

Summary
Interleukin‐8 (IL‐8) is a pro‐angiogenic cytokine associated with aggressive prostate cancer (CaP). We detected high levels of IL‐8 in sera from patients with CaP compared with healthy controls and patients with benign prostatic hypertrophy. This study examines the role of IL‐8 in the pathogenesis of metastatic prostate cancer. We developed a biocompatible, cationic polylactide (CPLA) nanocarrier to complex with and efficiently deliver IL‐8 small interfering RNA (siRNA) to CaP cells in vitro and in vivo. CPLA IL‐8 siRNA nanocomplexes (nanoplexes) protect siRNA from rapid degradation, are non‐toxic, have a prolonged lifetime in circulation, and their net positive charge facilitates penetration of cell membranes and subsequent intracellular trafficking. Administration of CPLA IL‐8 siRNA nanoplexes to immunodeficient mice bearing human CaP tumours produced significant antitumour activities with no adverse effects. Systemic (intravenous) or local intra‐tumour administration of IL‐8 siRNA nanoplexes resulted in significant inhibition of CaP growth. Magnetic resonance imaging and ultrasonography of experimental animals demonstrated reduction of tumour perfusion in vivo following nanoplex treatment. Staining of tumour sections for CD31 confirmed significant damage to tumour neovasculature after nanoplex therapy. These studies demonstrate the efficacy of IL‐8 siRNA nanotherapy for advanced, treatment‐resistant human CaP.
Keywords: angiogenesis, cytokines, interleukin‐8, metastasis, nanotherapy
Introduction
Prostate cancer (CaP) is the second leading cause of cancer‐related male mortality in the United States.1 The vast majority of these patients present with locally advanced or metastatic disease, rendering the cancer inoperable.1 Radiotherapy, alone or in combination with hormone ablation therapy, is a safe and effective treatment for patients with organ‐confined CaP. However, neither therapy is effective in improving the prognosis of advanced CaP, which is uniformly fatal. As many as 30% of patients treated with radiotherapy, chemotherapy and/or hormone ablation therapy develop a recurrence of their cancer. Bone metastasis, an incurable complication of CaP, was detected in > 90% of patients who died from CaP.2
Angiogenesis, the formation of new blood vessels, a feature of aggressive tumours, is highly regulated by a dynamic interplay between stimulators and inhibitors that control endothelial cell proliferation, migration and invasion.3 Of the stimulators, interleukin‐8 (IL‐8) has the greatest pro‐angiogenic activity on endothelial cells of the tumour, inducing neovascularization4 and prolonged survival and proliferation of CaP cells.4 Due to its cardinal role in angiogenesis, targeting IL‐8 is a novel strategy for CaP therapy.5 Humanized monoclonal antibodies against IL‐8 (e.g. ABX‐IL‐8) have been used to inhibit tumour development and progression.6, 7 Co‐administration of ABX‐IL‐8 potentiated the sensitivity of melanoma xenografts to dacarbazine, consistent with the role of IL‐8 in facilitating chemo‐resistance.8 Therefore suppressing IL‐8 expression within the tumour microenvironment halts tumour progression and increases the sensitivity of several solid tumours to chemotherapy.
RNA interference (RNAi), mediated by small interfering RNA (siRNA), is effective in selectively inhibiting the expression of specific genes.9 Compared with standard chemotherapy, siRNA may have more prolonged tumour suppressive effects in vivo.9 However, naked siRNA has a half‐life < 1 hr in circulation and is rapidly degraded by nucleases in plasma.10 Due to its high molecular weight, hydrophilicity and high charge density, naked siRNA minimally penetrates cell membranes.11 This led to the development of delivery vehicles for the therapeutic use of siRNA. The past two decades have seen an increase in viral vectors as gene delivery vehicles.12 However, some impediments such as limitations in large‐scale production, concerns about biosafety, and host immune responses, have diminished their value in this capacity.13 Rapid progress in the field of nanotechnology has resolved some of these problems by developing non‐viral gene delivery vectors.14 These include lipid‐based materials,15 cationic polymers,16 biodegradable polymers,17 and inorganic nanoparticles like gold nanorods.18 Lipid‐based materials used for siRNA delivery are mainly cationic liposomes, which manifest significant toxicity and have the ability to evoke strong immune or inflammatory responses.19 Polymers used for siRNA delivery such as polycations (e.g. poly vinylpyridine, poly l‐lysine, and polyethylenimine or polycation‐containing block polymers) form polymer–gene nanoplexes by electrostatic interactions with sizes of ~ 100 nm. Their application in gene delivery is well documented.20
We recently synthesized and analysed a well‐defined degradable cationic polymer prepared by ring open polymerization (ROP) techniques as a gene delivery scaffold.21, 22 We used a well‐defined, tertiary amine‐functionalized cationic polylactide (CPLA) with 26 mol% of tertiary amine‐functionalities relative to lactide‐based backbone repeating units, namely CPLA‐26, to deliver IL‐8 siRNA by way of CPLA‐26–IL‐8 siRNA nanoplexes for treatment of CaP in a xenograft animal model (Fig. 1). Because IL‐8 knock down inhibits tumour angiogenesis, metastases and proliferation23 we investigated the therapeutic potential of CPLA‐26–IL‐8 siRNA nanoplexes in our CaP model. We hypothesized that targeted inhibition of angiogenesis would significantly inhibit and possibly eliminate CaP tumours. To test this hypothesis we undertook studies in vitro using CaP cell lines and in vivo using a human xenograft CaP tumour animal model. Contrast enhanced (CE) ‐magnetic resonance imaging (MRI) and CE ultrasound (US) were used to characterize the effects of nanotherapy for IL‐8 knock down on the neovascularization of human CaP tumours in our animal model. Therapeutic efficacy was determined by tumour growth measurements and histopathological evaluation.
Figure 1.
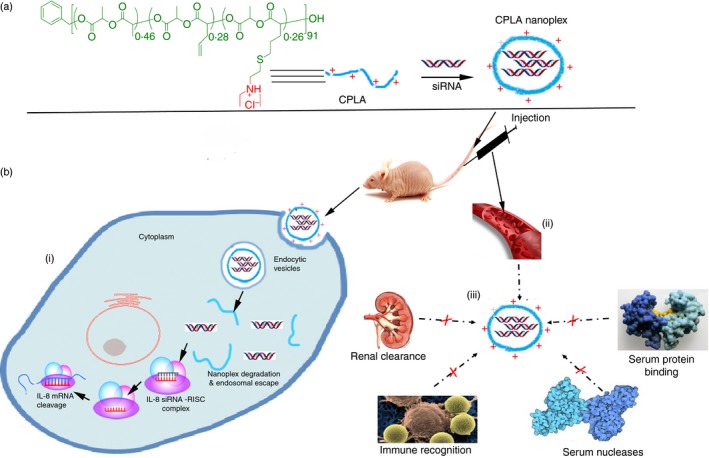
Schematic diagram of CPLA‐26 for delivery of interleukin‐8 (IL‐8) small interfering (si) RNA in prostate tumour model. (a) CPLA‐26 preparation by synthetic route via tandem living ring open polymerization (ROP) and click chemistry, further, the organocatalysed ROP process and thiol‐ene click reaction were used. (b) The schematic illustration of bloodstream circulation and uptake by tumour cells of CPLA in vivo (i) CPLAs can successfully deliver IL‐8 siRNA across cell membrane into cancer cells for synergistic antitumour therapy. (ii) CPLAs possess high stability and prolonged circulating time in bloodstream. (iii) CPLA protects siRNA from degradation by serum nucleases, immune recognition, renal clearance, and replacing by other substances in serum.
Materials and methods
Materials and reagents
Cells and culture conditions
Normal prostate epithelial cells were obtained from Clonetics (Walkersville, MD). PC‐3, DU‐145 and LNCaP cells were obtained from the American Type Culture Collection (Manassas, VA) and differ significantly in their aggressive phenotypes, as we previously demonstrated with PC‐3 > DU‐145 > LNCaP.24 DU‐145 and PC‐3 were isolated from metastases in brain and lumbar vertebrae, respectively. DU‐145 and PC‐3 are androgen independent. LNCaP is androgen responsive but growth does not depend upon androgen. PC‐3M and PC‐3MM2 cells were obtained from Dr Isaiah Fidler (The University of Texas MD Anderson Cancer Center, Houston, TX). PC‐3M is a subline of PC‐3 isolated from a liver metastasis from a patient that formed a solid tumour in a nude mouse. Cells were grown at 37° in a humidified atmosphere of 95% air and 5% CO2 and were maintained in RPMI‐1640 medium supplemented with non‐essential amino acids, l‐glutamine, a twofold vitamin solution (Life Technologies, Grand Island, NY), sodium pyruvate, Earle's balanced salt solution, 10% fetal bovine serum, and penicillin and streptomycin (Flow Labs, Rockville, MD). Cells were harvested by trypsinization. Normal prostate epithelial cells were maintained in proprietary medium as recommended by the manufacturer.
Patient samples
Patient serum was obtained after receiving informed consent under institutionally approved institutional review board protocols from: the Roswell Park Cancer Institute's Data Bank Biorepository (DBBR); collaborating urologists in western New York; or The Cancer and Leukemia Group B; NCI protocol # CALGB‐150201. Samples from controls (n = 56), benign prostatic hyperplasia (BPH; n = 50), localized CaP (n = 49) and castration‐resistant prostate cancer (CRPC; n = 109) were collected and all samples were de‐identified to ensure participant confidentiality.
Immunofluorescence
PC‐3 and PC‐3M cells were grown to 70% confluence in glass Petri dishes and fixed for 10 min at 37° in 4% formaldehyde, followed by permeabilization with ice‐cold 90% methanol. Cells were washed in 1 × PBS and treated with primary antibodies to IL‐8, CXCR‐1, CXCR‐2 and β‐actin (Santa Cruz Biotechnology, Inc., Dallas, TX) followed by staining with cy5‐labelled goat anti‐rabbit antibodies (Molecular Probes, Grand Island, NY) to determine the expression levels of these proteins.
Cell proliferation assay
PC‐3, PC‐3M or PC‐3MM2 cells were seeded in 96‐well plates at a density of 1 × 104 cells per well and allowed to adhere overnight. Cells were washed and re‐fed fresh medium alone or medium containing different treatments including recombinant human (rh) IL‐8 (R&D Systems, Minneapolis, MN), antisera to the IL‐8 receptor, or IL‐8 siRNA using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) transfection agent. At the end of incubation, viability was determined by the 3‐(4,5‐Dimethylthiazol‐2‐Yl)‐2,5‐Diphenyltetrazolium Bromide (MTT) assay25 using a microtitre plate reader (Bio‐Tek Instruments, Inc., Winooski, VT, UK) at 570 nm. Cell proliferation/inhibition was calculated as the percentage of proliferation/inhibition: [1 – (A/B)] × 100, where A is the absorbance of treated cells, and B is the absorbance of untreated control cells expressed as % of control cells.
Cell invasion assay
The effect of IL‐8 gene knock down on the invasive activity of CaP cells in vitro was tested using a Quantitative Cell Invasion Assay kit (Chemicon International, Inc., Temecula, CA). The invasion assay was performed using 24‐well, Transwell tissue‐culture plates. The insert contains an 8‐μm porous polycarbonate membrane pre‐coated with basement membrane proteins derived from the Engelbeth Holm–Swarm mouse tumour. Details of the methodology have been described previously.24
Western blotting for IL‐8 protein
Cell lysates were prepared by re‐suspending cells in lysis buffer [65 mmol/lTris–HCl (pH 7·4), 150 mmol/l NaCl, 1 mmol/l EDTA, 1% nonidet‐P40, 1% sodium deoxycholate, 1 μg/ml aprotinin, 100 μg/ml PMSF] for 30 min at 4° and cleared by centrifugation for 30 min at 13 000 g. Supernates were collected and stored at −80° until used. Total protein concentration was determined using Coomassie Protein Reagent (Bio‐Rad, Hercules, CA), fractionated by 10% SDS–PAGE, transferred to nitrocellulose membranes, incubated with primary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and horseradish peroxidase‐conjugated secondary antibodies, and revealed with Super Signal Pico West (Pierce, Rockford, IL). β‐Actin expression was used as an internal control.
Synthesis of CPLA‐26
CPLA‐26 was synthesized as described by us.21, 26 Briefly, allyl‐functionalized polylactide (M n NMR = 14 500, PDI GPC = 1·12, 54 mol% of allyl functionalities) was prepared by ROP of allyl‐functionalized lactide (50 eq) with l‐lactide (50 eq) by using benzyl alcohol (1·0 eq) as the initiator and 4‐dimethylaminopyridine (4·0 eq) as the organocatalyst in dichloromethane at 35° for 21 days (conversion of co‐monomers: ~ 80%). CPLA‐26 (M n NMR = 18 500, PDI GPC = 1·37, 26 mol% of tertiary amine functionalities) was synthesized by thiol‐ene functionalization of the allyl‐functionalized polylactide (1·0 eq of allyl functionality) using (diethylamino)ethanethiol hydrochloride (0·5 eq) as the thiol agent and 2,2′‐dimethoxy‐2‐phenylacetophenone (0·2 eq) as the photoinitiator in CDCl3 at room temperature under UV irradiation (λ max = 365 nm) for 30 min, followed by purification by dialysis against acetone for 10 days.
