

Summary
Multiple sclerosis (MS) is an incurable central nervous system autoimmune disease. Understanding MS pathogenesis is essential for the development of new MS therapies. In the present study, we identified a novel microRNA (miR) that regulates experimental autoimmune encephalomyelitis (EAE), an animal model of MS. Expression of miR223 was up‐regulated specifically in spinal cords and lymphoid organs but not in other examined tissues. A global miR223 knockout (miR223−/−) in mice led to a significant delay in EAE onset, reduction in spinal cord lesion, and lessening of neurological symptoms. These protective effects could be reproduced in bone marrow chimeras reconstituted with miR223−/− haematopoietic stem cells. We also found that miR223 deficiency reduced T helper type 1 (Th1) and Th17 infiltration into spinal cords. To address underlying mechanisms, we investigated the role of miR223 in regulating the function, development and interaction of the major immune cells. Expression of the genes associated with dendritic cell (DC) activation (CD86 and MHC II) and Th1 and Th17 differentiation [interleukin‐12 (IL‐12) and IL‐23, respectively] was significantly decreased in the spleens of miR223−/− mice bearing EAE. The miR223−/− DCs expressed significantly lower levels of basal and lipopolysaccharide‐induced IL‐12 and IL‐23 compared with the wild‐type DCs. These data are consistent with the observed lower efficiency of miR223−/− DCs to support Th1 and Th17 differentiation from naive T cells over‐expressing an EAE antigen‐specific T‐cell receptor. Our data suggest that miR223 promotes EAE, probably through enhancing DC activation and subsequently the differentiation of naive T cells toward Th1 and Th17 effector cells.
Keywords: autoinflammatory disease, experimental autoimmune encephalomyelitis/multiple sclerosis, neuroimmunology, T cells
Introduction
Multiple sclerosis (MS) is a central nervous system (CNS) autoimmune disease characterized by chronic inflammation, demyelination and axonal loss in the CNS. Despite vigorous efforts made in the past several decades, the mechanism underlying the pathogenesis of MS is still not well defined and could be better understood through identification of new genes modulating MS. In this regard, data from animal studies suggest that microRNAs (miRs), a class of non‐coding RNAs that modulate gene expression at the post‐transcriptional level, can either negatively or positively regulate this disease. Using mouse experimental autoimmune encephalomyelitis (EAE) as an animal model of MS, several miRs have been shown to enhance autoimmune inflammation in the CNS. For instance, a global ablation of miR155 in mice led to resistance to EAE.1, 2 In this study, miR155 deficiency markedly decreased the number of both T helper type 1 (Th1) and Th17 cells.1, 2 miR326 enhances EAE as delivery of miR326 locked nucleic acid to mice enhanced Th17 cell differentiation and aggravated EAE, whereas administration of miR326 antisense oligonucleotide produced beneficial effects.3 miR155 has been reported to promote EAE and regulate DC and T‐cell responses through repression of Arginase‐2 and Ets‐1.4, 5. More recently, it is reported that miR21 deficiency in mice or treatment with miR21 antisense oligonucleotide led to suppression of EAE.6 In contrast to these negative miRs, miR124 was shown to be neuroprotective, as administration of miR124 led to deactivation of microglia, reduced activation of myelin‐specific T cells, and suppression of the disease.7 The above data therefore convincingly demonstrate a critical role of certain miRs in modulating immune cells and EAE pathogenesis. However, to our knowledge, the effect of an EAE‐modulating miR on DCs and their interaction with T cells has not been studied.
In this study, our goals were to identify novel miR players in EAE as well as to determine the role of such miRs in orchestrating activation and differentiation of various immune cell subsets. Accordingly, among the miRs investigated, we found that miR223 showed dramatic change in expression. miR223 was initially identified as an miR that is preferentially expressed in haematopoietic tissues.8, 9, 10 miR223−/− mice showed an increase in the granulocytic compartment due to expansion of granulocyte progenitors.8 This finding was consistent with the observation that miR223−/− mice developed inflammatory lung pathology as a result of enhanced neutrophil maturation and activity, but incompatible with in vitro findings, which showed that miR223 promoted granulocytic differentiation.11, 12 Additionally, in a type II diabetes model, miR223−/− mice fed a high‐fat diet exhibited an increased severity of systemic insulin resistance accompanied by a significant increase in adipose tissue inflammation and inflammatory M1 macrophage infiltration.13 The investigators suggested that miR223 protected against adipose tissue inflammation by inhibiting M1 macrophage polarization. Finally, in a mouse model of stroke, miR223−/− mice were more sensitive to global ischaemia and excitotoxicity‐induced neuronal cell death, allowing the conclusion that miR223 is neuroprotective.14 Hence, the function of miR223 appears complex.
The complexity of the reported data suggests an incomplete understanding of the physiological function of miR223. We therefore performed a detailed investigation using miR223‐deficient mice as well as bone marrow chimeras that had miR223 deficiency in haematopoietic cells. We now provide compelling evidence that miR223 supports CNS autoimmune inflammation. Our data further suggest that the destructive role of miR223 in EAE is likely to be mediated by tipping immune cell differentiation towards pathogenic Th1 and Th17 cells.
Materials and methods
Mice and EAE induction
C57BL/6, miR223−/− (B6.Cg‐Ptprca Mir223tm1Fcam/J) and miR223+/+(B6.SJL‐Ptprca Pepcb/BoyJ) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed under pathogen‐free conditions. All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at the J.L. Pettis Memorial VA Medical Center and Loma Linda University. EAE was induced in 8‐ to 10‐week‐old female mice as previously described.15 The mice were monitored daily for clinical symptoms of disease, and disease severity was scored regularly according to previously described criteria.15 To determine motor function, the hanging wire grip test was performed. Mice were placed on top of a wire cage lid that was shaken gently three times, causing them to grip the wire. The wire cage lid was then inverted at a height of 20 cm above the cage floor to prevent the animal from easily climbing down. Latency to fall was recorded three times and the cut‐off time was set between 30 and 120 seconds.
Bone marrow transplantation
Bone marrow transplantation was performed as previously described.16 Five‐week‐old female miR223−/− mice or age‐ and gender‐matched wild‐type mice were used as donor mice and female C57BL/6 mice at the same age were used as recipients. Engraftment was assessed by analysis of miR223 level in peripheral blood mononuclear cells 4 weeks post‐bone marrow transplantation.
T‐cell isolation and polarization assay
Naive T cells were isolated from the spleen single‐cell suspension by CD4 and CD62L positive selection using magnetic bead methods according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA). Purified naive T cells were cultured in RPMI‐1640 medium (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 U/ml streptomycin. For Th0 polarization (T‐cell activation), naive T cells were cultured in complete RPMI‐1640 medium, plate‐bound CD3 antibodies and soluble CD28 antibodies (2 μg/ml). For effector T‐cell polarization, naive T cells were cultured under conditions described for Th0, with the addition of the following factors. Th1 cell polarization: interleukin‐12 (IL‐12; 20 ng/ml) and IL‐4 antibodies (20 μg/ml) (Biolegend, San Diego, CA). Th2 polarization: recombinant murine IL‐4 (50 ng/ml), anti‐IL‐12 (10 μg/ml) and anti‐interferon‐γ (IFN‐γ) (10 µg/ml). Th17 polarization: IL‐6 (20 ng/ml), IL‐23 (20 ng/ml) and transforming growth factor‐β (3 ng/ml) (Biolegend). Regulatory T‐cell polarization: transforming growth factor‐β1 (20 ng/ml), IL‐2 (20 ng/ml) and all trans‐retinoic acid (1 nm). Cells were harvested 4 days later for analysis by FACS and/or RNA isolation.
