co‐cultured with Huh‐7 cells without HCV expression (data not shown).
Figure 3.
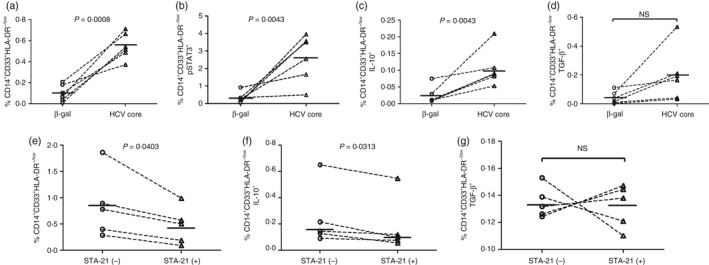
Hepatitis C virus (HCV) induces myeloid‐derived suppressor cell (MDSC) differentiation and regulatory cytokine expression via the signal transducer and activator of transcription 3 (STAT3) pathway. (a), (b), (c), and (d) HCV core protein induces monocytic MDSCs (M‐MDSCs) and their expression of pSTAT3, interleukin‐10 (IL‐10) and transforming growth factor‐β (TGF‐β). Healthy peripheral blood mononuclear cells (PBMCs) (n = 6) were incubated with HCV core or β‐gal control protein (1 μg/ml) in vitro for 3 days, stimulated with lipopolysaccharide (LPS)/R848 for 6 hr before harvesting, followed by flow cytometric analysis for the development of M‐MDSCs (a: CD14+ CD33+ HLA‐DR −/low cells) and the frequencies of pSTAT3+ M‐MDSCs (b), IL‐10+ M‐MDSCs (c), and TGF‐β + M‐MDSCs (d) in PBMCs. (e), (f) and (g) Involvement of STAT3 signalling in the development of MDSCs and expression of regulatory cytokines. PBMCs isolated from HCV patients (n = 5) were cultured ex vivo in the presence of the specific STAT3 inhibitor STA‐21 (20 μm) for 48 hr, stimulated by LPS/R848 for 6 hr, and Brefeldin A for 5 hr before harvest, then subjected to flow cytometric analysis for M‐MDSC frequency (e) as well as IL‐10+ M‐MDSCs (f) and TGF‐β + M‐MDSCs (g) in PBMCs. P values are shown in each panel from a paired t‐test or by a Wilcoxon signed rank test (f); NS, no significance.
Although MDSCs were increased in chronic HCV patients, the precise mechanisms involved in their differentiation and suppressive function remain elusive. Because the HCV‐induced transcription factor STAT3 activation is accompanied by the increases in IL‐10 and TGF‐β expression (Fig. 2a–c and Fig. 3b–d), we further investigated whether pSTAT3 promotes MDSC expansion and their production of IL‐10 or TGF‐β. To this end, we blocked STAT3 signalling ex vivo using the STAT3‐specific inhibitor STA‐2131 in PBMCs from HS and HCV‐infected patients, then analysed the frequencies of M‐MDSCs as well as IL‐10 and TGF‐β expression in these cells. As shown in Fig. 3(e), blocking STAT3 signalling significantly reduced the numbers of CD14+ CD33+ HLA‐DR−/low M‐MDSCs, which were increased during chronic HCV infection, whereas the percentage of M‐MDSCs in PBMCs from HS was not altered (data not shown). In addition, the expression levels of the suppressive cytokine IL‐10 (Fig. 3f), but not TGF‐β (Fig. 3g), in HCV M‐MDSCs were inhibited after blocking STAT3 signalling.