Preparation and characterization of siRNA–CPLA‐26 nanoplexes
Ten microlitres (10 μm) of IL‐8 siRNA or scrambled IL‐8 control siRNA (Life Technologies) was mixed with varying amounts of CPLA‐26 (weight ratio of IL‐8 siRNA : CPLA‐26) = 1 : 8, 1 : 16, 1 : 32, 1 : 64 and 1 : 128 and 1 : 256) with gentle vortexing. The mixtures were left for 30 min to form siRNA–CPLA‐26 nanoplexes, and then analysed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The z‐average hydrodynamic diameter (Dz‐ave), polydispersity index (PDI) and zeta potential (ζ) values of nanoplexes were measured by DLS on a nano‐ZS90 (Malvern Instruments, Malvern, UK) in water at 25°. All DLS measurements were conducted using a 4‐mW 633‐nm helium/neon laser as the light source at a fixed measuring angle of 90° to the incident laser beam. The correlation decay functions were analysed by a cumulants method coupled with Mie theory to obtain volume and number distribution. TEM images were obtained using a JEOL 100CX microscope. Samples for TEM were prepared by dip coating a 300‐mesh carbon‐coated copper grid with a 0·1 mg/ml sample solution, followed by negative staining using a 1% aqueous solution of uranyl acetate. The binding affinity of siRNA–CPLA‐26 nanoplexes was assayed by agarose gel electrophoresis. We used fluorescein amidite (FAM) ‐labelled IL‐8‐siRNA complexed with CPLA‐26. Nanoplexes were prepared with different weight ratios (IL‐8‐siRNAFAM : CPLA 26 in the ratios of 1 : 8, 1 : 16, 1 : 32, 1 : 64, 1 : 128 and 1 : 256) and resolved on a 1% agarose gel. Nanoplexes and unconjugated IL‐8‐siRNA were loaded into the wells in tris–acetate–EDTA solution and resolved for 40 min at 70 V. Retardation of the nanoplexes was visualized by the fluorescence of FAM.
Cytotoxicity of CPLA
Cytotoxicity of CPLA‐26 was determined by measuring the viability of cells after treatment with CPLA‐26 for 48 hr. Briefly, 5 × 103cells were plated in 96‐well plates. After incubation for 24 hr, medium was replaced with medium containing CPLA‐26 ranging from 2–300 μg/ml. Untreated cells served as controls. Following a further 48 hr of incubation, cells were incubated with MTT (5 mg/ml), added at 10% volume/volume, for 3 hr at 37°, 5% CO2. Medium plus MTT was aspirated, and the formazan reaction products were dissolved in DMSO and shaken for 1 hr. The optical density of the formazan solution was read on a Microplate Reader (BioTek Instruments, Inc., Winooski, VT) at 570 nm. Results are shown as a percentage of untreated cells with 100% viability for three independent experiments.
Confocal imaging
PC‐3 cells were seeded on a square glass coverslip in 35‐mm glass‐bottom dishes at 40–50% confluence, cultured at 37° in a humidified atmosphere containing 5% CO2 for 24 hr. Preparation of siRNA nanoplexes was performed by mixing FAM‐labelled IL‐8 siRNA and CPLA‐26 for 30 min. Cells were transfected with IL‐8 siRNAFAM–CPLA‐26 nanoplexes at a weight ratio of CPLA 26 : siRNAFAM of 64 : 1 and further cultured at 37° in a humidified atmosphere containing 5% CO2 for 24 hr. All dishes were rinsed with saline to wash out any untransfected nanoplexes. Cell membranes were stained sequentially with wheat germ agglutinin conjugated to Texas Red for 20 min followed by 4’,6‐diamidino‐2‐phenylindole (DAPI) as a nuclear probe and subjected confocal imaging using a Zeiss LS700 confocal microscope. The parameters of photodetector gain, pinhole size and exposure time were kept constant for all confocal images.
RNA extraction and real‐time quantitative PCR
Cytoplasmic RNA was extracted by an acid guanidinium‐thiocyanate‐phenol‐chloroform method,27 using the TRIzol® reagent (Invitrogen). The final RNA pellet was dried and resuspended in diethyl pyrocarbonate‐treated water and the concentration of RNA was determined by spectrophotometry at 260 nm. Relative expression of IL‐8 mRNA or other mRNAs of interest was assessed using the SYBR green master mix from Stratagene (La Jolla, CA) to perform quantitative PCR (qPCR) using the Stratagene MX3005B (La Jolla, CA). Differences in threshold cycle number are used to quantify the relative amount of PCR target contained within each tube.28 Relative mRNA species expression is quantified and expressed as transcript accumulation index (), calculated using the comparative C T method.29 All data were controlled for quantity of RNA input by performing measurements on a reference gene, β‐actin. In addition, results from RNA‐treated samples were normalized to results obtained from RNA of the control, untreated sample.
In vivo animal models
Experimental studies were carried out using protocols approved by our respective Institute Animal Care and Use Committees (IACUC). Four‐ to 6‐week‐old male athymic BALB/c mice (Charles River Laboratories, Wilmington, MA) were kept in a temperature‐controlled room on a 12/12 hr light/dark schedule with food and water ad libitum. We previously described subcutaneous implantation of CaP cells.30 Briefly, 1 × 106 PC‐3 cells harvested from non‐confluent monolayer cultures in 50 μl of medium were injected under the right flank and tumour growth was monitored. Tumour‐bearing mice (n = 4/group) were treated with IL‐8 siRNA nanoplexes (50 ng/g body weight) or an equal volume of PBS as control, either by tail vein (systemic) or intratumoral (local) injections every other day for ~45 days (local) and ~35 days (systemic). Mice were monitored for toxicity and body weight daily; tumour growth was measured three times weekly with calipers. Tumours were harvested at the end of the study, weighed and fixed in 10% normal buffered formalin. Samples (4 mm) were stained with haematoxylin & eosin for further analysis. Image acquisition was carried out using a Scanscope XT system (Aperio Imaging; Leica Biosystems, Nussloch, Germany) and analysed using imagescope software (Aperio Imaging).
Assays for murine angiogenic factors
Mouse serum was collected and analysed with the bead‐based multiplexing technique using the Luminex Assay system (Millipore, Billerica, MA). The system consists of an EMD Millipore MILLIPLEX® MAP mouse angiogenesis/growth factor magnetic bead panel assay, a 24‐plex kit used for simultaneous quantification of the following analytes: angiopoietin‐2, granulocyte colony‐stimulating factor (G‐CSF), sFas ligand, sAlk‐1, amphiregulin, leptin, IL‐1β, β‐cellulin, epidermal growth factor, IL‐6, endoglin, endothelin‐1, fibroblast growth factor‐2, follistatin, hepatocyte growth factor, platelet endothelial cell adhesion molecule‐1 (PECAM‐1), IL‐17, platelet‐derived gowth factor‐2, keratinocyte‐derived chemokine, monocyte chemoattractant protein‐1, prolactin, macrophage inflammatory protein‐1α, stromal‐cell‐derived factor‐1, vascular endothelial growth factor C (VEGF‐C), VEGF‐D, VEGF‐A and tumour necrosis factor‐α (TNF‐α). All experiments consisted of at least three biological replicates independently performed at least three times.
Magnetic resonance imaging
Magnetic resonance imaging was conducted using a 4·7‐T/33‐cm horizontal bore magnet (GE NMR Instruments, Fremont, CA) incorporating AVANCE digital electronics (Bruker Biospec, ParaVision; Bruker Medical, Billerica, MA) using protocols previously described by us.31 Briefly, animals were anaesthetized by isoflurane inhalation (2–3% in oxygen) and placed on an acrylic sled equipped with respiratory and temperature sensors and positioned within the magnet. Preliminary scout images were acquired on the sagittal and axial planes to assist in slice prescription for subsequent scans. Multi‐slice non‐contrast enhanced T2‐weighted images were acquired on the axial plane with the following parameters: TEeff/TR = 41/2500 ms, matrix size 256 × 192, 1‐mm thick slices, RARE/echoes = 8/8, FOV 3·2 × 3·2 cm, NEX = 4. Three‐dimensional spoiled gradient echo images were acquired before and after administration of the MR contrast agent, albumin‐(GdDTPA)35 (0·05 mmol/kg), using the following parameters: matrix size 192 × 96 × 96; FOV 4·8 × 3·2 × 3·2cm, flip angle = 40°, acquisition time = 2 min 18 seconds. Raw image data sets were transferred to a workstation for image processing and analysis. Regions of interest (ROI) of tumours, kidneys and muscle tissues were manually drawn in the images and object maps of the ROI were constructed. Signal intensity (SI) values from different ROI were obtained and used to calculate tumour enhancement (E).32 Relative intensity (RI) values were calculated by normalizing the SI values to the phantom. Per cent tumour enhancement (%E) was then calculated from the RI in pre‐ and post‐contrast images using the formula E = (RIpost – RIpre)/RIpre.33
Ultrasound
Ultrasound imaging was performed using the Vevo 2100 high‐frequency ultrasound system (VisualSonics, Toronto, ON, Canada). Anaesthetized mice were positioned on a heated platform (THM 150; Indus Instruments, Webster, TX) equipped with integrated temperature sensor and ECG electrodes for imaging. An ultrasound acoustic gel (Aquasonic 100, Parker Laboratories Inc., Fairfield, NJ) was applied to facilitate ultrasound transmission from the transducer to the skin. A solid‐state transducer (MS‐250SC) was placed on the tumour and held in position by a clamp mounted on the Vevo Rail System. The transducer has a centre frequency of 18 MHz and an axial resolution of 75 lm at an elevation of 8 mm. Non‐linear contrast mode imaging was employed to detect the presence of the ultrasound contrast agent, (Vevo MicroMarker; VisualSonics).31 The agent consists of phospholipid shell microbubbles filled with perflurobutane and nitrogen with a diameter ranging from 2·3 to 2·9 lm. A bolus injection of 50 μl was delivered via tail vein. Non‐linear detection of the contrast signal was done in three dimensions by stepping the transducer through the volume of the tumour. Acquisition parameters were kept constant at 4% power, 28 dB contrast gain, gate size of 6, high line density, and standard beam width. Tumour volume was determined by manually outlining the borders of the tumour in several images throughout the image stack acquired in thre‐dimensional scanning. The per cent agent (PA) value was calculated using Vevo 2100 three‐dimensional software as the percentage of pixels within the tumour volume that have a non‐linear contrast overlay instead of a grey‐scale B‐mode pixel. Baseline PA values were acquired before contrast agent delivery and compared with PA value after delivery, while the contrast agent circulates through the subcutaneous tumour. A differential PA value was calculated by subtracting the baseline PA from the post‐injection PA; this value was used as a measure of relative blood volume.
Immunohistochemistry
Immunostaining of tissue sections for the endothelial cell adhesion molecule, CD31,33 and the proliferation marker, ki67, was performed as described by Folaron et al.34 Briefly, excised tissues were placed in zinc–formalin fixative for 18 hr and transferred to 70% ethanol, dehydrated and embedded in paraffin. Sections (5 μm) were stained with rat anti‐mouse CD31 monoclonal antibody (PECAM‐1; MEC 13.3; BD Biosciences, San Jose, CA) at 10 μg/ml concentration for 60 min at 37°. Duplicate slides stained with an isotype match control (rat IgG2a also at 10 μg/ml) instead of the primary antibody were used as negative controls. All slides were scanned and digitized using the ScanScope XT system and imagescope software (Aperio Technologies, Version 9·1; Vista, CA) at a magnification of 10 ×. To obtain an estimate of the microvessel density, CD31+ endothelial clusters (brown) with clearly visible lumina were counted in three randomly selected fields at 10 × magnification using imagescope software (Aperio Technologies). Tissue sections from two or three samples per group were analysed for statistical significance using an unpaired t‐test.
Assays for cytokines
Interleukin‐8, VEGF and interferon‐γ (IFN‐γ) protein levels in mouse serum were quantified using ELISA kits from R&D Systems according to the manufacturer's protocol. Assay performance and inter‐ and intra‐assay variations were within the limits defined by the manufacturer.
Preparation and characterization of nanoparticles
As naked siRNA is highly susceptible to enzymatic degradation and cannot penetrate the cell membrane easily and efficiently, we designed a nanotherapeutic system based on CPLA. The concept of gene delivery using CPLA is illustrated in Fig. 1. Negatively charged IL‐8 siRNA is complexed with CPLA by electrostatic interaction. A portion of the positive charges on CPLA is neutralized by IL‐8 siRNA, and the nanoplexes formed provide protection of siRNA and possess a net positive charge that can enhance cellular internalization through endocytosis. When the nanoplex is transported into the cytoplasm, the tertiary amine groups of CPLA appear on the nanoplex surface to facilitate the intracellular ‘proton‐sponge’ effect, whereby extra protons are transported and absorbed into the endosome, inducing osmotic swelling and rupture.13, 35 The nanoplexes eventually escape from the endosome and the IL‐8 siRNA is released upon degradation of CPLA. To achieve efficient delivery of siRNA by this method, several factors must be taken into account. As compared with step polymerization, ROP can yield well‐defined polymers with degradable in‐chain functionalities.36 Recent developments in click chemistry provide powerful new tools for polymer synthesis and functionalization.36 Therefore, synthesis through tandem living ROP and click chemistry was designed to achieve well‐defined CPLA. Further, the organocatalysed ROP process and thiol‐ene click reaction were selected, because they can be conducted under mild conditions, without using toxic metal‐based catalysts.37, 38
Statistical analyses
All experiments were repeated at least three times. Values are expressed as the mean ± SE. The significance of the difference between the control and each experimental test was analysed by unpaired t‐test, and a value of P < 0·05 was considered statistically significant.