Dendritic cell isolation, activation and co‐culture with naive T cells
Dendritic cells (DCs) were derived from bone marrow cells as described elsewhere.17 Briefly, bone marrow cells depleted of red blood cells were cultured in the RPMI‐1640 medium containing granulocyte–macrophage colony‐stimulating factor (50 ng/ml) and IL‐4 (20 ng/ml) for 6 days. Half of the medium was replaced with fresh medium every 2 days. On day 6, immature non‐adherent DCs were collected and purified further with CD11c‐antibody‐conjugated magnetic beads (Miltenyi Biotec). To induce maturation, the DCs were incubated with 100 ng/ml of lipopolysaccharide (LPS) for 24 hr. For non‐specific DC–T‐cell interaction assays, LPS‐stimulated DCs or naive T cells derived from C57BL/6 mice were co‐cultured at a ratio of 1 : 5. After 5 days, cells were subjected to FACS or real‐time quantitative PCR (qPCR) analysis. For myelin oligodendrocyte glycoprotein (MOG)‐specific assays, naive T cells were isolated from the spleens of 2D2 mice that over‐express a MOG‐specific T‐cell receptor. The purified 2D2 T cells were co‐cultured with LPS‐activated DCs for 5 days in the presence of 2·5 µg/ml of MOG before FACS analysis.
Analysis of mRNA level
RNA extraction, cDNA synthesis and real‐time quantitative PCR were completed as described previously.16 Specific primers for each gene‐of‐interest are given in Table 1. Real‐time quantitative PCR analyses of miRs were carried out using TaqMan miRNA assays (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions. Relative expression was normalized to the ubiquitously expressed U6 transcripts and calculated using the ΔΔCT method.
Table 1.
Sequences of primers used in this study
| Forward | Reverse | |
|---|---|---|
| GAPDH | TGGCAAAGTGGAGATTGTTGCC | AAGATGGTGATGGGCTTCCCG |
| CD11c | CTGGATAGCCTTTCTTCTGCTG | GCACACTGTGTCCGAACTCA |
| CD32 | AATCCTGCCGTTCCTACTGATC | GTGTCACCGTGTCTTCCTTGAG |
| CD86 | TTGTGTGTGTTCTGGAAACGGAG | AACTTAGAGGCTGTGTTGCTGGG |
| IFN‐γ | GATGCATTCATGAGTATTGCCAAGT | GTGGACCACTCGGATGAGCTC |
| IL‐1β | TTGACGGACCCCAAAAGAT | GAAGCTGGATGCTCTCATCAG |
| IL‐4 | GGTCTCAACCCCCAGCTAGT | GCCGATGATCTCTCTCAAGTGAT |
| IL‐5 | TGCCTGGAGCAGCTGGAT | GTGGCTGGCTCTCATTCACA |
| IL‐6 | AGGATACCACTCCCAACAGAC | CAAGTGCATCATCGTTGTTCA |
| IL‐12 | AAATGAAGCTCTGCATCCTGC | TCACCCTGTTGATGGTCACG |
| IL‐17a | CTCCAGAAGGCCCTCAGACTAC | GGGTCTTCATTGCGGTGG |
| IL‐23 | TGGCATCGAGAAACTGTGAGA | TCAGTTCGTATTGGTAGTCCTGTTA |
| iNOS | CCCTTCAATGGTTGGTACATGG | ACATTGATCTCCGTGACAGCC |
| MHC Class II | GAGGCTCAACTTGTCCCAAAAC | GCAGCCGTGAACTTGTTGAAC |
| TNF‐α | GAACTCCAGGCGGTGCCTAT | TCGGCTGGCACCACTAGTTG |
Flow cytometry
Mice were perfused with PBS before the dissection of spinal cords and spleens. Spleens and lymph nodes were gently pressed through cell strainers to produce single‐cell suspensions as previously described.15 Isolated cells were stained with antibodies that recognize CD3, CD4, CD11c, CD86, IFN‐γ, IL‐17a and MHC class II (eBioscience, San Diego, CA). For intracellular cytokine staining, cells were pretreated with Brefeldin A for 4 hr at 37°, fixed and permeabilized in Perm‐Fix solution (eBioscience) for 1 hr at 4°, washed twice in Perm‐Wash buffer (eBioscience) and stained with the appropriate monoclonal antibody for 2 hr. Isotype primary conjugated antibodies served as a negative control. All analyses were performed following gating of viable cells.
Histological analysis and immunostaining
The spinal cords from EAE mice were prepared as described previously.15 Eight‐micrometre‐thick frozen sections were stained with haematoxylin & eosin (Sigma‐Aldrich, St Louis, MO) and luxol fast blue (IHC‐WORLD, Woodstock, MD). Slides were blocked in 1% goat and donkey serum in PBS for 30 min. Primary antibodies were diluted in incubation buffer (1% BSA, 1% animal serum, 0·3% Triton X‐100 and 0·01% sodium azide in PBS) overnight at 4°. Slides were washed and incubated for 60 min at room temperature with secondary antibodies diluted in incubation buffer. After washing, a drop of DAPI solution (1 μg/μl) was added to slides and incubated for 5 min at room temperature. Slides were rinsed once with PBS and mounted with ProLong Gold antifade medium (Invitrogen). Images were taken on a Nikon fluorescent microscope. The following antibodies were used: anti‐goat IFN‐γ, anti‐rabbit IL‐17, anti‐goat FITC, anti‐rabbit FITC, anti‐rat Texas Red (all from Santa Cruz Biotechnology, Pasa Robles, CA).
Statistical analysis
Results are expressed as mean ± SEM and statistically analysed by Student's t‐test or one‐way analysis of variance. A value of P < 0·05 was considered statistically significant.
Results
miR223 is elevated specifically in CNS and peripheral immune organs in mice with active EAE
To identify novel miRNAs that are involved in EAE, we compared the expression levels of eight relevant miRs in EAE‐bearing mice with those in healthy mice. Consistent with previous reports,1, 2, 7 miR155 expression was increased in EAE spinal cords compared with control spinal cords (~10‐fold) (Fig. 1a). On the other hand, expression of the neuroprotective miR124 in spinal cords was decreased by ~50% during EAE (Fig. 1b). In contrast to the moderate changes of these two miRs, which have previously been shown to modulate EAE,4, 7 miR223 expression was elevated more than 30‐fold in the spinal cords of EAE mice (Fig. 1a). In addition, miR223 expression was significantly increased not only in the spinal cord but also in the peripheral immune system, such as spleen, lymph nodes and bone marrow. In contrast, miR223 level remained unchanged in non‐lymphoid tissues including muscle and colon (Fig. 1c). Furthermore, we found that miR223 expression in CD4+ and CD11b+ cells isolated from spleens was significantly increased in EAE‐bearing mice compared with healthy control mice (Fig. 1d).
Figure 1.