MDSCs regulate T‐cell differentiation and function in chronic HCV infection
In addition to producing inhibitory proteins, it has been suggested that MDSCs exert their immunosuppressive functions by inducing CD4+ CD25+ Foxp3+ Treg cell development in cancer or transplant patients.12, 15, 32 Therefore, we next sought to determine whether HCV‐associated MDSCs can induce Foxp3+ Treg cell generation. To this end, we first co‐cultured healthy PBMCs with or without CD33+ myeloid cells derived from HCV patients for 5 days, and then analysed Foxp3+ Treg cell development by flow cytometry. As shown in Fig. 4(a), the representative dot plots of flow cytometry and summary data of co‐culture experiments, healthy PBMCs co‐cultured with HCV CD33+ myeloid cells (enriched in MDSCs) induced a significant increase in CD4+ CD25+ Foxp3+ Treg cells when compared with those cultured without HCV myeloid cells. To address the concern of allo‐stimulation in the co‐culture system, we also observed Treg induction using healthy CD4+ T cells incubated with autologous myeloid cells isolated from the same PBMCs treated with HCV‐core protein for 5 days in vitro. As shown in Fig. 4(b), CD33+ myeloid cells (but not CD33− non‐myeloid cells) derived from healthy PBMCs treated with HCV core protein, but not from PBMCs exposed to β‐gal protein, can significantly induce Foxp3+ Treg cell development. In addition, purified HCV CD4+ T cells incubated with autologous HCV CD33+ myeloid cells led to a high number of Foxp3+ Treg cells; whereas HCV CD4+ T cells (enriched in Treg cells, but lacking MDSCs) incubated alone without the presence of HCV CD33+ myeloid cells resulted in a significant decrease in Foxp3+ Treg cells (Fig. 4c). We also tested whether depletion of CD33+ myeloid cells from HCV PBMCs might lead to a decrease in Foxp3+ Treg cells that were expanded during chronic HCV infection. As shown in Fig. 4(d), the representative dot plots of flow cytometry and summary data of depletion experiments, depletion of CD33+ myeloid cells from HCV PBMCs resulted in a significant decrease in the numbers of CD4+ CD25+ Foxp3+ Treg cells when compared with HCV PBMCs with MDSCs in culture, suggesting that generation and maintenance of Treg cells need the presence of MDSCs.
Figure 4.
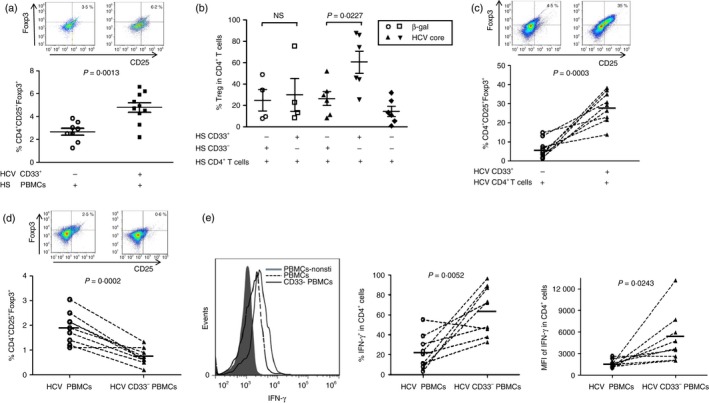
Myeloid‐derived suppressor cells (MDSCs) from hepatitis C virus (HCV) patients promote Foxp3+ regulatory T (Treg) cell differentiation and suppress interferon‐γ (IFN‐γ) expression by effector T (Teff) cells. (a) CD33+ myeloid cells from HCV patients induce Treg cell generation in healthy peripheral blood mononuclear cells (PBMCs). Healthy PBMCs were co‐cultured with or without CD33+ myeloid cells derived from HCV patients for 5 days, followed by immunostaining for CD4, CD25, Foxp3 and flow cytometric analysis for Treg cell development. Representative dot plots and cumulative results from 10 independent experiments are shown. (b) CD33+ myeloid cells from healthy PBMCs treated with HCV core protein promote Treg cell generation. Healthy CD4+ T cells were co‐cultured with CD33+ or CD33− cells isolated from PBMCs exposed to HCV core or β‐gal control protein for 3 days, followed by flow cytometric anlysis for Foxp3+ Treg cell frequencies. Cumulative results from four independent experiments are shown. (c) Induction of Treg cell development by co‐culture of CD4+ T cells with CD33+ myeloid cells derived from HCV patients. CD4+ T cells isolated from HCV patients were co‐cultured with or without autologous CD33+ myeloid cells for 3 days, respectively, followed by immune staining and flow analysis for CD4+ CD25+ Foxp3+ Treg cells. Representative dot plot and cumulative results from nine HCV patients are shown. (d) Depletion of CD33+ myeloid cells from PBMCs of HCV patients reduces Treg cells accumulated during HCV infection. HCV PBMCs, with or without CD33+ myeloid cell depletion, were cultured ex vivo for 5 days, followed by immune staining and flow analysis for CD4+ CD25+ Foxp3+ Treg cells. Representative dot plot and cumulative results from nine HCV patients are shown. (e) Depletion of CD33+ myeloid cells from PBMCs of HCV patients boosts the IFN‐γ production by CD4+ Teff cells. PBMCs isolated from chronic HCV patients, with or without depletion of CD33+ myeloid cells, were cultured ex vivo in the presence of anti‐CD3/CD28 stimulation, followed by flow cytometric analysis for IFN‐γ expression in CD4+ T cells. Representative overlaid histogram (left panel) and cumulative results for the percentage (middle panel) as well as the mean fluorescence intensity (MFI) (right panel) of IFN‐γ expression in CD4+ T cells from nine HCV patients are shown. P‐values are shown in each panel.