Results
Correlation of IL‐8, CXCR‐1, and CXCR‐2 expression by CaP cells with an aggressive phenotype
We conducted in vitro experiments on the metastatic CaP cell lines, PC‐3 and PC‐3M, to examine the role of IL‐8 and IL‐8 receptors in their aggressive phenotypes. Consistent with our earlier report of increased IL‐8 secretion by PC‐3 cells,24 we observed similar results confirming our previous finding and extending it to the more aggressive PC‐3M cells (Fig. 2a). In addition, we selected a derivative of PC‐3M cells, PC3‐MM2, which are characterized as even more aggressive than PC‐3M cells in our previous report24 and found even greater secretion of IL‐8 compared with PC‐3 parental cells (Fig. 2b). The level of IL‐8 secreted by the PC‐3MM2 cells (309·8 ± 12·3 pg/ml/106 cells/24 hr; n = 8) was significantly (P < 0·001) higher than that detected with PC‐3 cells (200·4 ± 8·2 ng/ml/106 cells/24 hr; n = 4). As the IL‐8 signal is mediated in these cells by their corresponding IL‐8 receptors, we determined the expression of the IL‐8 receptors, CXCR‐1 and CXCR‐2, on PC‐3 and PC‐3M cells and found significantly elevated expression levels by immunofluorescence. Abundant CXCR‐1 expression but weak CXCR‐2 expression was detected on the surface of PC‐3 and PC‐3M cells (Fig. 2c). Conversely, negligible expression of either CXCR‐1 or CXCR‐2 was detected on the surface of normal prostate and LNCaP cells, which do not manifest an aggressive phenotype (see Supplementary material, Fig. S1 panel 1 and 2). Additionally gene expression analysis of CXCR‐1 and CXCR‐2 receptors by all cell lines show similar pattern (see Supplementary material, Fig. S2). We also analysed serum from patients with BPH, localized CaP, CRPC and normal subjects. The results shown in Fig. 2(d) clearly document that IL‐8 levels are significantly higher in the sera of localized CaP and CRPC patients compared with normal and BPH subjects.
Figure 2.
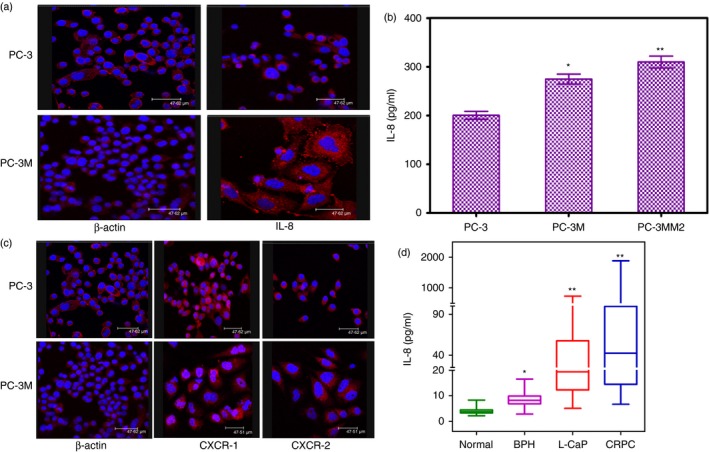
Constitutive expression of interleukin‐8 (IL‐8) and its receptor CXCR‐1 and CXCR‐2 by aggressive prostate cancer (CaP) cell lines and CaP patients. (a) Immunofluorescence of PC‐3 cells upper panel and PC‐3M cells lower panel of β‐actin and IL‐8, (b) basal protein levels of IL‐8 in PC‐3, PC‐3M and PC‐3MM2 cells. Tumour cells (3 × 106) were cultured for 48 hr, the culture supernates were collected and used for quantifying the levels of IL‐8 by ELISA. Values are mean ± SE of four experiments. *P < 0·05 and **P < 0·01 compared with PC‐3 cells (c) CXCR‐1 and CXCR‐2 immunofluorescence of PC‐3 cells upper panel and PC‐3M cells lower panel of β‐actin, CXCR‐1 and CXCR‐2 (d) Serum levels of IL‐8 in CaP patients compared with normal subjects and a different category of CaP patients. Blood samples from controls (n = 46), benign prostatic hyperplasia (BPH) (n = 50), Localized CaP (n = 49), and castration‐resistant protstate cancer (CRPC) (n = 109) were collected, serum was separated and stored until analysis. IL‐8 in serum was measured in triplicate by ELISA. Values are mean ± SE. *P < 0·05 and **P < 0·01.
Effect of rhIL‐8 on CaP cell proliferation and survival in vitro
To verify whether IL‐8 has a direct mitogenic role in CaP cell proliferation, we examined the effect of treating PC‐3 or PC‐3M cells with exogenous rhIL‐8 on proliferation. We observed concentration‐dependent increases in the proliferation of PC‐3 and PC‐3M cells in response to rhIL‐8 (Fig. 3a). The response plateaued at 1 nm of rhIL‐8 and total cell number increased by 60% over 72 hr compared with untreated cells. The maximal efficacy of 1 nm rhIL‐8 on proliferation of PC‐3 and PC‐3M cells correlates well with the known high affinity of this ligand for each of its receptors. However, when non‐aggressive LNCaP or normal prostate epithelial cells were similarly treated with rhIL‐8, no significant effect on proliferation was observed. When DU‐145 cells were treated similarly, a significant increase in proliferation was seen starting at 2·5 nm rhIL‐8 onwards (see Supplementary material, Fig. S3a).
Figure 3.
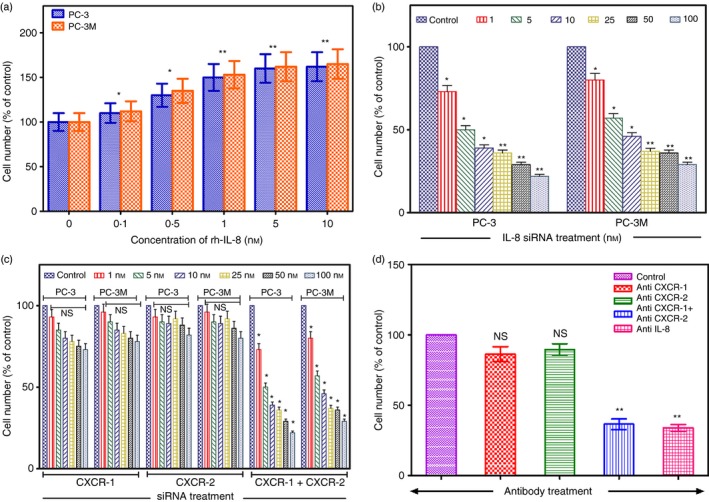
Recombinant human interleukin‐8 (rh‐IL‐8) induces CaP cell proliferation, which is inhibited by silencing the genes for IL‐8 or its receptors. (a) PC‐3 and PC‐3M cells were treated with different concentrations of rhIL‐8 and cell counts were determined after 72 hr. Results are the mean ± SE of six independent experiments done in triplicate. (b) PC‐3 and PC‐3M cells were treated with different concentrations of IL‐8 small interfering (si) RNA + Lipofectamine 2000™ as described in the Materials and methods and cell counts were determined after 48 hr. Results are the mean ± SE of three independent experiments done in triplicate. (c) PC‐3 and PC‐3M cells were treated with different concentrations of CXCR‐1 or CXCR‐2 siRNA individually or in combination as described and cell counts were determined after 48 hr. Results are the mean ± SE of three independent experiments done in triplicate. For (b) or (c) above treatment with the relevant scrambled siRNA or had no measurable effect on cell proliferation and was comparable to untreated control cells. (d) PC‐3 cells were treated with neutralizing antibodies (6 μg/ml) to IL‐8, CXCR‐1 or CXCR‐2 and cell counts were determined after 48 hr. Results are the mean ± SE of three individual experiments done in triplicate. Statistical significance:*P < 0·05, **P < 0·01, NS = not significant compared to untreated controls.
Effect of abrogation of IL‐8 signalling in CaP cells
Since IL‐8 showed a direct role in CaP cell proliferation and stimulation of the production of proteins involved in cell survival pathways, we performed further experiments to see if abrogation of IL‐8 expression could reverse CaP cell growth and proliferation in vitro. We transfected PC‐3 and PC‐3M cells with the siRNA specific for IL‐8 (IL‐8 siRNA) using Lipofectamine‐2000 and compared its effect to a control siRNA. Figure 3(b) shows that cells treated with increasing doses of IL‐8 siRNA (1–100 nm) manifested decreased proliferation in a dose‐dependent manner as determined by an MTT assay performed 72 hr post‐treatment. Scrambled siRNA control did not have any effect on proliferation of CaP cells. This experiment was performed with several concentrations of fetal bovine serum (0·5, 1, 5 and 10%) with the same outcome showing that decreased CaP cell proliferation was consistently associated with IL‐8 siRNA and not due to a confounder. Similar results were obtained for DU‐145 cells, which have an intermediate aggressive phenotype (see Supplementary material, Fig. S3b). However with non‐aggressive LNCaP cells and normal prostate epithelial cells no measurable effects of IL‐8 siRNA were seen (see Supplementary material, Fig. S3b). These data demonstrate that the growth of CaP cells in vitro is modulated by IL‐8. These data also demonstrate that growth of highly aggressive (PC‐3MM2, PC‐3M, PC‐3) and intermediately aggressive (DU‐145) CaP cells prone to metastasis to bone could be suppressed by silencing the expression of the IL‐8 gene, whereas non‐aggressive (LNCaP) CaP cells and normal prostate epithelial cells were not affected by IL‐8 siRNA. We performed additional experiments to examine the role of IL‐8 receptors in CaP cell growth. We observed that when gene expression of either IL‐8 receptor, CXCR‐1 or CXCR‐2, was individually silenced with specific siRNAs, inhibition of proliferation of PC‐3 cells was minimal. However, when siRNAs specific to both IL‐8 receptors were used together, suppression of PC‐3 growth was significant (Fig. 3c). The silencing of CXCR‐1 or CXCR‐2 genes either alone or in combination did not inhibit the proliferation of NP and LNCaP cells and had a minimal effect on moderately aggressive DU‐145 cells (see Supplementary material, Fig. S4). When PC‐3 cells were treated with polyclonal mouse antisera to CXCR‐1 and CXCR‐2 independently we did not observe any significant decrease in cell proliferation (Fig. 3d), However, when anti‐IL‐8 polyclonal mouse sera or a combination of CXCR‐1 and CXCR‐2 monoclonal antibodies were used we observed significant inhibition of proliferation (Fig. 3d).
Inhibition of the invasive activity of PC‐3 cells by siRNA specific for IL‐8
We next analysed if knock down of the expression of IL‐8 by siRNA affects the ability of CaP cells to invade the basement membrane, an important step in the process of tumour metastasis. CaP cells and normal prostate epithelial cells were transfected with IL‐8 siRNA or scrambled control siRNA using lipofectamine‐2000 for 48 hr. Confirmation of the knockdown effect was assessed by Western blotting (Fig. 4a). The IL‐8‐specific siRNA suppressed IL‐8 synthesis almost completely, whereas scrambled siRNA had no effect on IL‐8 expression (Fig. 4a upper panel and b). A set of mock transfected cells showed no decrease in IL‐8 synthesis by CaP cells (Fig. 4a upper panel and b). Actin expression by the controls was unaffected by treatment with either IL‐8‐specific or scrambled siRNA. Next, we assessed the effect of IL‐8 knock down on cell invasion. For each cell type the efficiency of IL‐8 knock down was also assessed by quantifying the level of IL‐8 in culture supernates by ELISA to confirm the effect of gene knock down (see Supplementary material, Fig. S5). Knockdown‐confirmed IL‐8 siRNA‐treated cells were placed in the upper compartment of a dual‐chamber culture plate separated by a polycarbonate membrane precoated with basement membrane proteins derived from the Engelbeth Holm–Swarm mouse tumour. After 24 hr of incubation, cells invading the lower surface of the filter were stained and counted following the removal of non‐invading cells. Results of this study are shown in Fig. 4(a, lower panel and c). All CaP cells including highly aggressive PC‐3M and PC‐3 cells, moderately aggressive DU‐145 cells, and minimally aggressive LNCaP cells when treated with IL‐8 siRNA exhibited significant (P < 0·01) decreases in their invasive activity with the least effect on the minimally aggressive LNCaP cells (Fig. 4c). By comparison, the invasive activities of control cells, either untreated or treated with scrambled siRNA, were unaffected. Normal prostate epithelial cells showed no invasive activity and also were unaffected by IL‐8 siRNA.
Figure 4.