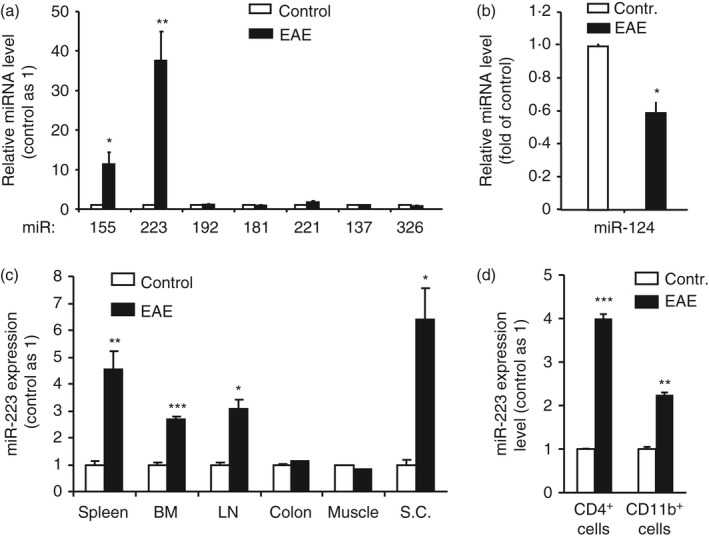
MicroRNA (miR) 223 was significantly up‐regulated in spinal cords and peripheral lymphoid tissues but not in non‐lymphoid tissues in experimental autoimmune encephalomyelitis (EAE)‐bearing mice. At day 14 post‐MOG immunization, mice were killed and tissues were collected for cell or RNA isolation followed by quantification of miRs by real‐time quantitative PCR. (a) Expression of miR223 and miR155 in spinal cords was induced during EAE analysis. (b) miR124 expression was suppressed during EAE. (c) miR223 expression was induced by EAE in central nervous system and peripheral lymphoid tissues. (d) miR223 expression in CD4+ and CD11b+ cells purified from the spleens was increased during EAE. Results are representative of three independent experiments. Data represent the mean ± SEM (n = 4, *P < 0·05, **P < 0·01, ***P < 0·001).
Global miR223 deficiency led to resistance to EAE
To establish a functional link between miR223 and EAE pathogenesis, we first investigated whether the course of EAE was modulated in mice deficient in miR223 globally. Compared with wild‐type (WT) control mice, miR223 knockout (KO) mice exhibited a delayed onset of the paralytic disease and a significant reduction in EAE severity (Fig. 2a). In addition, miR223−/− mice showed stronger motor function, determined by a hanging wire test, at day 13 and 16 following disease induction (Fig. 2b). For example, at day 13, miR223−/− mice were able to remain on the cage cover for several minutes as normal mice did, consistent with a lack of disease symptoms at this time‐point. We further performed histological analysis on spinal cord tissues. The data demonstrated significantly less cellular infiltration in miR223−/−, compared with WT mice, at day 22 (Fig. 2c).
Figure 2.
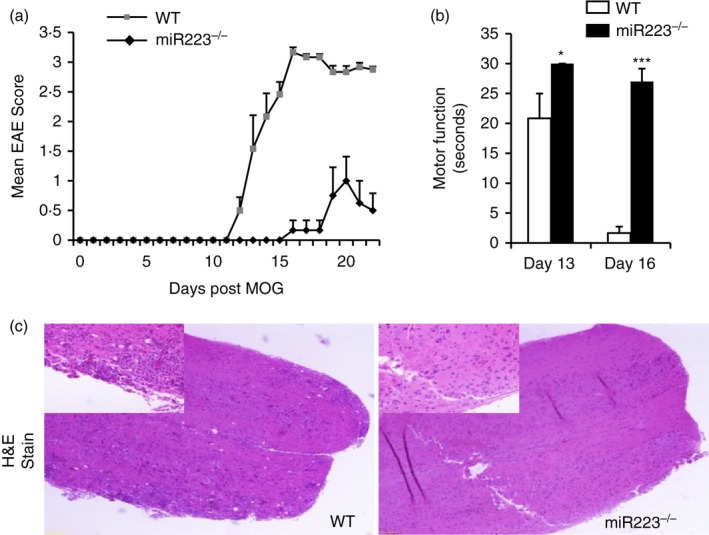
Mice whose microRNA (miR) 223 was globally deficient are resistant to experimental autoimmune encephalomyelitis (EAE) induction. (a) Clinical scores of EAE and (b) motor function (30‐second maximum recorded) were determined at various times after MOG immunization. (c) Histological analysis of spinal cord sections 22 days after MOG immunization. Each panel shows the representative image of three mice analysed per group. Original magnification ×4 (larger) and ×20 (inset). Similar results were obtained in two independent experiments. Data represent the mean ± SEM (*P < 0·05, ***P < 0·001).
miR223 deficiency in haematopoietic stem cells (HSCs) led to resistance to EAE
To determine whether EAE resistance in miR223−/− mice resulted primarily from miR223 deficiency in immune cells or CNS‐resident cells, we generated bone marrow chimeras (BMCs) wherein miR223 deficiency was restricted to haematopoietic stem cell (HSC) ‐derived cell lineages. Hence, lethally irradiated C57BL/6 mice were reconstituted with either miR223+/+ or miR223−/− Sca1+ HSCs. At week 4 post‐HSC infusion, the expression of miR223 was extremely low in the blood cells collected from BMCs reconstituted with miR223−/− HSCs (BMC‐miR223−/−) compared with BMCs reconstituted with WT HSCs (BMC‐WT) (Fig. 3a). These data demonstrated that the HSC‐derived cells in the recipient mice have been successfully replaced with the miR223−/− donor HSCs. Following successful reconstitution, the animals were immunized with MOG35–55 for EAE. Interestingly, BMC‐WT mice began to show neurological symptoms as early as day 7 post‐MOG immunization (Fig. 3b). By day 13, all of the BMC‐WT mice developed EAE. In contrast, none of the BMC‐miR223−/− mice developed disease at this stage (Fig. 3c). Although 50% of BMC‐miR223−/− mice eventually developed EAE at day 19, the peak disease activity was remarkably lower compared with BMC‐WT mice (Fig. 3b). In addition, BMC‐miR223−/− mice compared with BMC‐WT mice displayed a significant improvement in motor function, not only at disease onset but also at the time of disease recovery (Fig. 3d). Furthermore, histopathological analysis at day 25 revealed a reduction in spinal cord lesion and demyelination in the BMC‐miR223−/− mice compared with BMC‐WT mice (Fig. 3e,f).
Figure 3.