As HCV‐induced MDSCs were able to produce high levels of IL‐10 (Fig. 2) that are capable of triggering Treg cell induction, we tested whether Treg cell induction by MDSC is merely a cytokine‐dependent event. To this end, we incubated HCV CD33+ myeloid cells or CD33− non‐myeloid cells with autologous CD4+ T cells in the presence of anti‐IL‐10 blocking antibody or IgG control (Biolegend, San Diego, CA) for 3 days, followed by flow cytometric analysis for Treg cell induction. Not surprisingly, simply blocking cytokine IL‐10 alone could not abrogate the HCV MDSC‐induced Treg cell induction (data not shown). To determine if direct contact of MDSCs with naive T cells is required for differentiation into Treg cells, we isolated CD33+ myeloid cells (CD33− non‐myeloid cells as control) and naive CD4+ T cells from HCV patients and co‐cultured in the same conditions (1 : 3 cell ratio, stimulated by anti‐CD3/CD28 for 3 days) using a transwell plate (Corning, Corning, NY). Interestingly, the induction of Treg cells by HCV CD33+ myeloid cells was not observed in this transwell system when compared with CD33− non‐myeloid cells (data not shown), suggesting that direct contact of myeloid cells with naive T cells is required for differentiation into Treg cells in HCV patients.
In addition to inducing or maintaining the differentiation of Treg cells, we also studied whether MDSCs can inhibit the function of Teff cells that are usually suppressed or exhausted during chronic viral infections.33, 34 To this end, PBMCs derived from chronically HCV‐infected patients, with or without depletion of CD33+ myeloid cells, were stimulated with anti‐CD3 and anti‐CD28 for 72 hr, followed by intracellular staining for the IFN‐γ expression in CD4+ T cells. As shown in Fig. 4(e), the representative overlaid histogram (left panel) and summary data (middle and right panel) from the flow cytometric analysis, depletion of CD33+ myeloid cells from PBMCs of chronic HCV patients significantly increased the IFN‐γ production by CD4+ T cells. This result holds true by analysing both the percentage of positive cell frequency and mean fluorescence intensity of IFN‐γ production in CD4+ T cells.