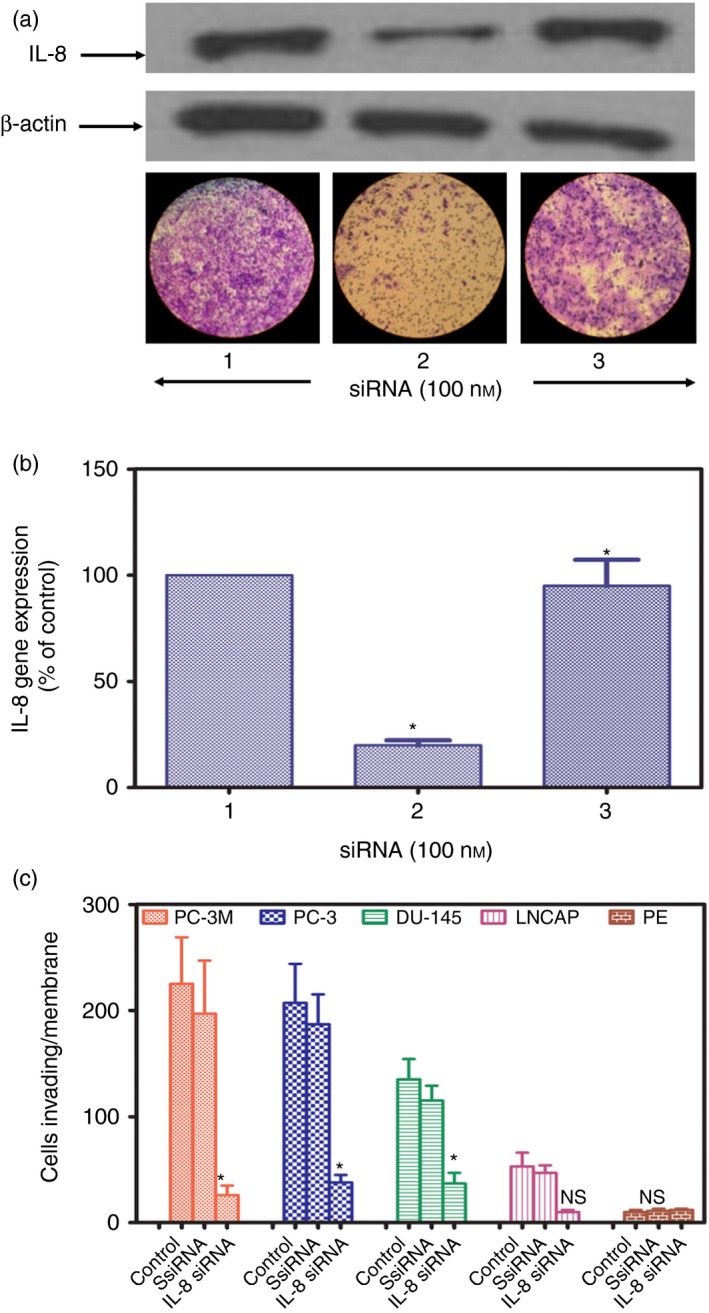
Knockdown of interleukin‐8 (IL‐8) gene expression inhibits the invasive activity of prostate cancer (CaP) cells. Representative PC‐3 cells were transfected for 72 hr using Lipofectamine 2000™ with either 100 nm IL‐8 small interfering (si) RNA, or 1000 nm scrambled IL‐8 siRNA, or an equal volume of buffer control. (a) Upper panel. Lane 1, PC‐3 cells transfected with scrambled IL‐8siR. Lane 2, PC‐3 cells transfected with IL‐8 siRNA. Lane 3, PC‐3 cells treated with buffer control alone. Cells were lysed, lysates were run on 4% SDS reducing gel electrophoresis and transferred to PVDF membranes for Western blotting for IL‐8 and β‐actin as described. Representative Western blots from three separate experiments done in triplicate yielded similar results. IL‐8 siRNA induced marked suppression of IL‐8 synthesis by PC‐3 cells, whereas β‐actin synthesis was unaffected. Lower panel. Representative photographs a cell invasion assay at 100 × magnification using PC‐3 cells transfected, with IL‐8 siRNA (Lane 1), scrambled siRNA (Lane 2) or untransfected (Lane 3) applied to the upper surface of a filter coated with basement membrane proteins from the Engelbeth Holm–Swarm mouse tumour. After incubation for 24 hr, the upper surface of the filter was scrubbed free of cells and basement membrane, the filter was fixed and stained and the lower surface was photographed. Note the marked inhibition of invasion due to knockdown of IL‐8 expression. (b) Confirmation of decreased IL‐8 gene expression by PC‐3 cells transfected with IL‐8 siRNA. Results are mean of three experiments done in triplicate. (c) Suppression of invasive activities of different CaP cell lines and normal prostate epithelial cells (PE) transfected with IL‐8 siRNA compared with scrambled (Ss) IL‐8 siRNA and untransfected controls. *P < 0·001 compared with control; NS = not significant.
Cytotoxicity of CPLA‐26
Any nanodelivery agent for use in clinical applications must be safe and non‐toxic. The MTS cell viability assay (Promega, Madison, WI) was used to determine the effect of CPLA‐26 on cell viability in vitro. Dose‐dependent cytotoxicity studies using CPLA were carried out with PC‐3 cells. Untreated PC‐3 cells served as the reference (i.e. 100% viability). Results from the MTS assay (Fig. 5a) show that the treatment of PC‐3 cells with a broad range of concentrations of CPLA‐26 for 48 hr did not produce any cytotoxicity. Similar studies on normal prostate epithelial cells also did not produce any evidence of toxicity (see Supplementary material, Fig. S6a). A dosage of 300 μg/ml of CPLA‐26 is very high compared with the maximum safe dosages of only 10 μg/ml for poly ethylenimine reported in the literature.39 Hence the much higher concentrations of CPLA, which did not produce any cytotoxicity, demonstrate that CPLA‐26 is safe to use in biological systems.
Figure 5.
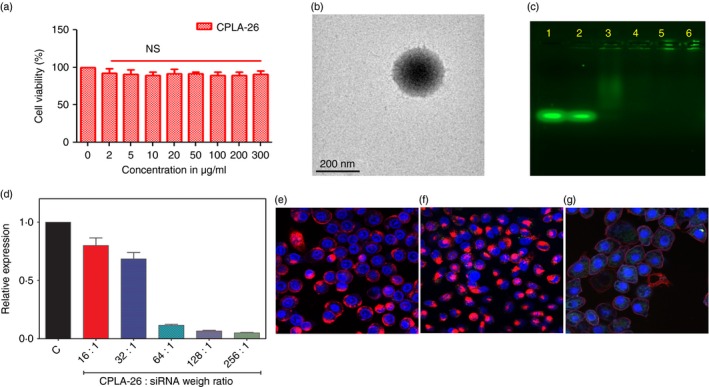
Characterization of CPLA‐26 nanoparticles. (a) Cytotoxicity assay for CPLA‐26. PC‐3 cells were treated with different concentrations of CPLA‐26, incubated for 48 hr and viability was determined. (b) Transmission electron microscopy (TEM) of CPLA‐26. Samples were prepared by dip coating a 300‐mesh carbon‐coated copper grid with a 0·1 mg/ml solution of CPLA‐26 followed by negative staining with 1% aqueous uranyl acetate. Images were obtained with a JEOL 100CX electron microscope. (c) Encapsulation of small interfering (si) RNAFAM by CPLA‐26 as evaluated by agarose gel electrophoresis. FAM labelled interleukin‐8 (IL‐8) siRNA was mixed at varying weight ratios of CPLA‐26 : IL‐8 siRNA and resolved on 1% agarose gel. Lane 1, free siRNAFAM. Lanes 2–6, CPLA‐26:siRNA nanoplexesFAM at ratios of 1 : 16 (lane 2), 1 : 32 (lane 3), 1 : 64 (lane 4), 1 : 128 (lane 5) and 1 : 256 (lane 6). In Lanes 3–6 encapsulation of the IL‐8 siRNAFAM by the highly cationic CPLA‐26 nanoparticles retarded the nanocomplex from entering the gel and moving toward the anode. (d) Ability of CPLA‐26 IL‐8 siRNA nanoplexes to knockdown gene expression of IL‐8 by PC‐3 cells. Cells were treated with IL‐8 siRNA nanoplexes in serum‐free media for 2 hr and reconstituted to 10% fetal bovine serum. After 48 hr mRNA was extracted and the expression of IL‐8 was measured by quantitative PCR. Results are normalized to the control gene, β‐actin, and are expressed as relative fold expression compared with untreated control cells. (e–g) Confocal images of internalization of CPLA‐26 IL‐8 siRNAFAM by PC‐3 cells. Nanoplexes were prepared by mixing FAM‐labelled IL‐8 siRNA and CPLA‐26 for 30 min. Cells were treated with either free CPLA‐26, free IL‐8 siRNAFAM or CPLA‐26 IL‐8siRNAFAM naoplexes. After 24 hr cells were rinsed and stained with wheat germ agglutinin conjugated to Texas Red followed by DAPI as a nuclear probe and subjected to confocal imaging. (e) Free CPLA‐26. (f) Free IL‐8 siRNAFAM. (g) CPLA‐26 IL‐8‐siRNAFAM nanoplexes.
Physical characterization of CPLA‐26–IL‐8 siRNA nanoplexes
Zeta potential (ζ) of CPLA‐26–IL‐8 siRNA nanoplexes (WCPLA/WsiRNA = 32, 64, 128) was measured by DLS. With positively charged CPLA‐26 as the major component, these nanoplexes possessed positive surface charge densities increasing with the mass fraction of CPLA‐26 (Table 1). The nanoplexes with the highest WCPLA/WsiRNA of 128 (ζ = 55·7 ± 1·3 mV) showed only slightly lower surface charge density than naked CPLA‐26 (ζ = 63·5 ± 1·3 mV). Selected as the representative sample, CPLA‐26‐IL‐8 siRNA nanoplexes with a weight ratio WCPLA : WsiRNA of 128 were characterized by TEM (Fig. 5b). Spherical morphologies of these nanoplexes were revealed. Statistical analysis of multiple nanoplex particles on TEM images indicated an average diameter (Da) of 226 ± 28 nm, which is in the order of magnitude of their Dz‐ave value (326 ± 6 nm) measured by DLS analysis of their aqueous solution. Agarose gel retardation assays were used to determine the minimum amount of CPLA‐26 required to interact completely with siRNA. Results shown in Fig. 5(c) demonstrate that CPLA‐26 starts to minimally retard IL‐8sIRNAFAM at a 16 : 1 CPLA:siRNA weight ratio (Fig. 5c, lane 2). Increased binding can be observed at higher ratios (lanes 3–6) with complete retardation at 1 : 64 (Fig. 5c, lane 4). All weight ratios (2 : 1 to 128 : 1) were screened by PCR for the ability of the IL‐8 siRNA nanoplexes to knock down expression of IL‐8 by PC‐3 cells. Results shown in Fig. 5(d) demonstrate that CPLA‐26–IL‐8 siRNA nanoplexes show increasing efficiency of knock down of the IL‐8 gene with increasing weight ratio. The maximum effect, 92% inhibition, was observed with CPLA siRNA nanoplexes at a weight ratio of 128 : 1, which is higher than the efficiency of all commercially available transfection agents (data not shown).
Table 1.
Dynamic light scattering analysis of CPLA‐26–interleukin‐8 small interfering RNA nanoplexes
| WCPLA/WsiRNA | Dz‐ave (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| 32 : 1 | 399 ± 2 | 0·33 ± 0·06 | 17·4 ± 0·4 |
| 64 : 1 | 253 ± 2 | 0·26 ± 0·03 | 32·3 ± 1·3 |
| 128 : 1 | 326 ± 6 | 0·36 ± 0·05 | 55·7 ± 1·3 |
Confocal imaging
To visualize the delivery of IL‐8 siRNA nanoplexes into cells, CPLA‐26 was complexed with FAM‐labelled IL‐8 siRNA to form CPLA‐26–IL‐8 siRNAFAM nanoplexes and its internalization into PC‐3 cells was monitored by confocal microscopy (Fig. 5e–g). After incubation of PC‐3 cells with CPLA‐26–IL‐8 siRNAFAM for 4 hr, strong green fluorescence signals from the IL‐8 siRNAFAM were observed (Fig. 5g). The overlaid fluorescence and bright‐field images further revealed that most of CPLA‐26–IL‐8FAM was taken up into the cytoplasm by endocytosis. Free CPLA‐26 and naked IL‐8 siRNAFAM were used as controls in the cell internalization study. Experiments using free CPLA‐26 showed no fluorescence. Under the same conditions, only minimal uptake of naked IL‐8 siRNAFAM was observed. Similar studies performed with normal prostate epithelial cells also show an uptake pattern comparable to PC‐3 cells (see Supplementary material, Fig. S5).
Silencing of IL‐8 gene expression by CPLA‐26–IL‐8‐siRNA nanoplexes has therapeutic effects on the growth of localized tumours in a model of human CaP
These studies progress from our in vitro observation that knockdown of IL‐8 gene expression results in down‐regulation of pro‐angiogenic proteins and decreased invasiveness, proliferation and viability of CaP cells. We observed that IL‐8 siRNA nanoplexes administered to nude mice bearing PC‐3 tumours indeed can significantly suppress tumour growth. Five‐week‐old athymic BALB/c nude mice were inoculated subcutaneously with 1 × 106 PC‐3 cells in the flank region. Forty‐eight hours after tumour cell inoculation, animals were treated with of IL‐8 siRNA nanoplexes (50 ng siRNA/g body weight), administered subcutaneously within the vicinity of tumour cell inoculation. Nanoplexes were administered on alternate days for a total of 7 weeks. Previously we detected circulating levels of labelled siRNA nanoplexes up to 48 hr after administration either subcutaneously or intravenously.21 The dose of siRNA in the nanoplexes was selected to attain an siRNA level in the vicinity of the tumour similar to the in vitro levels required for knockdown of the IL‐8 gene. Tumours began to grow in control animals at the inoculated site by the first week, whereas only small palpable tumours were seen in animals treated with nanoplexes at this time (Fig. 6a). Tumour growth was followed for 7 weeks until termination of the experiment. Tumour volumes in both groups were monitored periodically either by measuring with a caliper (Fig. 6a) or by MRI (Fig 6b). The mean tumour volume of the control animals by the end of 7 weeks was 128·6 ± 14·5 mm3, whereas that of treated animals was 79·3 ± 19·7 mm3 (P < 0·001). Mean tumour volumes measured by MRI midpoint in the experiment were also significantly lower in IL‐8 nanoplex‐treated mice (Fig. 6b). The mean tumour volume of the control animals by MRI at mid‐point was 64·2 ± 12·4 mm3, whereas that of treated animals was 15·9 ± 2·8 mm3 (P < 0·001). At termination of the experiment after 7 weeks, tumours were removed, weighed and snap frozen for RNA extraction. The mean tumour weight of the treated mice was 432·0 ± 27·5 mg, whereas that of the untreated control animals was 777·2 ± 48·6 mg (P < 0·001; Fig. 6c).
Figure 6.