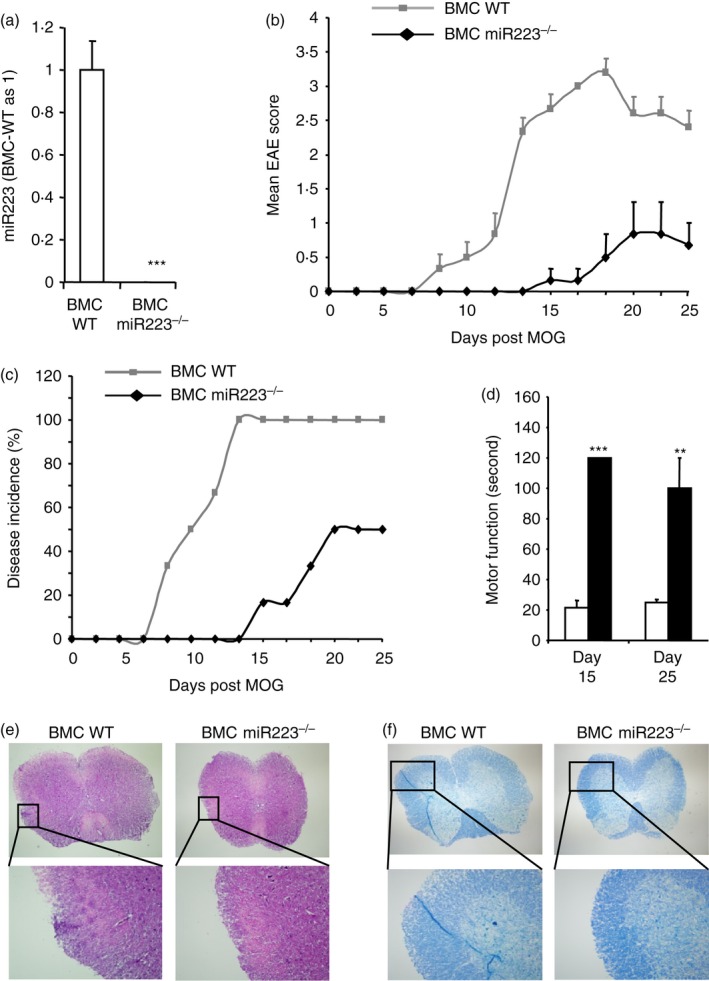
Mice whose microRNA (miR) 223 was deficient in miR223 in haematopoietic stem cells (HSCs) are resistant to experimental autoimmune encephalomyelitis (EAE) induction. C57BL/6J mice were lethally irradiated and reconstituted with miR223+/+ or miR223−/− Sca1+ HSCs. (a) At week 4 post bone marrow transplantation, miR223 level in the peripheral blood cells was determined by real‐time quantitative PCR (mean ± SEM, n = 6). (b, c) At week 5 following bone marrow transplantation, EAE was induced in the wild‐type (WT) and miR223−/− bone marrow chimeras (BMC). Clinical scores (b) and disease incidence (c) were recorded over 25 days (mean ± SEM, n = 6). (d) Motor function was evaluated at day 15 and day 25 after MOG immunization (mean ± SEM, n = 6). (e, f) Spinal cords were isolated at day 25 after MOG immunization. Spinal cord frozen sections were stained with haematoxylin & eosin (e) and luxol fast blue (f) to evaluate the extent of inflammation and demyelination, respectively. Myelin sheath is stained light blue. Each panel shows the representative image of three mice analysed per group. Original magnification ×10 (upper) and ×20 (lower). **P < 0·01, ***P < 0·001.
miR223 deficiency in mice led to suppression of signature cytokines associated with Th1 and Th17 cells
To begin to study the mechanism by which miR223 deficiency led to EAE suppression, we first evaluated the effect of miR223 deficiency on the expression of the IL‐17 and IFN‐γ, the signature cytokines produced by the key pathogenic effector T‐cell subset, Th17a and Th1, respectively. We found that the spinal cord mRNA level of IL‐17a and IFN‐γ was significantly reduced in miR223 global knockout mice compared with that of WT mice (Fig. 4a). It is important to note that the spinal cord expression of these cytokines did not differ significantly between healthy WT and healthy miR223−/− mice that were not subjected to EAE induction (Fig. 4b). Besides IL‐17 and IFN‐γ, expression of other inflammatory cytokines including tumour necrosis factor‐α, IL‐1β and IL‐6 were also remarkably reduced in the spinal cords of miR223‐deficient mice (Fig. S1). Interestingly, miR223 deficiency did not significantly increase the expression of Th2 cytokines IL‐4 and IL‐5 (see Supplementary material, Fig. S1). Consistent with the qPCR measurement of mRNA levels, immmunohistological analysis showed a decrease in the abundance of IL‐17‐expressing and IFN‐γ‐expressing cells in the spinal cord of global miR223 knockout mice compared with wild‐type mice (Fig. 4c–e). Reduced Th1 and Th17 cell infiltration to spinal cords was also observed in bone marrow chimeras that lack miR223 in haematopoietic cells. FACS analysis of the spinal cord single cell suspension revealed a decrease in the percentage of CD4+ IL17+ Th17 cells (WT 8·44%; KO 2·49%, average of two mice) and IFN‐ϒ + Th1 cells (WT 8·2%; KO 5·3%, average of two mice). Together, these data suggest that miR223 regulates the profile of pathogenic effector T cells.
Figure 4.
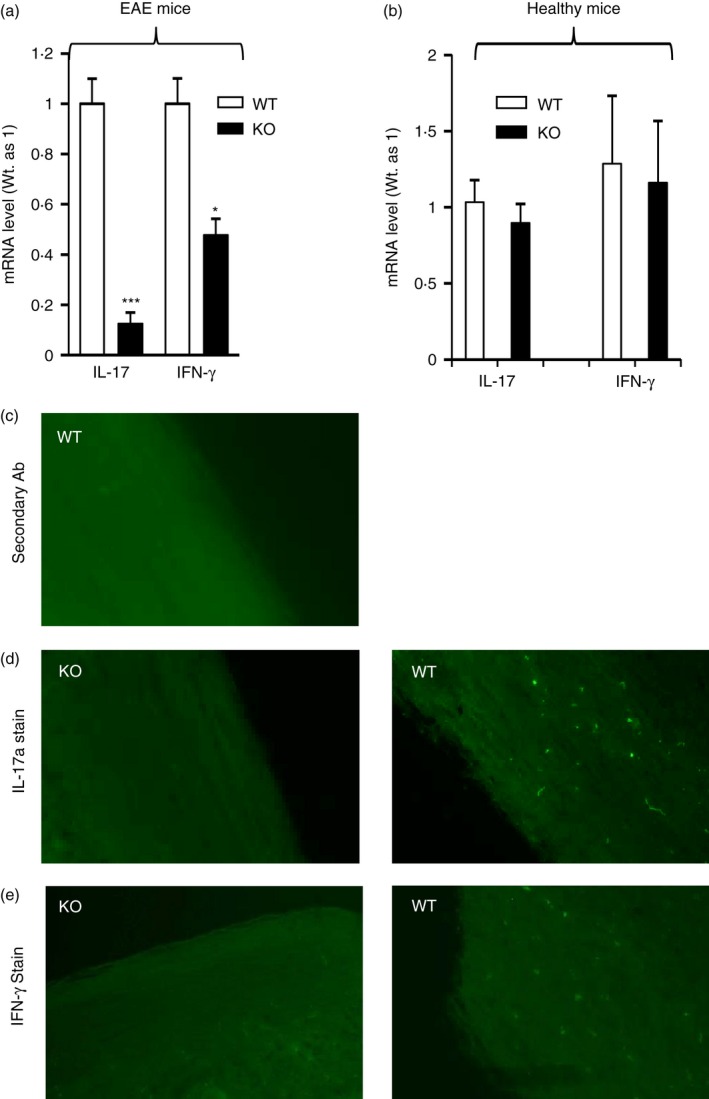
T helper type 1 (Th1) and Th17 differentiation in mice whose microRNA (miR) 223 was deficient globally is impaired following experimental autoimmune encephalomyelitis (EAE) induction. (a) At day 22‐post MOG‐immunization, animals were killed and spinal cords were isolated. RNAs isolated from spinal cords were subjected to quantitative PCR analysis of Th1 cytokine interferon‐γ (IFN‐γ) and Th17 cytokine interleukin‐17a (IL17a) mRNA levels (n = 5). (b) Wild‐type and miR223−/− mice (n = 4) at an age similar to the mice used in (a) but not immunized with MOG were killed and spinal cords were collected for RNA isolation. The mRNA levels of IFN‐γ and IL‐17a mRNA levels were determined by quantitative PCR. (c–e) Spinal cords sections were subjected to immunostaining with secondary antibody only (negative control) (c) or IL‐17a antibody (d) or IFN‐γ antibody (e). *P < 0·05, ***P < 0·001.