Discussion
Compelling studies clearly implicate an important role for MDSCs in tumour progression and the antitumour immune response; however, the significance of MDSCs in viral infections is far from clear. In this study, we demonstrate a significant increase in the monocytic subset of myeloid cells (CD14+ CD33+ CD11b+) that are immature (HLA‐DR−/low) and immunosuppressive in patients with chronic HCV infection. These HCV‐induced M‐MDSCs express high levels of pSTAT3‐mediated IL‐10, since blocking STAT3 signalling reduces the expression of IL‐10 and decreases the numbers of M‐MDSCs developed in chronic HCV infection. In addition, we demonstrated that HCV‐induced MDSCs promote Foxp3+ Treg cell development and inhibit CD4+ Teff cells producing IFN‐γ. Of note, both Treg cell expansion and Teff cell inhibition are associated with viral persistence.1, 2
The mechanism underlying MDSC expansion in virus‐infected individuals remains to be determined. It has been suggested that MDSCs can be induced or expanded by the virus itself or its coding proteins, including HIV, HCV and lymphocytic choriomeningitis viruses.21, 23, 27 We and others have demonstrated that HCV core, the first protein to be secreted from HCV‐infected hepatocytes and circulating in the blood of virus‐infected patients, can activate STAT3 phosphorylation, prevent myeloid cell maturation and promote M‐MDSC development, an effect that can be abrogated by adding STAT3 inhibitor in vitro 35 (Fig. 3). These results indicate that HCV can regulate MDSC differentiation and suppressive functions via the STAT3 signalling pathway. Indeed, ablation of STAT3 using conditional knockout mice reduces the expansion of MDSCs and improves T‐cell responses in tumour‐bearing mice.36 Additionally, myeloid cells expressing toll‐like receptor 3 that can interact with double‐stranded RNA released from non‐viable virus in the blood may serve as a plausible mechanism for MDSC differentiation by HCV. Another factor that might contribute to the increases in numbers of MDSCs during HCV infection is an inflamed liver, a microenvironment where active HCV replication and persistent inflammation occurs and so may modify myeloid cell differentiation, resulting in expansion of MDSCs.
Although dysregulated myelopoiesis by inflammatory cytokines, such as IL‐6, IL‐10, TGF‐β and prostaglandin E2 that are up‐regulated during chronic HCV infection,37, 38, 39, 40, 41, 42, 43 may partially explain the expansion of MDSCs, the molecular nature of this process is unclear. Previous studies have shown that increased expression of arginase 1, nitric oxide synthase or reactive oxygen species is the primary mechanism by which MDSCs promote immunosuppression during chronic HCV infection.23, 24, 25 HCV core‐treated CD33+ cells exhibit a CD14+ CD11b+ HLA‐DR−/low phenotype with up‐regulated expression of p47phox, a component of the NOX2 complex that is critical for reactive oxygen species production,23 and STAT3 has been shown to directly regulate the production of immunosuppressive mediators, such as IL‐6, IL‐10, IL‐4Rα and p47phox.31, 35, 36 Our results showed that the MDSCs that have accumulated during chronic HCV infection expressed higher levels of pSTAT3 and IL‐10 regulatory molecules that are essential and not redundant to their suppressive functions, a feature not seen in MDSCs from healthy controls.44, 45 The detailed mechanism of how STAT3‐mediated signalling pathways are activated and regulate the MDSC functions during chronic HCV infection requires further investigation. Recently, Condamine and Gabrilovich46 proposed two overlapping STAT3‐mediated pathways that regulate the distinct features of MDSCs: one signalling pathway of STAT3, along with survival factors, such as c‐Myc, Cyclin D and Survivin, promotes proliferation but prevents differentiation and maturation of immature myeloid progenitors, which may contribute to MDSC expansion in mouse. A second signalling pathway uses STAT‐3, phosphatidyl inositol 3‐kinase and nuclear factor‐κB transcription factors to induce production of the immunosuppressive mediators arginase 1, reactive oxygen species, IL‐10 and TGF‐β in MDSCs. These factors produced at inflammation sites or in a tumour microenvironment may expand MDSCs and enhance their immunosuppressive functions.7
We and others have previously shown that HCV (core protein) has direct effects on T‐cell differentiation and responses.29, 30, 33, 34, 47, 48 Based on our new findings of an HCV‐induced, STAT3/IL‐10‐mediated MDSC expansion and regulation of T‐cell differentiation and function, we propose a model, as depicted in Fig. 5, that represents a novel mechanism by which HCV suppresses immune responses, in particular T‐cell differentiation and function, during chronic viral infection. HCV‐induced MDSCs may trigger naive CD4 T‐cell differentiation into Foxp3+ Treg cells, and they may also promote natural occurring and adaptive Treg cell expansion. The mechanisms regarding how HCV induces pSTAT3 expression in MDSCs to dampen immune responses to viral infection and antiviral activities are far from clear, and we continue to investigate these potential mechanisms in our laboratory, focusing on microRNA‐based regulatory mechanisms. Nevertheless, this study demonstrates that HCV‐induced MDSCs control T‐cell activities through STAT3 signalling, shedding new light on the features of MDSCs and providing a novel mechanism for T‐cell immune suppression during chronic viral infection. Hence, targeting STAT3 signalling and/or MDSC expansion may represent a promising strategy for immunotherapy to treat human viral diseases.