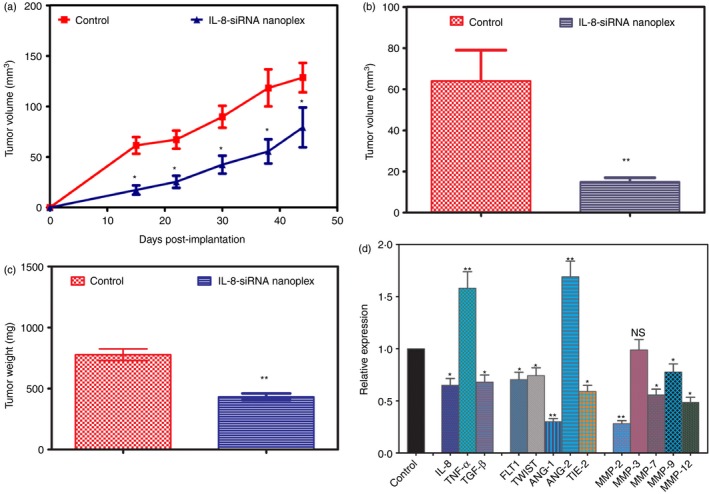
Response of PC‐3 xenografts to intralesionally administered CPLA‐26 interleukin‐8 (IL‐8) small interfering (si) RNA naoplexes in vivo. PC‐3 cells (1 × 106) were implanted in the flank of nude mice and 2 days later mice were injected either with IL‐8 siRNA nanoplexes (50 ng/kg body weight) or an equal volume of saline on alternate days for ~7 weeks. (a) Growth curves of PC‐3 tumours in control and IL‐8 siRNA nanoplex‐treated mice. Serial caliper tumour measurements were done on alternate days for 7 weeks and tumour volume was calculated by the formula L/2 × W2. (b) Midpoint tumour volume measured by MRI. (c) Endpoint tumour weight ex vivo. (d) Relative fold expression of tumour angiogenesis‐related genes performed by quantitative PCR from tumours resected on termination of experiment. Gene expression is presented as fold increase in ΔCT. Gene expression is presented as fold expression compared with gene expression of the corresponding gene in the control group as unity. *P < 0·05, **P < 0·001 compared with control, NS = not significant.
Suppression of tumour growth observed in response to IL‐8 nanoplex treatment may be the result of changes in the expression of various growth factors. To validate this hypothesis we performed gene expression analysis on tumour samples resected from the treatment groups. We selected a panel of genes known for their involvement in tumour angiogenesis, including cytokines and matrix metalloproteinase (MMPs), and carried out gene expression studies using qPCR. We examined the relative expression of these genes in tumours resected from IL‐8 siRNA nanoplex treated animals and compared it with controls. RNA was reverse‐transcribed and cDNA‐amplified by qPCR using primers specific for the genes of interest and the housekeeping gene, β‐actin. Experiments were repeated three times and results are shown in Fig. 6(d). Treatment of tumour‐bearing animals with IL‐8 siRNA nanoplexes for 7 weeks significantly down‐regulated the gene expression of IL‐8, transforming growth factor‐β, fms‐like tyrosine kinase, epithelial–mesenchymal transition‐inducing factor, tyrosine kinase with immunoglobulin‐like and EGF‐like domains‐2, angiopoietin‐1 genes, and up‐regulated angiopoietin‐2 and TNF‐α genes. All the MMPs studied except MMP‐3 were also down‐regulated significantly but MMP‐3 showed no change compared with controls.
Inhibition of prostate tumour growth by systemic administration of IL‐8 siRNA nanoplexes in a model of CaP
Inhibition of tumour growth in vivo was investigated by tail vein administration of IL‐8 siRNA nanoplexes. BALB/c nude mice with PC‐3 subcutaneous xenografts were randomly divided into control and IL‐8 siRNA nanoplex‐treated groups. Three days after tumour inoculation we initiated IL‐8 siRNA nanoplex treatment via tail vein at the same dose described above and followed the treatment every other day for 4 weeks. Tumours in the control group grew rapidly (Fig. 7a) compared with growth of tumours in the IL‐8 siRNA nanoplex‐treated group. Tumour volumes by caliper measurements were significantly lower at every point measured in the IL‐8 siRNA nanoplex‐treated group. After killing the mice, tumours were collected and weighed. Consistent with the growth curves, the mean weight of the tumours in the IL‐8 siRNA nanoplex‐treated group was significantly less than the saline‐treated group (Fig. 7b). To demonstrate the role of IL‐8 knockdown in the interference of tumour growth, we sectioned tumour xenografts and analysed the levels of IL‐8 gene expression and IL‐8 protein in the tissue homogenates by qPCR and ELISA, respectively. As shown in Fig. 7(c,d) control xenografts demonstrated substantial levels of expression of IL‐8 gene and protein, which was significantly suppressed in the IL‐8 siRNA nanoplex‐treated group.
Figure 7.
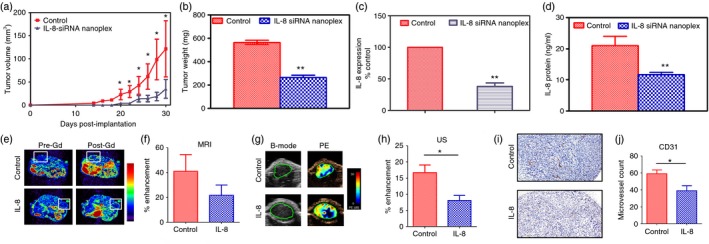
Response of PC‐3 xenografts to CPLA‐26 interleukin‐8 (IL‐8) small interfering (si) RNA nanoplex treatment administered intravenously to nude mice. PC‐3 (1 × 106) cells were implanted in the flank and 2 days later mice were injected with IL‐8 siRNA nanoplexes by tail vein on alternate days for 5 weeks. (a) Serial caliper tumour measurements were done biweekly on control and IL‐8 siRNA nanoplex‐treated animals. (b) End‐point tumour weight ex vivo for both groups, (c) IL‐8 gene expression in resected tumours from control and IL‐8 siRNA nanoplex‐treated animals. (d) Quantitative levels of IL‐8 protein in tumours from mice treated with IL‐8 siRNA nanoplexes and untreated controls. (e) Axial magnetic resonance images of control and CPLA‐26 IL‐8 siRNA nanoplex‐treated tumours (box outline) before (Pre‐Gd) and after (Post‐Gd) administration of the contrast agent, albumin‐GdDTPA, (f) Calculated values of % contrast enhancement in tumours from control and CPLA‐26 IL‐8 siRNA nanoplex‐treated animals. (g) B‐mode ultrasound (US) and % enhancement (PE) maps of tumours from control and treated animals. (h) Quantification of % enhancement based on contrast‐enhanced ultrasound of control and CPLA‐26 IL‐8 siRNA nanoplex‐treated tumours. (i) Photomicrographs of CD31‐stained tumour sections (20× magnification) from control and treated animals showing vascular damage following IL‐8 siRNA nanoplex treatment. (j) Calculated values of microvessel counts (3 10× fields per individual tumour; controls n = 4, IL‐8 siRNA nanoplex treated n = 4) showing reduction in vessel count in treated tumours compared with untreated controls. *P < 0·05, **P < 0·01 compared with controls.
Effects of silencing IL‐8 signalling on tumour angiogenesis
To assess the impact of silencing the IL‐8 gene on tumour angiogenesis, we examined the vascularity of control and siRNA IL8 nanoplex‐treated tumours. Quantitative estimates of MRI and ultrasound contrast agents were used as an indirect measure of blood volume (Fig. 7e–g). As shown in Fig. 7(e) compared with volume‐matched controls, the IL‐8 siRNA nanoplex‐treated group showed a reduction in contrast enhancement within the tumour but this reduction did not reach statistical significance. However, when indirect blood volume measurements were made using ultrasound contrast agents, IL‐8 siRNA nanoplex‐treated tumours showed a significant reduction in contrast enhancement (P < 0·05) compared with volume‐matched control tumours (Fig. 7f,g). In addition to non‐invasive imaging, immunostaining of the tumour sections for the endothelial cell marker, CD31, was also performed. As shown in Fig. 7(h,i), a significant reduction in microvessel density (P < 0·05) was seen in IL‐8 siRNA nanoplex‐treated tumours compared with control tumours. Additionally, when staining of the proliferation marker ki 67 was performed we did not observe significant differences between the study groups (see Supplementary material, Fig. S7).
Effect of IL‐8 siRNA nanoplexes on the expression of angiogenic factors associated with tumour resistance in tumour‐bearing mice
Although inhibition of angiogenesis as an approach to treat malignant tumours has provided clinical benefit, there is increasing recognition of the development of resistance to this mode of treatment. Although ample evidence indicates that such resistance can be influenced by the tumour microenvironment, the underlying mechanisms remain incompletely understood with speculations that tumours achieve resistance through secreted factors. To determine if the expression of such factors is modulated by treatment with IL‐8 siRNA nanoplexes, we measured serum levels in our treatment group and compared them to those in controls. We found that serum levels of the cytokine IL‐17A, which confers resistance of tumours to therapy, were significantly lower in the treated group compared with controls (Fig. 8a). Interleukin‐17A, the canonical cytokine of the T helper type 17 subset of T cells also known as CTLA8, is a pro‐inflammatory cytokine that has been implicated in autoimmunity, inflammation and cancer. We next examined amounts of pro‐inflammatory factors known to be induced by IL‐17A and found that serum levels of G‐CSF and IL‐6 were significantly decreased in IL‐8 siRNA nanoplex‐treated mice compared with controls (Fig. 8b,c), whereas the amounts of other IL‐17‐inducible factors, such as chemokine (C‐X‐C motif) ligand 1 (CXCL1), ligand 2 (CXCL2), TNF‐α, and IL‐1β, were unchanged compared with controls (data not shown). The IL‐8 siRNA nanoplex‐treated mice also demonstrated significantly decreased levels of three different isoforms of vascular endothelial growth factors, VEGF‐A, VEGF‐C and VEGF‐D (Fig. 8d–f). Levels of basic fibroblast growth factor‐2, a member of the fibroblast growth factor family known to be involved in angiogenesis, and levels of follistatin, another important angiogenic factor, were also significantly lower in IL‐8 siRNA nanoplex‐treated animals as compared with controls (Fig. 8g,h). Together, these data demonstrate that the expression of IL‐17A and other factors involved in inflammation and angiogenesis regulated by IL‐17A are all suppressed in tumour‐bearing animals treated with IL‐8 siRNA nanoplexes compared with controls. Over the course of 4 weeks of treatment with IL‐8 siRNA nanoplexes no resistance to antiangiogenic therapy was observed.
Figure 8.
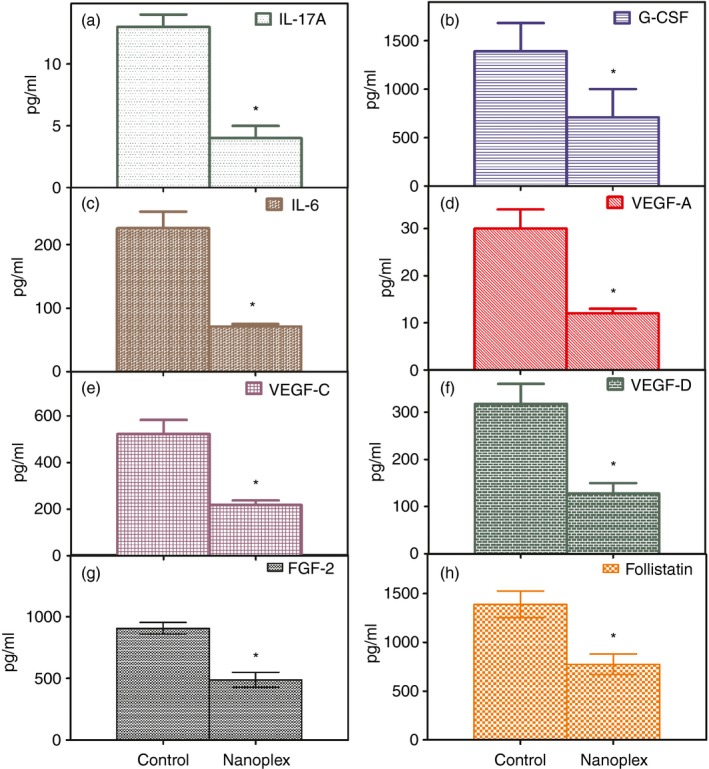
Levels of protein factors associated with mediating tumour resistance to antiangiogenic treatment in serum of treated and control mouse. PC‐3 cells (1 × 106) were implanted in the flank of nude mice and 2 days later mice were injected either with IL‐8 siRNA nanoplexes (50 ng/kg body weight) or an equal volume of saline on alternate days for ~5 weeks when the study was terminated. On termination the animals were anaesthetized, blood was collected by cardiac puncture, serum was separated and stored at −20° until further analyses. The serum levels of protein factors were quantified by bead‐based multiplexing technique using the Luminex Assay system as described in the methods section. Shown are serum concentrations of interleukin‐17A (IL‐17A) (a), granulocyte colony‐stimulating factor (G‐CSF) (b), IL‐6 (c), vascular endothelial cell growth factor A (VEGF‐A) (d), VEGF‐C (e), VEGF‐D (f), fibroblast growth factor‐2 (FGF‐2) (g) and follistatin in control groups compared with CPLA‐26–IL‐8 siRNA nanoplex‐treated groups (n = 4 per group). The data are shown as the mean ± SEM. *P < 0·05 compared with controls.