miR223 deficiency compromised DC activation and pathogenic T‐cell‐supporting cytokine expression both in vivo and in vitro
Differentiation of naive CD4+ T cells into various T‐cell lineages is dependent upon their interaction with DCs, the major antigen‐presenting cells in the peripheral immune system. This interaction involves the release of specific cytokines by the DCs and a physical contact that occurs between the DCs and the naive CD4+ T cells.18 To determine if miR223 affects the DC activation and maturation, WT and miR223−/− mice were immunized with MOG and their tissues were harvested at an earlier time‐point, 3 days after EAE induction, when DC activation occurs. FACS analysis revealed that abundant CD11c+ MHC II+ DCs were present among spinal cord cells isolated from WT mice with EAE (Fig. 5a). In contrast, CD11c+ MHC II+ DCs were essentially undetectable in the spinal cord cells derived from miR223−/− mice at this early disease stage (Fig. 5a). FACS analysis also indicated that the intensity of CD86 and MHC II staining, reflecting DC maturation/activation, was significantly reduced in the spinal cord of miR223−/− mice compared with that of WT mice (Fig. 5b). In splenocytes, although the abundance of CD11c+ MHC II+ DCs was not reduced in miR223−/− mice (Fig. 5a), the intensity of MHC II and CD86 staining was significantly reduced (Fig. 5c). Consistent with the FACS analysis data, qPCR analysis also revealed a significant reduction in the mRNA levels of DC activation markers (CD86 and MHC II) in the spleens of miR223−/− mice compared with those of WT mice (Fig. 5d). Importantly, the cytokines supporting differentiation of Th1 and Th17, i.e. IL‐6, IL‐12 and IL‐23 as well as other general inflammatory cytokines, were significantly reduced in the spleens of miR223−/− mice (Fig. 5d).
Figure 5.
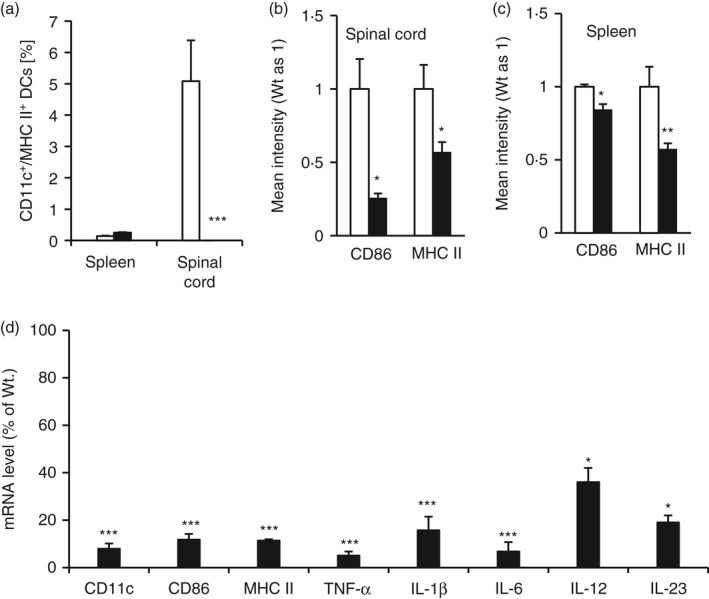
MicroRNA (miR) 223 deficiency suppressed expression of the genes associated dendritic cell (DC) activation and T helper type 1 (Th1)/Th17 cell differentiation in vivo. Mice were killed at day 3 post‐MOG immunization. Single cell suspension was prepared from indicated tissues and subjected to FACS analysis of the DC marker genes (a–c). RNAs isolated from an aliquot of splenocyte preparations were subjected to quantitative PCR analysis of DC activation markers and cell differentiation cytokines (d). All data are presented as means ± SEM (n = 4). *P < 0·05, **P < 0·01, ***P < 0·001.
miR223 deficiency compromised the capability of mature DCs to support pathogenic effector T‐cell development
Based on our in vivo finding that expression of the genes associated with DC activation and Th1 and Th17 cell differentiation was reduced in miR223−/− mice immunized with MOG, we next sought to test the hypothesis that miR223 deficiency in DCs impairs DC activation, which in turn compromises effector T‐cell polarization. We found that activation of immature WT DCs with LPS resulted in a significant increase in miR223 expression (Fig. 6a) and a variety of inflammatory cytokines including Th1 and Th17‐differntiating cytokines (IL‐12 and IL‐23) (Fig. 6b–f). The LPS‐induced expression of the DC activation markers (CD86, MHC II) and the key T‐cell‐supporting cytokines (IL‐12 and IL‐23) were significantly suppressed in miR223−/− DCs. These in vitro data provide additional evidence that miR223 deficiency impairs DC activation and maturation.
Figure 6.
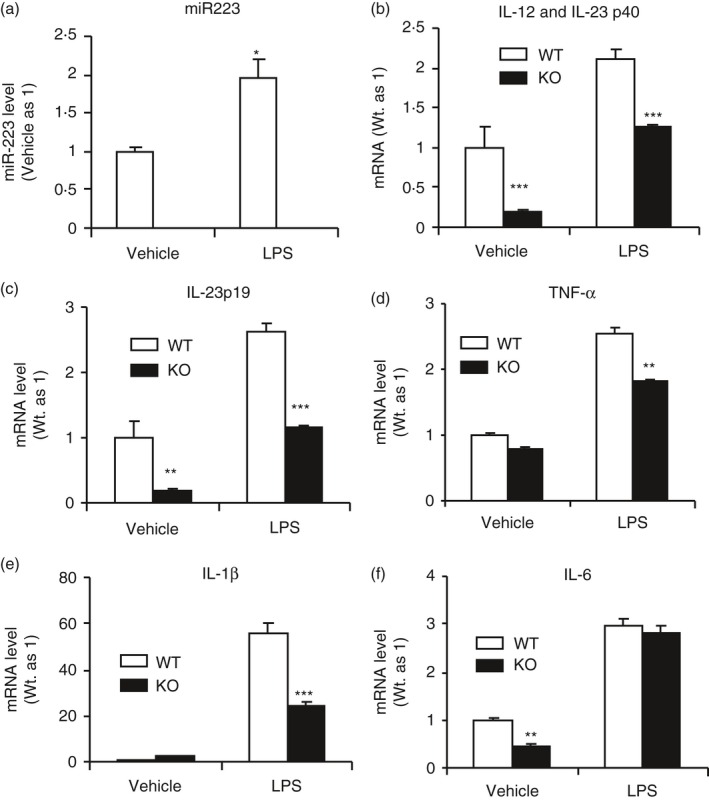
Dendritic cells (DCs) deficient in microRNA (miR) 223 expressed significantly reduced amount of interleukin‐12 (IL‐12) and IL‐23, the T helper type 1 (Th1)‐ and Th17‐differentiating cytokines respectively. Immature DCs were prepared from bone marrow of wild‐type (WT) and miR223−/− mice as indicated and stimulated with 100 ng/ml lipopolysaccharide (LPS) for 20 hr. Expression of miR223 (a) and various cytokines (b–f) was measured by real‐time quantitative PCR. All data are presented as means ± SEM (n = 4). *P < 0·05, **P < 0·01, ***P < 0·001.
We then developed a co‐culture system consisting of LPS‐activated DCs and naive T cells to evaluate if miR223−/− DCs and wild‐type DCs differ in their ability to regulate T‐cell differentiation. Activated DCs and naive T cells were cultured at a ratio of 1 : 5 and allowed to interact for 5 days. FACS analysis demonstrated a decline in IL‐17‐producing cells in the gated CD3+ CD4+ T cells (Fig. 7a,b). To determine if miR223 deficiency also regulates other T‐cell subtype differentiation and if the response occurs in a MOG‐specific environment, an additional experiment using 2D2 mouse‐derived naive T cells, which over‐express a MOG‐specific T‐cell receptor (Fig. 7c). miR223 deficiency in DCs led to a significant decline in the percentage of IL‐17+ cells and also a less‐pronounced decrease in the percentage of IFN‐γ + cells among the gated CD3+ CD4+ population (Fig. 7c). These results demonstrate that miR223 deficiency inhibits the formation of pathogenic Th17 cells, and to a lesser extent, the Th1 cells. Interestingly, polarization of IL‐4+ IL‐5+ Th2 cells and FOXP3+ regulatory T cells was not significantly affected by miR223 deficiency in DCs (Fig. 7c).