Figure 5.
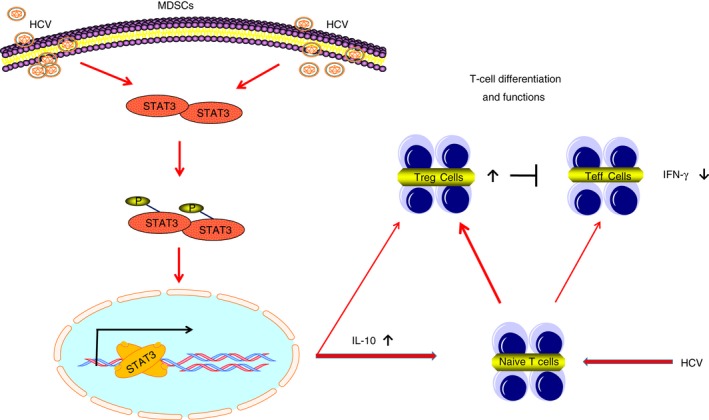
A schematic model for hepatitis C virus (HCV) ‐induced myeloid‐derived suppressor cells (MDSCs) regulate T‐cell differentiation and function via signal transducer and activator of transcription 3 (STAT3) signalling. HCV infection activates STAT3 phosphorylation and interleukin‐10 (IL‐10) expression and drives MDSC expansion, which in turn, promotes CD4+ CD25+ Foxp3+ regulatory T (Treg) cell differentiation and inhibits CD4+ effector T (Teff) cell interferon‐γ (IFN‐γ) production. Therefore, inhibition of the STAT3/IL‐10 pathway and MDSC development may provide a novel approach for HCV immunotherapy. Of note, we and others have previously shown that HCV can directly induce Treg cell differentiation and suppress Teff cell functions.29, 30, 33, 34, 47, 48 Hence, direct antiviral therapy is the key to controlling infection and correcting HCV‐induced immune dysregulation.
Disclosure
The authors declare no competing interests.
Acknowledgements
This work was supported by the National Institute of Health grants R01DK093526 and R01AI114748 (to Z.Q.Y. and J.P.M.). This publication is the result of work supported with resources and the use of facilities at the James H. Quillen Veterans Affairs Medical Center. The contents in this publication do not represent the views of the Department of Veterans Affairs or the United States Government. The authors declare no conflict of interests of this paper.
References
- 1. Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity 2014; 40:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosen HR. Emerging concepts in immunity to hepatitis C virus infection. J Clin Invest 2013; 123:4121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Au JS, Pockros PJ. Novel therapeutic approaches for hepatitis C. Clin Pharmacol Ther 2014; 95:78–88. [DOI] [PubMed] [Google Scholar]
- 4. Manns MP, von Hahn T. Novel therapies for hepatitis C – one pill fits all? Nat Rev Drug Discov 2013; 12:595–610. [DOI] [PubMed] [Google Scholar]
- 5. Ostrand‐Rosenberg S, Sinha P. Myeloid‐derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dai J, El Gazzar M, Li GY, Moorman JP, Yao ZQ. Myeloid‐derived suppressor cells: paradoxical roles in infection and immunity. J Innate Immun 2014; 7:116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E et al Mechanism regulating reactive oxygen species in tumor‐induced myeloid‐derived suppressor cells. J Immunol 2009; 182:5693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH et al Population alterations of L‐arginase‐ and inducible nitric oxide synthase‐expressed CD11b+/CD14–/CD15+/CD33+ myeloid‐derived suppressor cells and CD8+ T lymphocytes in patients with advanced‐stage non‐small cell lung cancer. J Cancer Res Clin Oncol 2010; 136:35–45. [DOI] [PubMed] [Google Scholar]
- 10. Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA‐DR–/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC‐sign. Cancer Res 2010; 70:4335–45. [DOI] [PubMed] [Google Scholar]
- 11. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M et al Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte–macrophage colony‐stimulation factor‐based antitumor vaccine. J Clin Oncol 2007; 25:2546–53. [DOI] [PubMed] [Google Scholar]
- 12. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP et al A new population of myeloid‐derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology 2008; 135:234–43. [DOI] [PubMed] [Google Scholar]
- 13. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C et al Multipeptide immune response to cancer vaccine IMA901 after single‐dose cyclophosphamide associates with longer patient survival. Nat Med 2012; 18:1254–61. [DOI] [PubMed] [Google Scholar]