Effects of free and nanocomplexed IL‐8 siRNA and the CPLA‐26 nanocarrier on the expression of pro‐inflammatory cytokines in vivo
Normal BALB/c nude mice were injected via the tail vein with saline, CPLA‐26 (300 μg/ml) alone, free IL‐8 siRNA (50 ng/g body weight) and IL‐8 siRNA nanoplexes (50 ng siRNA/g body weight). One hour and 3 hr post‐treatment, serum was obtained and assayed by ELISA for the pro‐inflammatory cytokines, IL‐6 and IFN‐γ. As shown in Fig. 9(a,b), free IL‐8 siRNA was very effective in inducing both IL‐6 and IFN‐γ in normal animals. Injection of the free nanocarrier CPLA‐26 produced no effect on the expression of these cytokines. However, encapsulation of the IL‐8 siRNA within the CPLA nanocarrier inhibited the ability of IL‐8 siRNA to induce expression of pro‐inflammatory cytokines in vivo. These observations demonstrate that complexing siRNA with nanoparticles not only protects the RNA from degradation in vivo but also has the surprising effect of protecting the host recipient from activation of pro‐inflammatory cytokines.
Figure 9.
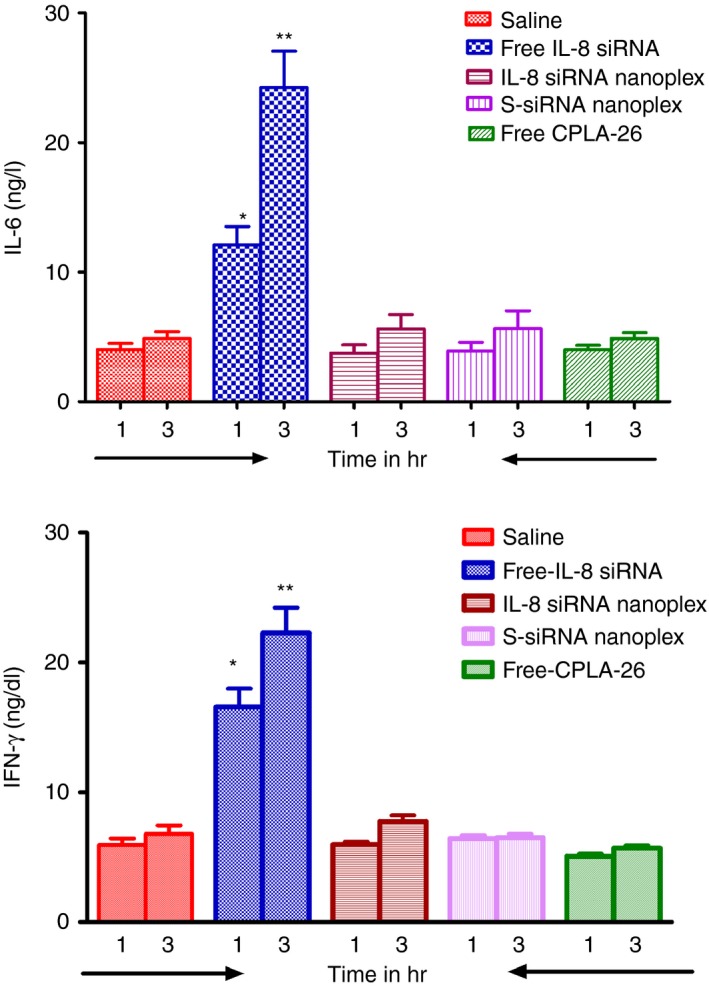
Levels of interleukin‐6 (IL‐6) and interferon‐γ (IFN‐γ) in sera from normal BALB/c mice treated with IL‐8 small interfering (si)RNA nanoplexes. *P < 0·05 versus control.
Discussion
Although treatment of CaP has improved, CRPC remains a serious clinical problem because chemoresistance develops rapidly. Despite initial response rates of 20–50%, most patients with CRPC die from their disease with a median survival of < 2 years.40 Therefore new therapies to replace or complement current agents are a major goal for patients with CRPC.41 We previously reported that CaP cells have different aggressive/metastatic phenotypes, which directly correlate with the gene expression and protein synthesis of pro‐angiogenic factors.24 This provided the first direct evidence linking the metastatic potential of CaP cells and their expression of pro‐angiogenic cytokines, suggesting that silencing any of these genes could yield a new treatment for metastatic CaP. Quoting our work, Lehrer et al.,42 measured the serum levels of angiogenic factors in men with CaP and noted significant elevation of IL‐8 in patients with bone metastases compared with men with local disease. We also reported that in lieu of using prostate‐specific antigen as a biomarker for CaP, a combination of cytokine measurements in serum could be used to diagnose aggressive CaP and a cytokine significantly associated with aggressive tumours in this study was again IL‐8.43 Concluding that IL‐8 is a cardinal cytokine contributing to CaP metastasis and angiogenesis we undertook the present study to further understand the role of IL‐8 in CaP aggressiveness and to determine if a new antiangiogenic therapy targeting the IL‐8 gene could be of benefit in treating CaP. In the current study we observed differential expression of IL‐8 and its receptors, CXCR‐1 and CXCR‐2, in human CaP cells with varying aggressive phenotypes (Fig. 2). Interleukin‐8 protein was significantly higher in the most invasive and highly metastatic CaP cells, PC‐3MM2 > PC‐3M > PC‐3 cells (Fig. 2b), demonstrating a direct correlation with aggressive phenotype. Malignant cells secrete factors that contribute to their survival. In addition, they express receptors that support proliferation and tumour growth. Interleukin‐8 is a known survival factor for a variety of cell types including malignant cells44 and is abundantly expressed by several carcinomas including CaP.4, 45, 46 Also, IL‐8 has angiogenic and mitogenic activities in a wide variety of human solid tumours.47 Though the mechanism(s) by which IL‐8 promotes tumour growth remains to be determined, some studies suggest an autocrine role of IL‐8 in modulating tumour cell survival and proliferation.48 In clinical studies, IL‐8 mRNA transcripts were shown to be significantly higher in neoplasms compared with normal tissues.49 Increased IL‐8 expression in metastasis‐prone cell lines has been observed and is believed to be the result of an atypical epigenetic mechanism whereby upstream CpG methylation rather than promoter methylation, results in increased IL‐8 production.50 Our data associating IL‐8 with increased growth and invasiveness of CaP cells in general is consistent with observations that IL‐8 facilitates the progression of LNCaP cells toward androgen‐independence.4 In addition to the expression of IL‐8 we also observed similar increases in the expression of CXCR‐1 and CXCR‐2 protein in PC‐3M and PC‐3MM2 cells compared with less aggressive LNCaP cells (Fig. 2c), suggesting that CaP cells are subject to continuous autocrine/paracrine IL‐8 stimulation, which was greatest in the most aggressive CaP cells, PC‐MM2. This observation is in agreement with evidence that IL‐8 can bind two receptors, CXCR‐1 and CXCR‐2. Whereas CXCR‐1 only binds IL‐8, CXCR‐2 binds IL‐8 and several other CXC chemokines.51 These receptors play an important role in angiogenesis and tumour progression.52 Our data also confirm recent observations that show that IL‐8 can be an autocrine factor, in CaP angiogenesis, tumorigenicity and metastasis in athymic nude mice.4, 5, 53 In this study we observed that levels of IL‐8 were significantly higher in the sera of CaP patients compared with BPH patients and normal controls, in agreement with other reports.54, 55 We observed that exogenous rhIL‐8 induced cell proliferation of CaP cells (Fig. 3a). Silencing of the IL‐8 gene yielded significant suppression of cell proliferation of both PC‐3M and PC‐3MM2 cells (Fig 3b, c). Lastly, only the simultaneous silencing of both genes for IL‐8 receptors, CXCR‐1 and CXCR‐2, inhibited proliferation of CaP cells; independent silencing of either IL‐8 receptor gene did not have a significant effect on the growth CaP. These data demonstrate that IL‐8 may act as an autocrine or paracrine tumour growth factor and inhibition of IL‐8 production and/or activity can inhibit proliferation of CaP cells. Furthermore, our data show that neutralizing antibodies to IL‐8, CXCR‐1 and CXCR‐2 independently did not inhibit proliferation of PC‐3M and PC‐3MM2 cells (Fig. 3d), indicating that IL‐8 receptors present at locations other than the cell surface may mediate the autocrine proliferative effect of IL‐8. Hence, we conclude that IL‐8 acts upon CaP cells to induce an aggressive, metastatic phenotype. It is likely that secretion of IL‐8 from CaP cells can modulate the activity of other cell types within the tumour microenvironment because it is well known that the pro‐angiogenic activity of IL‐8 acts upon the endothelial compartment of the tumour to induce neovascularization. The chemotactic activity of IL‐8 has a profound effect in recruiting neutrophils and macrophages to the tumour site inducing their tumour‐promoting activity.56 Hence the contribution of IL‐8 to tumour growth and aggressiveness is based on its mitogenic, angiogenic and chemotactic properties as documented in the present study.
The second goal of this study was to see if knock down of IL‐8 gene expression could be a potential therapeutic strategy for controlling CaP growth in vivo. Knockdown of gene expression by specific siRNAs may shortly become a method for treating a variety of diseases.57 Therapeutic RNAs have advantages over other drugs. They are easily synthesized, highly specific and their pharmacokinetics are generally independent of their length and sequence. Because the RNAi pathway is present in all mammalian cells, the primary challenge for effective gene silencing in vivo is delivery of the siRNA to the specific sites. Systemic delivery is required for the widest distribution of RNAi therapeutics. To that end, effort has been focused on the development of siRNA formulations that confer ‘drug‐like’ properties including favourable delivery and uptake following systemic administration. To achieve knockdown of IL‐8 gene expression we used a specific siRNA duplex that was verified experimentally to inhibit the expression of its target gene. Each siRNA strand was designed using the same algorithm and the duplex was confirmed to reduce target gene expression by at least 80% when measured 48 hr post‐transfection. Our use of siRNA for therapy is consistent with other reported pre‐clinical58, 59 and clinical studies.60, 61
Systemic administration of siRNA is essential for therapy. However, problems like protection from nuclease degradation, rapid uptake by mononuclear phagocytic cells, and induction of IFN production must be solved to ensure the success of this approach.62 In most clinical studies delivery of siRNA was achieved by agents used for in vitro transfection, which tend to aggregate. In vitro, aggregation contributes to a higher rate of transfection; however, it is incompatible with in vivo use. For example, Lipofectamine–plasmid DNA complexes aggregate to form relatively large particles > 500 nm in diameter,63 which accumulate in the lungs, the first‐pass organ after intravenous injection.64 To target tumours, aggregation must be avoided. We synthesized a well‐defined, degradable cationic polymer, CPLA‐26, and successfully demonstrated its application as a gene delivery platform that does not aggregate.21, 22 In this study we used CPLA–IL‐8 siRNA nanoplexes for treatment of a xenograft model of CaP (Fig. 1). The negatively charged IL‐8 siRNA is complexed with CPLA by electrostatic interaction (Fig. 1). Some of the positive charges on CPLA are neutralized by IL‐8 siRNA, and the nanoplexes formed not only protect the siRNA but also possess a net positive charge that enhances cellular internalization via endocytosis. When the nanoplex is transported to the cytoplasm, the tertiary amine groups of CPLA on the nanoplex surface enhance the intracellular ‘proton‐sponge’ effect, whereby extra protons are transported and absorbed into the endosome, inducing osmotic swelling and rupture.13, 65 The nanoplexes eventually escape from the endosome and the IL‐8 siRNA is released upon the biodegradation of CPLA.
In this study we synthesized a well‐defined CPLA‐26 with 26 mol% of amine functionalities relative to polymer backbone repeat units and formed nanoplexes with IL‐8 siRNA.21, 22 These nanoplexes were readily taken up by PC‐3 cells and efficiently suppressed the expression of their IL‐8 gene, offering an innovative, new candidate for treatment of CaP satisfying the essential requirements for a safe and efficient nanocarrier for gene delivery.22 For use in clinical applications nanocarriers must be free of cytotoxic activity. CPLA‐26 expressed no cytotoxicity, has a particle diameter < 500 nm (Fig. 5a,b) and exhibited an overall positive charge, as confirmed by its inability to permeate into the agarose gel toward the anode electrode (Fig. 5c). TEM of CPLA‐26 also showing a particle diameter < 500 nm may account for it not being sequestered in the lungs. These data show that the particles form multilamellar vesicles whose aqueous regions include siRNA, substantiating its utility as an RNA delivery agent for the treatment of metastatic CaP.21, 22, 26 Additionally CPLA‐26 also has comparable transfection efficiency (Fig. 5e–g) compared with our series of other synthesized CPLAs.22 CPLA‐26 used in this study meets all the criteria required for its use in vivo for gene therapy.