Figure 7.
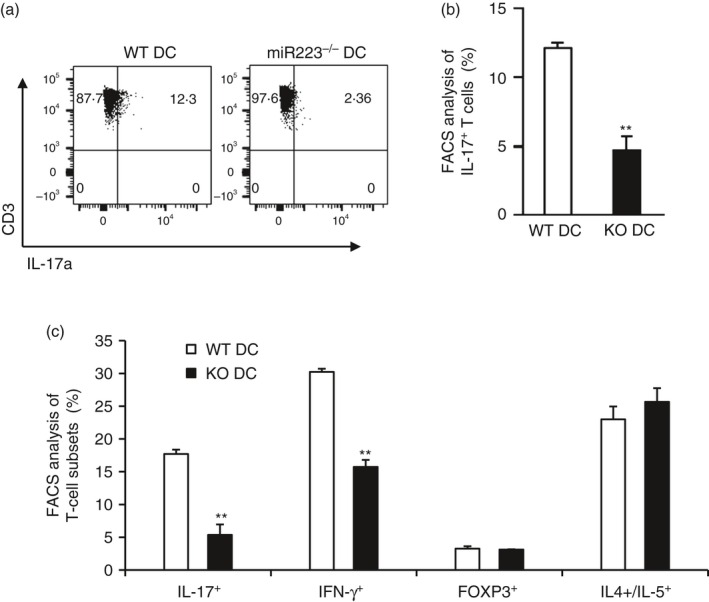
The ability of microRNA (miR) 223‐deficient dendritic cells (DCs) in differentiating T helper type 1 (Th1) and Th17 cells was significantly reduced. Isolated wild‐type (WT) DCs or miR223‐deficient DCs were activated with lipopolysaccharide (LPS) for 24 hr. Activated DCs were co‐cultured with naive T cells isolated from spleens of C57BL/6 mice (a) or 2D2 mice which over‐express a MOG‐specific T‐cell receptor (b). After 5 days, cells were harvested and subjected to FACS analysis of various effector T‐cell‐specific cytokines as indicated. All data are presented as means ± SEM (n = 4), **P < 0·01.
We also examined if miR223 deficiency in naive T cells per se could suppress the polarization of naive T cells into Th1 and Th17 effector T cells. Naive T cells isolated from the spleens of control, miR223−/−, or wild‐type mice were cultured under Th0, Th1 and Th17 polarization conditions. FACS analysis of the cultured T cells revealed that miR223 deficiency significantly reduced the abundance of Th17 (Fig. 8a,b). Consistent with FACS analysis, the mRNA levels of IL‐17a was significantly reduced in cultured miR223‐deficient T cells (Fig. 8c). In contrast, miR223−/− DCs and WT DCs did not differ significantly in their ability to induce Th1 differentiation as determined by either FACS (Fig. 8d,e) or qPCR analysis (Fig. 8f) of IFN‐γ expression. These in vitro data demonstrate that lack of miR223 in T cells could compromise specifically Th17 polarization, indicating that miR223 exhibits intrinsic promoting effect on Th17 development.
Figure 8.
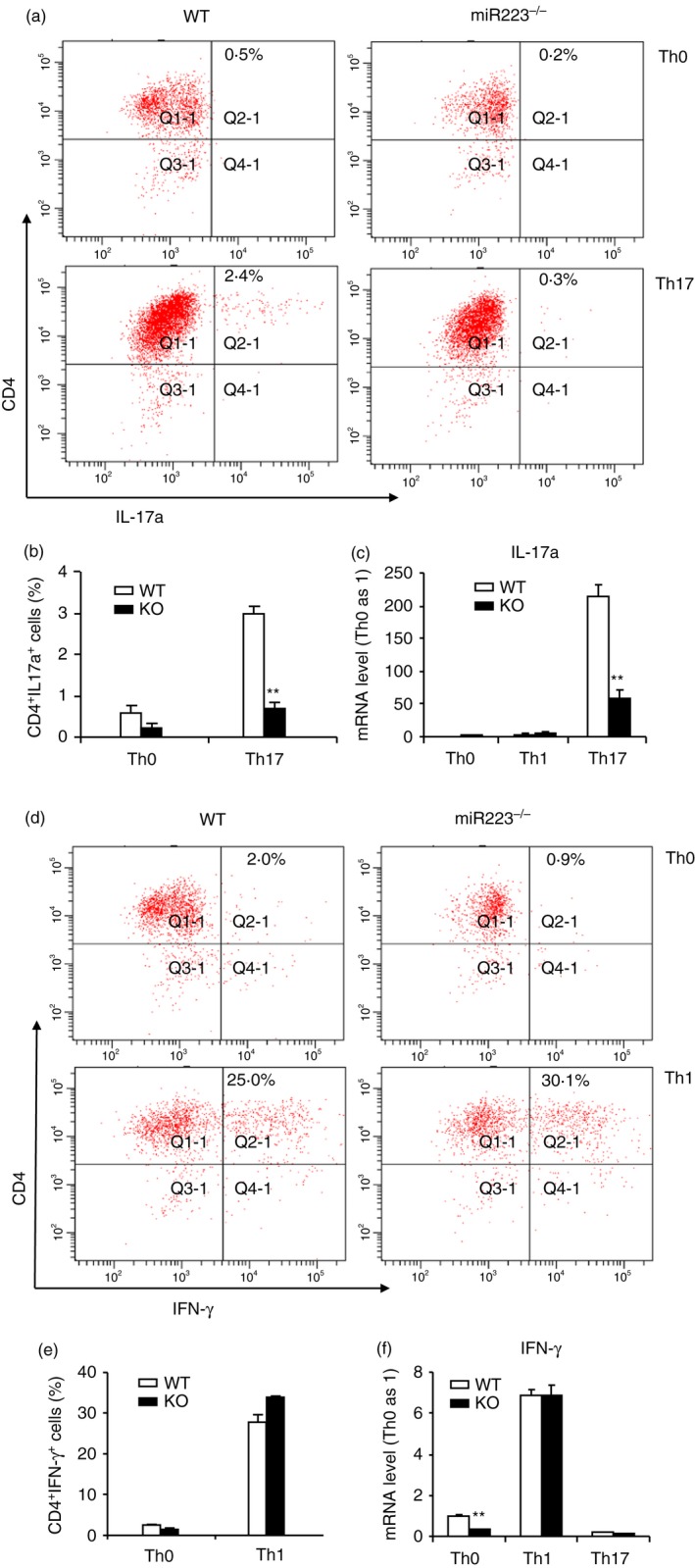
In the absence of dendritic cells (DCs), differentiation of microRNA (miR) 223‐deficient naive T cells into T helper type 17 (Th17) but not Th1 is intrinsically deficient. Naive CD4+ T cells were isolated from spleens of wild‐type (WT) or miR223−/− mice and cultured under various T‐cell polarization conditions. Polarized T cells were subjected to both FACS and real‐time quantitative PCR analysis 96 hr later. A representative Th17 FACS histogram was shown in (a) and statistical analysis in (b). Quantitative PCR measurement of interleukin‐17a (IL‐17a) mRNA level under Th17 polarization was shown in (c). A representative Th1 FACS histogram was shown in (d) and statistical analysis (e). Quantitative PCR analysis of interferon‐γ (IFN‐γ) mRNA level under Th1 polarization was shown in (f). Data shown here are representative of two independent experiments. Mean ± SEM (n = 4), **P < 0·01.
miR223 deficiency inhibits expression of the genes associated with M1 macrophages during EAE
As macrophages are important effector cells in EAE,19 we performed an additional in vivo experiment to evaluate if expression of the cytokines associated with the inflammatory M1 macrophages was suppressed by miR223 deficiency. Indeed, expression of the inflammatory M1 macrophage cytokines and marker genes (tumour necrosis factor‐α, CD82, CD86 and inducible nitric oxide synthase) declined significantly in both the spleens and spinal cords of miR223 knockout mice, compared with WT mice (see Supplementary material, Fig. S2).