- 14. Vasquez‐Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J et al STAT3 regulates arginase‐I in myeloid‐derived suppressor cells from cancer patients. J Clin Invest 2013; 123:1580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jitschin R, Braun M, Buttner M, Dettmer‐Wilde K, Bricks J, Berger J et al CLL‐cells induce IDOhi CD14+HLA‐DRlo myeloid‐derived suppressor cells that inhibit T‐cell responses and promote TRegs. Blood 2014; 124:750–60. [DOI] [PubMed] [Google Scholar]
- 16. Chen S, Akbar SM, Abe M, Hiasa Y, Onji M. Immunosuppressive functions of hepatic myeloid‐derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin Exp Immunol 2011; 166:134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang A, Zhang B, Yan WW, Wang B, Wei HF, Zhang F et al Myeloid‐derived suppressor cells regulate immune response in patients with chronic hepatitis B virus infection through PD‐1‐induced IL‐10. J Immunol 2014; 193:5461–9. [DOI] [PubMed] [Google Scholar]
- 18. Vollbrechi T, Striner R, Tufman A, Roider J, Huber RM, Bogner JR et al Chronic progressive HIV‐1 infection is associated with elevated levels of myeloid‐derived suppressor cells. AIDS 2012; 26:F31–7. [DOI] [PubMed] [Google Scholar]
- 19. Macatangay BJ, Landay AL, Rinaldo CR. MDSC: a new player in HIV immunopathogenesis. AIDS 2012; 26:1567–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qin A, Cai W, Pan T, Wu K, Yang Q, Wang N et al Expansion of monocytic myeloid‐derived suppressor cells dampens T cell function in HIV‐1‐seropositive individuals. J Virol 2013; 87:1477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garg A, Spector SA. HIV type 1 gp120‐induced expansion of myeloid derived suppressor cells is dependent on interleukin 6 and suppresses immunity. J Infect Dis 2014; 209: 441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gama L, Shirk EN, Russell JN, Carvalho KI, Li M, Queen SE et al Expansion of a subset of CD14highCD16negCCR2low/neg monocytes functionally similar to myeloid‐derived suppressor cells during SIV and HIV infection. J Leukoc Biol 2012; 91:803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tacke RS, Lee HC, Goh C, Courtney J, Polyak SJ, Rosen HR et al Myeloid suppressor cells induced by hepatitis C virus suppress T‐cell responses through the production of reactive oxygen species. Hepatology 2012; 55:343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zeng QL, Yang B, Sun HD, Feng GH, Jin L, Zou ZS et al Myeloid‐derived suppressor cells are associated with viral persistence and downregulation of TCR ζ chain expression on CD8+ T cells in chronic hepatitis C patients. Mol Cells 2014; 37:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai W, Qin A, Guo P, Yan D, Hu F, Yang Q et al Clinical significance and functional studies of myeloid‐derived suppressor cells in chronic hepatitis C patients. J Clin Immunol 2013; 33:798–808. [DOI] [PubMed] [Google Scholar]
- 26. Bowers NL, Helton ES, Huijbregts RP, Goepfert PA, Heath SL, Hel Z. Immune suppression by neutrophils in HIV‐1 infection: role of PD‐L1/PD‐1 pathway. PLoS Pathog 2014; 10:e1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Norris BA, Uebelhoer LS, Nakaya HI, Price AA, Grakoui A, Pulendran B. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral‐specific T cell immunity. Immunity 2013; 38:309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nonnenmann J, Stirner R, Roider J, Jung MC, Schrudl K, Bogner JR et al Lack of significant elevation of myeloid‐derived suppressor cells in peripheral blood of chronically hepatitis C virus‐infected individuals. J Virol 2014; 88:7678–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yao ZQ, Nguyen DT, Hiotellis AI, Hahn YS. Hepatitis C virus core protein inhibits human T lymphocyte responses by a complement‐dependent regulatory pathway. J Immunol 2001; 167:5264–72. [DOI] [PubMed] [Google Scholar]