As angiogenesis is critical for tumour growth, the development of antiangiogenic therapies is becoming an important treatment for various cancers.66 We sought to test the hypothesis that suppression of IL‐8 gene expression could be used to inhibit angiogenesis in aggressive, metastatic prostate cancers. We achieved this goal using CPLA–IL‐8 siRNA nanoplexes that could effectively knock down expression of the IL‐8 gene and inhibit proliferation and invasiveness of CaP cells. In vivo experiments using athymic nude mice bearing human CaP tumours demonstrated that treatment with 50 ng IL‐8 siRNA nanoplexes/g body weight every other day intravenously for 4 weeks significantly inhibited tumour growth compared with untreated control animals (Fig. 6a–c). The IL‐8 siRNA nanoplex treatment concomitantly suppressed gene expression of IL‐8, MMP‐2 and MMP‐9 in situ in resected tumours compared with tumours from untreated control animals (Fig. 6d). Our findings are consistent with another study using intralesional administration of an adenoviral vector containing antisense IL‐8 to treat athymic nude mice implanted with highly metastatic human transitional cell carcinoma cells.67 The investigators observed that tumour growth was significantly inhibited compared with controls. There also was a significant decrease in the expression of IL‐8, MMP9, and microvessel density. Other studies involving antisense IL‐8 transfection have shown similar results mediated through a decrease in MMP expression.68
We corroborated our results using functional molecular imaging of tumours.31 Two such methods, CE‐MRI and CE‐US, were used to characterize therapeutic effects of IL‐8 siRNA nanoplexes on prostate tumour xenografts in vivo. MRI can accurately delineate soft tissues including tumours,69 and is widely used for studying angiogenesis of human tumours.70 CaP is usually well vascularized, consequently targeting angiogenesis for treatment is actively being explored71, 72, 73 using monoclonal antibodies or small molecule inhibitors of tyrosine kinase to treat CaP. Until the present study, no one has used siRNA to silence the IL‐8 gene to inhibit angiogenesis for the treatment of CaP. Using CE‐MRI we confirmed that IL‐8 siRNA nanoplexes could efficiently cause regression of CaP tumours in vivo, which was correlated with significant inhibition of angiogenesis. A reduction in contrast enhancement compared with baseline measurements was seen following nanoplex treatment indicative of a reduction in blood flow (Fig. 7e,f). Although this reduction did not achieve statistical significance the trend was apparent. CE‐MRI is one of the most widely used imaging methods for assessment of angiogenesis in preclinical and clinical studies.70, 74, 75 Our MRI results showed a quantitative reduction in angiogenesis of CaP tumours after treatment with IL‐8 siRNA nanoplexes. Although our MRI studies did not reach statistical significance, CE‐US monitoring of nanoplex therapy of CaP tumours showed a significant reduction in contrast enhancement, a clear indication of treatment response (Fig. 7g,h). Ultrasound imaging offers a relatively inexpensive alternative to advanced imaging techniques such as MRI and computed tomography for non‐invasive imaging of tumour growth and vascular function.76 The use of microbubbles that range from a few microns in size considerably improves the imaging and diagnostic capabilities of US.76 Microbubbles enhance the acoustic signal of blood thereby enabling non‐invasive assessment of perfusion in tissues including tumours. The utility of CE‐US in determining tumour response to antiangiogenic and antivascular therapy has been demonstrated.77, 78 Immunohistochemistry analysis confirmed vascular damage following IL‐8 siRNA nanoplex treatment of tumour‐bearing animals. Quantitative estimates of microvessel density measured by CD31+ antigen levels were significantly lower in IL‐8 siRNA nanoplex‐treated animals compared with scrambled siRNA nanoplex‐treated controls (Fig. 7j,i). CD31 or PECAM‐1 is found in abundance on the surface of endothelial cells and plays a major role between adjacent endothelial cells during angiogenesis.79 Immunohistochemical detection of CD31 has been used to quantify angiogenesis in immunodeficient animals bearing various human tumours by calculation of microvessel density.80, 81, 82 A significantly decreased number of CD31+ cells and vascular damage demonstrated by CE‐MRI are strong evidence of the effectiveness of IL‐8 siRNA nanoplexes in the treatment of animals bearing human CaP tumours. However simultaneous staining of the proliferation marker, ki 67 did not show significant differences between the experimental and control animals. Such a paradoxical observation has been previously observed in lung cancer cells,83 although we believe that decrease in growth rate of our tumours in our study is secondary to the vascular damage induced by the IL‐8 siRNA nanoplex.
Inhibition of angiogenesis is a novel approach for treating solid tumours and currently available angiogenic inhibitors have provided clinical benefit as cancer therapy. However, there is increasing awareness that this treatment is associated with intrinsic refractoriness to the therapeutic agent(s).84 Probing the mechanism(s) of tumour resistance, a few studies suggest that the process is associated with either the tumour cells themselves or with their ability to hijack their microenvironment.84 A recent investigation identified a paracrine signalling network between the adaptive and innate immune systems was associated with resistance to antiangiogenic therapy in multiple tumour models.85 This study showed that IL‐17, a major effector cytokine of T heper type 17 cells, a subtype of adaptive immunity cells, modulated responses to antiangiogenic therapy and initiated tumour refractoriness.85 The authors found increases in circulating secreted factors upregulated by IL‐17 including G‐CSF, IL‐6, VEGF and Bv8, a G‐CSF–inducible angiogenic factor, in mice bearing refractory tumours. These factors are involved in promoting both inflammation and angiogenesis and are currently used as markers of treatment resistance. Since the present study involves an antiangiogenic strategy to treat CaP, we also measured the levels of these factors in the sera of mice treated with IL‐8sIRNA nanoplexes. Levels of these factors associated with resistance to antiangiogenic treatment were significantly lower in mice treated with IL‐8 siRNA nanoplexes than control animals (Fig. 8).
Using naked siRNA in vivo can induce Toll‐like receptor‐mediated IFN‐γ responses.62 This raises concerns about possible adverse effects. While IL‐8 siRNA could significantly induce IFN‐γ and IL‐6 in mice, complexing it with CPLA nanoparticles significantly prevented these potentially harmful side effects. Hence CPLA not only shielded the therapeutic siRNA from rapid degradation in vivo but also prevented adverse reactions to it. In conclusion, CPLA–IL‐8 siRNA nanoplexes administered to mice showed no evidence of toxicity or induction of inflammation. They also exhibited strong antitumour activity in a mouse model of human CaP after local or systemic administration. Our nanoplexes are a promising new tool for the safe and effective delivery of IL‐8 siRNA to CaP tumours to inhibit angiogenesis and concomitant tumour growth. We are eager to undertake the next steps to substantiate that IL‐8 siRNA nanoplexes can be used for the treatment of aggressive and metastatic CaP in humans.
Author contribution
RA and SAS were involved in the overall design of the study, RA and BN performed the experiments and analysis, RA wrote the manuscript and SAS edited it. SM, JL and DS and KC provided critical reagents, samples and input in the manuscript. CK, HZ, HS and CC were involved in the design and synthesis of nanoparticles. ST, KB and MS contributed to the imaging work and data interpretation, and reviewed the manuscript.
Disclosure
There are no conflicts of interest.
Supporting information
Figure S1. Basal expression of β‐actin, interleukin‐8 (IL‐8), CXCR‐1 and CXCR‐2 in normal prostate (NP) epithelial cells and prostate cancer (CaP) cells of varying aggressive phenotypes as quantitated by Immunofluorescence.
Figure S2. Quantitative (QT) real‐time(RT) PCR analysis of interleukin‐8 (IL‐8) (a), CXCR‐1 (b) and CXCR‐2 (c) in normal prostate (NP), and prostate cancer (CaP) cell lines of low (LNCaP), high (PC‐3) and highest (PC‐3M) metastatic potential, respectively.
Figure S3. Recombinant human interleukin‐8 (rhIL‐8) induces prostate cancer (CaP) cell proliferation which is inhibited by silencing the gene for IL‐8.
Figure S4. Inhibition of cell proliferation by silencing the genes of interleukin‐8 (IL‐8) receptors in normal prostate (NP) and prostate cancer (CaP) cells LNCaP, DU‐145 of mild and moderate aggressive potential.
Figure S5. Quantification of interleukin‐8 (IL‐8) in culture supernatant of normal prostate (NP) and prostate cancer (CaP) cells of varying aggressive potential by ELISA.
Figure S6. Representative images of internalization of CPLA‐26–IL‐8 siRNAFAM by normal prostate (NP) cells.
Figure S7. Histopathological assessment of PC‐3 xenografts response to interleukin‐8 (IL‐8) small interfering RNA nanoplex treatment.
Acknowledgements
The work was supported by grants from the National institute of Health grants (R21 CA 167177), the National Science Foundation (CBET‐1133737; DMR‐1206715) and the Margret Duffy and Robert Cameron Troup Memorial Fund for Cancer Research of the Kaleida Health Foundation.
References
- 1. Simard J, Dumont M, Soucy P, Labrie F. Perspective: prostate cancer susceptibility genes. Endocrinology 2002; 143:2029–40. [DOI] [PubMed] [Google Scholar]
- 2. Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem 2004; 91:718–29. [DOI] [PubMed] [Google Scholar]
- 3. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discovery 2007; 6:273–86. [DOI] [PubMed] [Google Scholar]
- 4. Inoue K, Slaton JW, Eve BY, Kim SJ, Perrotte P, Balbay MD et al Interleukin 8 expression regulates tumorigenicity and metastases in androgen‐independent prostate cancer. Clin Cancer Res 2000; 6:2104–19. [PubMed] [Google Scholar]
- 5. Kim SJ, Uehara H, Karashima T, McCarty M, Shih N, Fidler IJ. Expression of interleukin‐8 correlates with angiogenesis, tumorigenicity, and metastasis of human prostate cancer cells implanted orthotopically in nude mice. Neoplasia 2001; 3:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang S, Mills L, Mian B, Tellez C, McCarty M, Yang XD et al Fully humanized neutralizing antibodies to interleukin‐8 (ABX‐IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am J Pathol 2002; 161:125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mian BM, Dinney CP, Bermejo CE, Sweeney P, Tellez C, Yang XD et al Fully human anti‐interleukin 8 antibody inhibits tumor growth in orthotopic bladder cancer xenografts via down‐regulation of matrix metalloproteases and nuclear factor‐κB. Clin Cancer Res 2003; 9:3167–75. [PubMed] [Google Scholar]
- 8. Mills L, Tellez C, Huang S, Baker C, McCarty M, Green L et al Fully human antibodies to MCAM/MUC18 inhibit tumor growth and metastasis of human melanoma. Cancer Res 2002; 62:5106–14. [PubMed] [Google Scholar]
- 9. Kupferschmidt K. A lethal dose of RNA. Science 2013; 341:732–3. [DOI] [PubMed] [Google Scholar]
- 10. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater 2013; 12:967–77. [DOI] [PubMed] [Google Scholar]
- 11. Li CX, Parker A, Menocal E, Xiang S, Borodyansky L, Fruehauf JH. Delivery of RNA interference. Cell Cycle 2006; 5:2103–9. [DOI] [PubMed] [Google Scholar]
- 12. During MJ. Adeno‐associated virus as a gene delivery system. Adv Drug Deliv Rev 1997; 27:83–94. [DOI] [PubMed] [Google Scholar]
- 13. Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discovery 2005; 4:581–93. [DOI] [PubMed] [Google Scholar]
- 14. Musacchio T, Navarro G, Torchilin VP. Molecular assemblies for siRNA delivery. J Drug Deliv Sci Technol 2012; 22:5–16. [Google Scholar]
- 15. Li W, Szoka FC Jr. Lipid‐based nanoparticles for nucleic acid delivery. Pharm Res 2007; 24:438–49. [DOI] [PubMed] [Google Scholar]
- 16. Jeong JH, Kim SW, Park TG. Molecular design of functional polymers for gene therapy. Prog Polym Sci 2007; 32:1239–74. [Google Scholar]
- 17. Luten J, van Nostrum CF, De Smedt SC, Hennink WE. Biodegradable polymers as non‐viral carriers for plasmid DNA delivery. J Control Release 2008; 126:97–110. [DOI] [PubMed] [Google Scholar]
- 18. Bonoiu AC, Mahajan SD, Ding H, Roy I, Yong KT, Kumar R et al Nanotechnology approach for drug addiction therapy: gene silencing using delivery of gold nanorod‐siRNA nanoplex in dopaminergic neurons. Proc Natl Acad Sci USA 2009; 106:5546–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oh YK, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Deliv Rev 2009; 61:850–62. [DOI] [PubMed] [Google Scholar]
- 20. Gabrielson NP, Lu H, Yin L, Li D, Wang F, Cheng J. Reactive and bioactive cationic α‐helical polypeptide template for nonviral gene delivery. Angew Chem 2012; 51:1143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen CK, Law WC, Aalinkeel R, Yu Y, Nair B, Wu JC et al Biodegradable cationic polymeric nanocapsules for overcoming multidrug resistance and enabling drug‐gene co‐delivery to cancer cells. Nanoscale 2014; 6:1567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen CK, Law WC, Aalinkeel R, Nair B, Kopwitthaya A, Mahajan SD et al Well‐defined degradable cationic polylactide as nanocarrier for the delivery of siRNA to silence angiogenesis in prostate cancer. Adv Healthc Mater 2012; 1:751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waugh DJ, Wilson C. The interleukin‐8 pathway in cancer. Clin Cancer Res 2008; 14:6735–41. [DOI] [PubMed] [Google Scholar]
- 24. Aalinkeel R, Nair MP, Sufrin G, Mahajan SD, Chadha KC, Chawda RP et al Gene expression of angiogenic factors correlates with metastatic potential of prostate cancer cells. Cancer Res 2004; 64:5311–21. [DOI] [PubMed] [Google Scholar]