Discussion
Since 2008, the role of miR223 has been studied using various disease models.8, 13, 14, 20 Whether miR223 acts to protect or enhance inflammatory responses appears to depend on the nature of the disease studied. To date, the role of miR223 in CNS autoimmune inflammation has not been investigated. Here we have reported that genetic ablation of miR223, either globally or in HSC lineages, remarkably delayed the onset of EAE and alleviated neuropathology in mice immunized with MOG. We have obtained strong in vivo and in vitro evidence that while miR223 deficiency in autoreactive T cells per se specifically suppresses Th17 cell differentiation, it also compromises DC activation, leading to suppression of both Th1 and Th17 cell differentiation.
miR223 represents one of the most highly up‐regulated miRs in the spinal cord and lymphoid organs upon EAE induction in mice (Fig. 1) and has previously been reported to be up‐regulated in the brain of patients with MS.21 These data prompted us to further investigate the role of miR223 in the pathogenesis of EAE. Our data clearly demonstrate that miR223 promotes CNS autoimmunity in the EAE model. miR223 global knockout mice showed a significant delay in EAE onset and a significant reduction in peak disease clinical score (Fig. 2). Similarly, this disease phenotype was successfully reproduced in control mice reconstituted with miR223‐deficient HSCs (Fig. 3). Together these findings provide strong evidence for a causal relationship, rather than a mere association, between up‐regulation of miR223 and CNS autoimmunity in EAE mice.
Although the mechanism by which miR223 deficiency leads to amelioration of EAE remains to be studied further, our data clearly indicate an important role of miR223 in immune cell differentiation, activation, function and interaction. Previous studies have demonstrated miR223′s involvement in the differentiation of haematopoietic lineages, with much of the focus on myeloid differentiation. In fact, the first in vivo study using miR223 knockout mice demonstrated that miR223 negatively regulates granulocyte differentiation and progenitor proliferation through post‐transcriptional targeting of mef2c mRNA.8 Surprisingly, little work has been done on miR223′s role in lymphoid differentiation and mechanism of action, even though over‐expression of miR223 in HSCs resulted in increased expression of Thy‐1.2, a T‐cell marker.9
Aberrant regulation of T‐cell differentiation results in tissue inflammation and autoimmunity, with myelin‐specific Th17 being the major effector subset contributing to axonal loss and demyelination in MS and EAE.22, 23 This process requires DCs, as they direct T‐cell polarization. In fact, DCs isolated from patients with MS produce large amounts of Th17 polarizing cytokines.24 Additionally, DCs are known to promote entry of T cells into the CNS and can also promote the spreading of autoimmunity to different CNS epitopes.25, 26 To our knowledge, the effect of an EAE‐modulating miR on DCs and their interaction with T cells has not been studied. Our study demonstrates that miR223 not only plays an important role in DC maturation and T‐cell polarization, but also the interaction between these two cell types.
The remarkable decline in spinal cord mRNA levels of IL‐17 and IFN‐γ together with reduced abundance of IL‐17‐positive and –IFN‐γ‐positive cells in the spinal cord suggest that infiltration of both T‐cell subsets is inhibited in miR223−/− mice (Fig. 4) and BMCs lacking miR223 in haematopoietic cells. Reduced presence of these two effector T cells could be due to (1) impaired differentiation from naive T cells in the lymphoid organs and/or (2) reduced homing of the differentiation of T cells into the CNS. Although the latter possibility cannot be excluded and needs to be studied in future, our data support that differentiation of naive T cells into effector T cells is indeed compromised by miR223 deficiency. First, DC activation, the initial event leading to T‐cell differentiation, is inhibited by miR223 deficiency both in vivo and in vitro. This is evidenced by our finding showing that expression of the genes associated with DC activation is significantly decreased in the spleen of miR223−/− mice following EAE induction and in the purified miR223‐deficient DCs activated by LPS (Figs 5 and 6). Second, miR223‐deficient naive T‐cells showed a significant reduction in their ability to differentiate into Th17 cells, although Th1 polarization is not affected (Fig. 8). Third, most importantly, miR223‐deficient DCs activated by LPS were significantly less effective in supporting differentiation of the MOG‐specific naive T cells into both Th1 and Th17, even though differentiation of other T‐cell subtypes such as the protective Th2 and regulatory T cells were not affected (Fig. 7).
Although the pathogenic Th1 and Th17 cells, produced in the peripheral lymphoid organs and subsequently migrated into the CNS, are necessary to initiate EAE, other recruited cells, e.g. macrophages, could play a major role in the tissue damage.27 In this regard, activated inflammatory M1 macrophages can strip myelin from axons and secrete numerous cytokines including IL‐1 and tumour necrosis factor‐α, which can perpetuate inflammatory reactions and contribute to tissue damage.27 We therefore investigated the impact of miR223 deficiency on M1 macrophage in both CNS and peripheral lymphoid tissue. Our data showed that expression of the cytokines associated with M1 macrophages was significantly decreased in both spinal cords and spleens of miR223 knockout mice compared with those of wild‐type control mice (see Supplementary material, Fig. S2). These data are in contrast to a recent report that miR223 knockout mice, challenged with high‐fat‐diet, exhibited more severe adipose tissue inflammation, enhanced systemic insulin resistance, and increased pro‐inflammatory M1 macrophage infiltration into adipose tissues compared with wild‐type mice.13 Hence, while miR223 may suppress M1 macrophage polarization in a type II diabetes model, it enhances M1 macrophage polarization in the EAE model. The mechanisms underlying the different role of miR223 in M1 macrophage activation and differentiation in type II diabetes and EAE warrant further investigation.
In summary, we have identified miR223 as a novel miR that promotes pathogenesis of EAE. The underlying mechanism may be due to miR223′s role in tipping immune cell differentiation towards pathogenic Th1 and Th17 cells in the peripheral lymphoid organs. The resistance to EAE observed in miR223‐deficient mice is very likely to be mediated by suppressing the development and/or function of multiple immune cells, which at least include Th1, Th17, DCs and M1 macrophages. Future studies are needed to identify the miR223 target mRNA(s) which mediate the action of miR223 on immune cell development and function during EAE.
Disclosure
The authors declare no competing conflict of interest.