- 30. Yao ZQ, Eisen‐Vandervelde A, Waggoner S, Cale EM, Hahn YS. Direct binding of HCV core to gC1qR on CD4+ and CD8+ T cells leads to impaired activation of Lck and Akt. J Virol 2004; 78:6409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song H, Wang RX, Wang SM, Lin JY. A low‐molecular‐weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA 2005; 102:4700–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J et al Monocytic myeloid‐derived suppressor cells accumulate in renal transplant patients and mediate CD4+ Foxp3+ Treg expansion. Am J Transplant 2013; 13:3123–31. [DOI] [PubMed] [Google Scholar]
- 33. Moorman JP, Wang JM, Zhang Y, Ji XJ, Ma CJ, Wu XY et al Tim‐3 pathway controls regulatory and effector T cell balance during HCV infection. J Immunol 2012; 189:755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li GY, Zhou Y, Ying RS, Shi L, Cheng YQ, Ren JP et al Hepatitis C virus‐induced reduction in miR‐181a impairs CD4+ T‐cell responses through overexpression of DUSP6. Hepatology 2015; 61:1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tacke RS, Tosello‐Trampont A, Nguyen V, Mullins DW, Hahn SH. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL‐6 autocrine pathway. J Biol Chem 2011; 286:10847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korrylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon‐Thomas S et al Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11: 1314–21. [DOI] [PubMed] [Google Scholar]
- 37. Jabłońska J, Pawłowski T, Laskus T, Zalewska M, Inglot M, Osowska S et al The correlation between pretreatment cytokine expression patterns in peripheral blood mononuclear cells with chronic hepatitis C outcome. BMC Infect Dis 2015; 15:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pawlowski T, Radkowski M, Małyszczak K, Inglot M, Zalewska M, Jablonska J et al Depression and neuroticism in patients with chronic hepatitis C: correlation with peripheral blood mononuclear cells activation. J Clin Virol 2014; 60:105–11. [DOI] [PubMed] [Google Scholar]
- 39. Jain MK, Adams‐Huet B, Terekhova D, Kushner LE, Bedimo R, Li X et al Acute and chronic immune biomarker changes during interferon/ribavirin treatment in HIV/HCV co‐infected patients. J Viral Hepat 2015; 22:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marinho R, Pinto R, Santos ML, de M. Evidence for prostaglandin‐producing suppressor cells in HCV patients with normal ALT. Dig Dis Sci 2002; 47:556–61. [DOI] [PubMed] [Google Scholar]
- 41. Waris G, Siddigui A. Hepatitis C virus stimulates the expression of cyclooxygenase‐2 via oxidative stress: role of prostaglandin E2 in RNA replication. J Virol 2005; 79:9725–34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Lu L, Wei L, Peng G, Mu Y, Wu K, Kang L et al NS3 protein of hepatitis C virus regulates cyclooxygenase‐2 expression through multiple signaling pathways. Virology 2008; 371:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, Dominguez CX et al Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med 2015; 21:327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hart KM, Byrne KT, Molloy MJ, Usherwood EM, Berwin B. IL‐10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front Immunol 2011; 2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine‐induced myeloid‐derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol 2010; 185:2273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid‐derived suppressor cell differentiation and function. Trends Immunol 2011; 32:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ji XJ, Ma CJ, Wang JM, Wu XY, Niki T, Hirashima M et al HCV‐infected hepatocytes drive CD4+ CD25+ Foxp3+ regulatory T‐cell development through the Tim‐3/Gal‐9 pathway. Eur J Immunol 2013; 43:458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhai N, Chi X, Li T, Song H, Li H, Jin X et al Hepatitis C virus core protein triggers expansion and activation of CD4+ CD25+ regulatory T cells in chronic hepatitis C patients. Cell Mol Immunol 2015; 12:743–9. [DOI] [PMC free article] [PubMed] [Google Scholar]