- 25. Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL et al Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 1988; 48:589–601. [PubMed] [Google Scholar]
- 26. Jones CH, Chen CK, Jiang M, Fang L, Cheng C, Pfeifer BA. Synthesis of cationic polylactides with tunable charge densities as nanocarriers for effective gene delivery. Mol Pharm 2013; 10:1138–45. [DOI] [PubMed] [Google Scholar]
- 27. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Anal Biochem 1987; 162:156–9. [DOI] [PubMed] [Google Scholar]
- 28. Shively L, Chang L, LeBon JM, Liu Q, Riggs AD, Singer‐Sam J. Real‐time PCR assay for quantitative mismatch detection. Biotechniques 2003; 34:498–502, 4. [DOI] [PubMed] [Google Scholar]
- 29. Mahajan SD, Aalinkeel R, Schwartz SA, Chawda RP, Nair MP. Effector cell mediated cytotoxicity measured by intracellular Granzyme B release in HIV infected subjects. Biol Proced Online 2003; 5:182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bindukumar B, Schwartz SA, Nair MP, Aalinkeel R, Kawinski E, Chadha KC. Prostate‐specific antigen modulates the expression of genes involved in prostate tumor growth. Neoplasia 2005; 7:544. [PMC free article] [PubMed] [Google Scholar]
- 31. Seshadri M, Sacadura NT, Coulthard T. Monitoring antivascular therapy in head and neck cancer xenografts using contrast‐enhanced MR and US imaging. Angiogenesis 2011; 14:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seshadri M, Bellnier DA, Cheney RT. Assessment of the early effects of 5,6‐dimethylxanthenone‐4‐acetic acid using macromolecular contrast media‐enhanced magnetic resonance imaging: ectopic versus orthotopic tumors. Int J Radiat Oncol Biol Phys 2008; 72:1198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seshadri M, Mazurchuk R, Spernyak JA, Bhattacharya A, Rustum YM, Bellnier DA. Activity of the vascular‐disrupting agent 5,6‐dimethylxanthenone‐4‐acetic acid against human head and neck carcinoma xenografts. Neoplasia 2006; 8:534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Folaron M, Kalmuk J, Lockwood J, Frangou C, Vokes J, Turowski SG et al Vascular priming enhances chemotherapeutic efficacy against head and neck cancer. Oral Oncol 2013; 49:893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu H, Zou Y, Wang Y, Huang X, Huang G, Sumer BD et al Overcoming endosomal barrier by amphotericin B‐loaded dual pH‐responsive PDMA‐b‐PDPA micelleplexes for siRNA delivery. ACS Nano 2011; 5:9246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ. Applications of orthogonal “click” chemistries in the synthesis of functional soft materials. Chem Rev 2009; 109:5620–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nederberg F, Connor EF, Moller M, Glauser T, Hedrick JL. New paradigms for organic catalysts: the first organocatalytic living polymerization. Angew Chem 2001; 40:2712–5. [DOI] [PubMed] [Google Scholar]
- 38. Hoyle CE, Lee TY, Roper T. Thiol‐enes: chemistry of the past with promise for the future. J Polym Sci A Polym Chem 2004; 42:5301–38. [Google Scholar]
- 39. Wang Y, Zheng M, Meng F, Zhang J, Peng R, Zhong Z. Branched polyethylenimine derivatives with reductively cleavable periphery for safe and efficient in vitro gene transfer. Biomacromolecules 2011; 12:1032–40. [DOI] [PubMed] [Google Scholar]
- 40. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T et al EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration‐resistant prostate cancer. Eur Urol 2014; 65:467–79. [DOI] [PubMed] [Google Scholar]
- 41. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K et al Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012; 367:1187–97. [DOI] [PubMed] [Google Scholar]
- 42. Lehrer S, Diamond EJ, Mamkine B, Stone NN, Stock RG. Serum interleukin‐8 is elevated in men with prostate cancer and bone metastases. Technol Cancer Res Treat 2004; 3:411. [DOI] [PubMed] [Google Scholar]
- 43. Chadha KC, Miller A, Nair BB, Schwartz SA, Trump DL, Underwood W. New serum biomarkers for prostate cancer diagnosis. Clin Cancer Investig J 2014; 3:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kettritz R, Gaido ML, Haller H, Luft FC, Jennette CJ, Falk RJ. Interleukin‐8 delays spontaneous and tumor necrosis factor‐α‐mediated apoptosis of human neutrophils. Kidney Int 1998; 53:84–91. [DOI] [PubMed] [Google Scholar]
- 45. Ren Y, Poon RT, Tsui HT, Chen WH, Li Z, Lau C et al Interleukin‐8 serum levels in patients with hepatocellular carcinoma: correlations with clinicopathological features and prognosis. Clin Cancer Res 2003; 9(16 Pt 1):5996–6001. [PubMed] [Google Scholar]
- 46. Haraguchi M, Komuta K, Akashi A, Matsuzaki S, Furui J, Kanematsu T. Elevated IL‐8 levels in the drainage vein of resectable Dukes’ C colorectal cancer indicate high risk for developing hepatic metastasis. Oncol Rep 2002; 9:159–65. [PubMed] [Google Scholar]
- 47. Fujimoto J, Aoki I, Khatun S, Toyoki H, Tamaya T. Clinical implications of expression of interleukin‐8 related to myometrial invasion with angiogenesis in uterine endometrial cancers. Ann Oncol 2002; 13:430–4. [DOI] [PubMed] [Google Scholar]
- 48. Wang JM, Deng X, Gong W, Su S. Chemokines and their role in tumor growth and metastasis. J Immunol Methods 1998; 220:1–17. [DOI] [PubMed] [Google Scholar]
- 49. Green AR, Green VL, White MC, Speirs V. Expression of cytokine messenger RNA in normal and neoplastic human breast tissue: identification of interleukin‐8 as a potential regulatory factor in breast tumours. Int J Cancer 1997; 72:937–41. [DOI] [PubMed] [Google Scholar]
- 50. De Larco JE, Wuertz BR, Yee D, Rickert BL, Furcht LT. Atypical methylation of the interleukin‐8 gene correlates strongly with the metastatic potential of breast carcinoma cells. Proc Natl Acad Sci USA 2003; 100:13988–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Holmes WE, Lee J, Kuang WJ, Rice GC, Wood WI. Structure and functional expression of a human interleukin‐8 receptor. Science 1991; 253:1278–80. [DOI] [PubMed] [Google Scholar]
- 52. Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol 2000; 18:217–42. [DOI] [PubMed] [Google Scholar]
- 53. Singh RK, Varney ML. IL‐8 expression in malignant melanoma: implications in growth and metastasis. Histol Histopathol 2000; 15:843–9. [DOI] [PubMed] [Google Scholar]
- 54. Veltri RW, Miller MC, Zhao G, Ng A, Marley GM, Wright GL Jr et al Interleukin‐8 serum levels in patients with benign prostatic hyperplasia and prostate cancer. Urology 1999; 53:139–47. [DOI] [PubMed] [Google Scholar]
- 55. Pfitzenmaier J, Vessella R, Higano CS, Noteboom JL, Wallace D Jr, Corey E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer 2003; 97:1211–6. [DOI] [PubMed] [Google Scholar]
- 56. De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin‐8. Clin Cancer Res 2004; 10:4895–900. [DOI] [PubMed] [Google Scholar]
- 57. Tseng YC, Mozumdar S, Huang L. Lipid‐based systemic delivery of siRNA. Adv Drug Deliv Rev 2009; 61:721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL et al Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci USA 2007; 104:12982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hu‐Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence‐specific knockdown of EWS‐FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res 2005; 65:8984–92. [DOI] [PubMed] [Google Scholar]
- 60. Shen J, Samul R, Silva RL, Akiyama H, Liu H, Saishin Y et al Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther 2006; 13:225–34. [DOI] [PubMed] [Google Scholar]
- 61. Lu PY, Xie FY, Woodle MC. Modulation of angiogenesis with siRNA inhibitors for novel therapeutics. Trends Mol Med 2005; 11:104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hornung V, Guenthner‐Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S et al Sequence‐specific potent induction of IFN‐α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 2005; 11:263–70. [DOI] [PubMed] [Google Scholar]
- 63. Narang AS, Thoma L, Miller DD, Mahato RI. Cationic lipids with increased DNA binding affinity for nonviral gene transfer in dividing and nondividing cells. Bioconjug Chem 2005; 16:156–68. [DOI] [PubMed] [Google Scholar]
- 64. Li S, Rizzo MA, Bhattacharya S, Huang L. Characterization of cationic lipid–protamine–DNA (LPD) complexes for intravenous gene delivery. Gene Ther 1998; 5:930–7. [DOI] [PubMed] [Google Scholar]
- 65. Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B et al A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 1995; 92:7297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist 2015; 20:660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Inoue K, Wood CG, Slaton JW, Karashima T, Sweeney P, Dinney CP. Adenoviral‐mediated gene therapy of human bladder cancer with antisense interleukin‐8. Oncol Rep 2001; 8:955–64. [DOI] [PubMed] [Google Scholar]
- 68. Bar‐Eli M. Role of interleukin‐8 in tumor growth and metastasis of human melanoma. Pathobiology 1999; 67:12–8. [DOI] [PubMed] [Google Scholar]
- 69. Yanagi Y, Asaumi J, Unetsubo T, Ashida M, Takenobu T, Hisatomi M et al Usefulness of MRI and dynamic contrast‐enhanced MRI for differential diagnosis of simple bone cysts from true cysts in the jaw. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010; 110:364–9. [DOI] [PubMed] [Google Scholar]
- 70. Unetsubo T, Konouchi H, Yanagi Y, Murakami J, Fujii M, Matsuzaki H et al Dynamic contrast‐enhanced magnetic resonance imaging for estimating tumor proliferation and microvessel density of oral squamous cell carcinomas. Oral Oncol 2009; 45:621–6. [DOI] [PubMed] [Google Scholar]
- 71. Mukherji D, Temraz S, Wehbe D, Shamseddine A. Angiogenesis and anti‐angiogenic therapy in prostate cancer. Crit Rev Oncol Hematol 2013; 87:122–31. [DOI] [PubMed] [Google Scholar]
- 72. Kluetz PG, Figg WD, Dahut WL. Angiogenesis inhibitors in the treatment of prostate cancer. Expert Opin Pharmacother 2010; 11:233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Figg WD, Dahut W, Duray P, Hamilton M, Tompkins A, Steinberg SM et al A randomized phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgen‐independent prostate cancer. Clin Cancer Res 2001; 7:1888–93. [PubMed] [Google Scholar]
- 74. Hayes C, Padhani AR, Leach MO. Assessing changes in tumour vascular function using dynamic contrast‐enhanced magnetic resonance imaging. NMR Biomed 2002; 15:154–63. [DOI] [PubMed] [Google Scholar]
- 75. Farace P, Merigo F, Fiorini S, Nicolato E, Tambalo S, Daducci A et al DCE‐MRI using small‐molecular and albumin‐binding contrast agents in experimental carcinomas with different stromal content. Eur J Radiol 2011; 78:52–9. [DOI] [PubMed] [Google Scholar]
- 76. Sullivan JC, Wang B, Boesen EI, D'Angelo G, Pollock JS, Pollock DM. Novel use of ultrasound to examine regional blood flow in the mouse kidney. Am J Physiol Renal Physiol 2009; 297:F228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. O'Connor JP, Carano RA, Clamp AR, Ross J, Ho CC, Jackson A et al Quantifying antivascular effects of monoclonal antibodies to vascular endothelial growth factor: insights from imaging. Clin Cancer Res 2009; 15:6674–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Goertz DE, Yu JL, Kerbel RS, Burns PN, Foster FS. High‐frequency Doppler ultrasound monitors the effects of antivascular therapy on tumor blood flow. Cancer Res 2002; 62:6371–5. [PubMed] [Google Scholar]
- 79. Muller AM, Hermanns MI, Skrzynski C, Nesslinger M, Muller KM, Kirkpatrick CJ. Expression of the endothelial markers PECAM‐1, vWf, and CD34 in vivo and in vitro . Exp Mol Pathol 2002; 72:221–9. [DOI] [PubMed] [Google Scholar]
- 80. Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat 1995; 36:169–80. [DOI] [PubMed] [Google Scholar]
- 81. Norrby K, Ridell B. Tumour‐type‐specific capillary endothelial cell stainability in malignant B‐cell lymphomas using antibodies against CD31, CD34 and Factor VIII. APMIS 2003; 111:483–9. [DOI] [PubMed] [Google Scholar]
- 82. Ushijima C, Tsukamoto S, Yamazaki K, Yoshino I, Sugio K, Sugimachi K. High vascularity in the peripheral region of non‐small cell lung cancer tissue is associated with tumor progression. Lung Cancer 2001; 34:233–41. [DOI] [PubMed] [Google Scholar]
- 83. Mattern J, Volm M. Imbalance of cell proliferation and apoptosis during progression of lung carcinomas. Anticancer Res 2004; 24:4243–6. [PubMed] [Google Scholar]
- 84. Loges S, Schmidt T, Carmeliet P. Mechanisms of resistance to anti‐angiogenic therapy and development of third‐generation anti‐angiogenic drug candidates. Genes Cancer 2010; 1:12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J et al An interleukin‐17‐mediated paracrine network promotes tumor resistance to anti‐angiogenic therapy. Nat Med 2013; 19:1114–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Basal expression of β‐actin, interleukin‐8 (IL‐8), CXCR‐1 and CXCR‐2 in normal prostate (NP) epithelial cells and prostate cancer (CaP) cells of varying aggressive phenotypes as quantitated by Immunofluorescence.
Figure S2. Quantitative (QT) real‐time(RT) PCR analysis of interleukin‐8 (IL‐8) (a), CXCR‐1 (b) and CXCR‐2 (c) in normal prostate (NP), and prostate cancer (CaP) cell lines of low (LNCaP), high (PC‐3) and highest (PC‐3M) metastatic potential, respectively.
Figure S3. Recombinant human interleukin‐8 (rhIL‐8) induces prostate cancer (CaP) cell proliferation which is inhibited by silencing the gene for IL‐8.
Figure S4. Inhibition of cell proliferation by silencing the genes of interleukin‐8 (IL‐8) receptors in normal prostate (NP) and prostate cancer (CaP) cells LNCaP, DU‐145 of mild and moderate aggressive potential.
Figure S5. Quantification of interleukin‐8 (IL‐8) in culture supernatant of normal prostate (NP) and prostate cancer (CaP) cells of varying aggressive potential by ELISA.
Figure S6. Representative images of internalization of CPLA‐26–IL‐8 siRNAFAM by normal prostate (NP) cells.
Figure S7. Histopathological assessment of PC‐3 xenografts response to interleukin‐8 (IL‐8) small interfering RNA nanoplex treatment.