Supporting information
Figure S1. MicroRNA (miR) 223 deficiency led to reduced expression of cytokine and marker genes associated with various immune cells. (a) Bone marrow chimeras (BMCs) reconstituted with wild‐type haematopoietic stem cells (HSCs) (WT BMCs) or miR223−/− HSCs were killed at day 25 post‐MOG immunization. Total RNAs were isolated from spinal cords and subjected to real‐time quantitative PCR analysis in duplicates using Mouse Inflammatory Cytokines RT² Profiler PCR Array. Data shown are the average value measured from two pooled RNA samples, each of which contained RNAs derived from three wild‐type (WT) or three miR223 knockout (KO) mice. (b) Global miR223 knockout mice and control mice were subjected to experimental autoimmune encephalomyelitis (EAE) induction. At day 18 post‐MOG immunization, total RNAs were isolated from spinal cords and subjected to real‐time quantitative PCR analysis of the mRNA levels of the indicated genes. Mean ± SEM (n = 5), **P < 0·01, ***P < 0·001.
Figure S2. Expression of M1 macrophage markers was reduced in the spinal cord and spleen of microRNA (miR)223 knockout mice. Spleens and spinal cords were isolated 18 days after MOG immunization. Expression of genes associated with M1 macrophages in the spleen (a) and spinal cord (b) were determined by real‐time quantitative PCR (means ± SEM, n = 4). *P < 0·05, **P < 0·01, ***P < 0·001.
Acknowledgements
We thank Deb Chandra for characterization of the EAE model, Fatima Rajaallah and Christina Zhou for assistance with animal work, Nancy Lowen for technical support in histological analysis. This study was supported by funding from Loma Linda University and Department of Veterans Affairs (to XQ) and US Department of Defense (to DJB).
References
- 1. Zhang J, Cheng Y, Cui W, Li M, Li B, Guo L. MicroRNA‐155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol 2014; 266:56–63. [DOI] [PubMed] [Google Scholar]
- 2. Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA‐155 ameliorates experimental autoimmune encephalomyelitis. J Immunol 2011; 187:2213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S et al MicroRNA miR‐326 regulates TH‐17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 2009; 10:1252–9. [DOI] [PubMed] [Google Scholar]
- 4. Hu R, Huffaker TB, Kagele DA, Runtsch MC, Bake E, Chaudhuri AA et al MicroRNA‐155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J Immunol 2013; 190:5972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dunand‐Sauthier I, Irla M, Carnesecchi S, Seguin‐Estevez Q, Vejnar CE, Zdobnov EM et al Repression of arginase‐2 expression in dendritic cells by microRNA‐155 is critical for promoting T cell proliferation. J Immunol 2014; 193:1690–700. [DOI] [PubMed] [Google Scholar]
- 6. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S et al MicroRNA‐21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest 2015; 125:1069–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA‐124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP‐α‐PU.1 pathway. Nat Med 2011; 17:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnnidis JB, Harris MH, Wheeler RT, Stehling‐Sun S, Lam MH, Kirak O et al Regulation of progenitor cell proliferation and granulocyte function by microRNA‐223. Nature 2008; 451:1125–9. [DOI] [PubMed] [Google Scholar]
- 9. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004; 303:83–6. [DOI] [PubMed] [Google Scholar]
- 10. Felli N, Pedini F, Romania P, Biffoni M, Morsilli O, Castelli G et al MicroRNA 223‐dependent expression of LMO2 regulates normal erythropoiesis. Haematologica 2009; 94:479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y et al An evolutionarily conserved mechanism for microRNA‐223 expression revealed by microRNA gene profiling. Cell 2007; 129:617–31. [DOI] [PubMed] [Google Scholar]
- 12. Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C et al A minicircuitry comprised of microRNA‐223 and transcription factors NFI‐A and C/EBPα regulates human granulopoiesis. Cell 2005; 123:819–31. [DOI] [PubMed] [Google Scholar]
- 13. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H et al A novel regulator of macrophage activation: miR‐223 in obesity‐associated adipose tissue inflammation. Circulation 2012; 125:2892–903. [DOI] [PubMed] [Google Scholar]
- 14. Harraz MM, Eacker SM, Wang X, Dawson TM, Dawson VL. MicroRNA‐223 is neuroprotective by targeting glutamate receptors. Proc Natl Acad Sci U S A 2012; 109:18962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li B, Baylink DJ, Deb C, Zannetti C, Rajaallah F, Xing W et al 1,25‐Dihydroxyvitamin D3 suppresses TLR8 expression and TLR8‐mediated inflammatory responses in monocytes in vitro and experimental autoimmune encephalomyelitis in vivo . PLoS ONE 2013; 8:e58808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li B, Baylink DJ, Walter MH, Lau KH, Meng X, Wang J et al Targeted 25‐hydroxyvitamin D3 1α‐hydroxylase adoptive gene therapy ameliorates DSS‐induced colitis without causing hypercalcemia in mice. Mol Ther 2015; 23:339–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Son YI, Egawa S, Tatsumi T, Redlinger RE Jr, Kalinski P, Kanto T. A novel bulk‐culture method for generating mature dendritic cells from mouse bone marrow cells. J Immunol Methods 2002; 262:145–57. [DOI] [PubMed] [Google Scholar]
- 18. Kapsenberg ML. Dendritic‐cell control of pathogen‐driven T‐cell polarization. Nat Rev Immunol 2003; 3:984–93. [DOI] [PubMed] [Google Scholar]
- 19. Berger T, Weerth S, Kojima K, Linington C, Wekerle H, Lassmann H. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab Invest 1997; 76:355–64. [PubMed] [Google Scholar]
- 20. Li YT, Chen SY, Wang CR, Liu MF, Lin CC, Jou IM et al Brief report: amelioration of collagen‐induced arthritis in mice by lentivirus‐mediated silencing of microRNA‐223. Arthritis Rheum 2012; 64:3240–5. [DOI] [PubMed] [Google Scholar]
- 21. Ma X, Zhou J, Zhong Y, Jiang L, Mu P, Li Y et al Expression, regulation and function of microRNAs in multiple sclerosis. Int J Med Sci 2014; 11:810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest 2012; 122:1180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev 2012; 248:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Comabella M, Montalban X, Munz C, Lunemann JD. Targeting dendritic cells to treat multiple sclerosis. Nat Rev Neurol 2010; 6:499–507. [DOI] [PubMed] [Google Scholar]
- 25. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T et al Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 2005; 11:328–34. [DOI] [PubMed] [Google Scholar]
- 26. McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med 2005; 11:335–9. [DOI] [PubMed] [Google Scholar]
- 27. Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006; 129:1953–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. MicroRNA (miR) 223 deficiency led to reduced expression of cytokine and marker genes associated with various immune cells. (a) Bone marrow chimeras (BMCs) reconstituted with wild‐type haematopoietic stem cells (HSCs) (WT BMCs) or miR223−/− HSCs were killed at day 25 post‐MOG immunization. Total RNAs were isolated from spinal cords and subjected to real‐time quantitative PCR analysis in duplicates using Mouse Inflammatory Cytokines RT² Profiler PCR Array. Data shown are the average value measured from two pooled RNA samples, each of which contained RNAs derived from three wild‐type (WT) or three miR223 knockout (KO) mice. (b) Global miR223 knockout mice and control mice were subjected to experimental autoimmune encephalomyelitis (EAE) induction. At day 18 post‐MOG immunization, total RNAs were isolated from spinal cords and subjected to real‐time quantitative PCR analysis of the mRNA levels of the indicated genes. Mean ± SEM (n = 5), **P < 0·01, ***P < 0·001.
Figure S2. Expression of M1 macrophage markers was reduced in the spinal cord and spleen of microRNA (miR)223 knockout mice. Spleens and spinal cords were isolated 18 days after MOG immunization. Expression of genes associated with M1 macrophages in the spleen (a) and spinal cord (b) were determined by real‐time quantitative PCR (means ± SEM, n = 4). *P < 0·05, **P < 0·01, ***P < 0·001.