

Summary
Sexual stages of Plasmodium are critical for malaria transmission and stage‐specific antigens are important targets for development of malaria transmission‐blocking vaccines. Plasmodium falciparum gamete surface antigen (Pfs48/45) is important for male gamete fertility and is being pursued as a candidate vaccine antigen. Vaccine‐induced transmission‐blocking antibodies recognize reduction‐sensitive conformational epitopes in Pfs48/45. Processing and presentation of such disulphide‐bond‐constrained epitopes is critical for eliciting the desired immune responses. Mice lacking interferon‐γ‐inducible lysosomal thiol reductase (GILT), an enzyme that mediates reduction of S‐S bonds during antigen processing, were employed to investigate immunogenicity of Pfs48/45. It has been well established that the ability to reduce S‐S bonds in antigens guides effective T‐cell immune responses; however, involvement of GILT in the induction of subsequent B‐cell responses has not been explored. We hypothesized that the ability to reduce S‐S bonds in Pfs48/45 will impact the generation of T‐cell epitopes, and so influence helper T‐cell responses required for specific B‐cell responses. Non‐reduced and reduced and alkylated forms of Pfs48/45 were employed to evaluate immune responses in wild‐type and GILT knockout mice and studies revealed important differences in several immune response parameters, including differences in putative T‐cell epitope recognition, faster kinetics of waning of Pfs48/45‐specific IgG1 antibodies in knockout mice, differential patterns of interferon‐γ and interleukin‐4 secretions by splenocytes, and possible effects of GILT on induction of long‐lived plasma cells and memory B cells responsible for antigen‐recall responses. These studies emphasize the importance of antigen structural features that significantly influence the development of effective immune responses.
Keywords: antigen processing, antigen structure, interferon‐γ‐inducible lysosomal thiol reductase, malaria, Pfs48/45, vaccine
Abbreviations
- APC
antigen‐presenting cell
- CPM
counts per minute
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- GILT
interferon‐γ‐inducible lysosomal thiol reductase
- IAA
iodoacetamide
- IFA
immunofluorescence assay
- IFN‐γ
interferon‐γ
- IL‐4
interleukin‐4
- KO
knockout
- LLPC
long‐lived plasma cell
- MBC
memory B cell
- NR
non‐reduced
- RA
reduced/alkylated
- RT
room temperature
- S‐S
disulphide‐bond
- WT
wild‐type
Introduction
Various developmental stages in the life cycle of Plasmodium, the causative agent of malaria, which afflicts more than half of the world's population with approximately 214 million new clinical infections and ~ 438 000 deaths estimated in 2015, offer unique antigenic targets for the development of vaccines.1 Transmission‐blocking vaccines target the sexual stage development of the parasite and are considered essential tools for the eventual elimination of malaria. Transmission‐blocking vaccines will induce antibodies that, when ingested by the blood‐feeding mosquitoes, will impede the sexual reproduction of the parasite within the mosquito vector, hence effectively preventing the parasite from infecting a new naive host.2 Efforts in our laboratory are focusing on one of the key target antigens, Plasmodium falciparum gamete surface antigen (Pfs48/45), as a transmission‐blocking vaccine. The Pfs48/45 protein is expressed in male and female gametocytes of P. falciparum, is recognized by transmission blocking antibodies and gene knockout approaches showed that it is critical for male gamete fertility in the mosquito midgut.3 Recently, recombinant full‐length Pfs48/45 expressed in Escherichia coli was shown to elicit potent transmission blocking antibodies in pre‐clinical evaluation in mice and non‐human primates.4 The expressed Pfs48/45 retains partial native conformation as revealed by monoclonal antibody recognition of specific conformational epitopes. Pfs48/45 contains 15 cysteine residues within its 427 amino acids, and previous studies have used various monoclonal antibodies in a competitive ELISA to identify six epitopes.5 Almost all of the monoclonal antibodies, which block gamete fertilization in the mosquito vector, have been shown to recognize reduction‐sensitive conformational epitopes.4, 6, 7, 8 As the protein has not been crystallized, the precise location of the disulphide bonds and the topology of conformational epitopes remain unknown. Efforts to solve crystal structure of Pfs48/45, including biochemical characterization based on site‐directed cysteine mutagenesis have been unsuccessful to date (Kumar et al., unpublished).
Protein disulphide bonds (S‐S) play an important role in protein conformation and necessitate specific mechanisms for processing by an antigen‐presenting cell (APC) for display of immune epitopes and induction of an appropriate immune response.9, 10 Indeed, it has been shown that when a protein antigen is processed in the endocytic pathway, the protein is reduced and proteolytically cleaved, allowing many potential epitopes to be formed, but only a few are selected for display on MHC molecules. This selection of epitopes for MHC loading, or epitope hierarchy, dictates which naive T cells are specifically activated to participate in mounting an immune response. Studies have shown that changes in S‐S bonding pattern in protein immunogens can result in strikingly skewed responses by lymphocytes.11, 12, 13, 23 Hence the development of vaccine antigens must consider not only the structural features of an immunogen, but also the ability of the immune system to process and present the appropriate epitopes required for activation of specific immune responses important for protection.
Interferon‐γ‐inducible lysosomal thiol reductase (GILT) is an exclusive thiol reductase that is constitutively expressed in APCs and can have up‐regulated expression in other cell types via interferon‐γ (IFN‐γ).14 This enzyme specifically catalyses the reduction of S‐S bonds in protein antigens and processing within the lysosome during antigen presentation. Development of a GILT−/− mouse line (C57BL/6 background) revealed a role for GILT‐mediated endocytic reduction of exogenous antigens for display of the peptides in the context of both MHC class I and II molecules on APCs.13 Studies using GILT knockout (KO) mice and several antigens containing S‐S bonds have revealed impaired T‐cell responses compared with wild‐type (WT) mice. Based on data highlighting the influence of the GILT enzyme in CD8+ and CD4+ T‐cell responses, and the importance of T‐cell help for expansion of functional B‐cell responses, it is reasonable to speculate that GILT‐mediated processing of S‐S bond‐containing proteins will likewise have a significant impact on antigen‐specific B‐cell responses.
Depending upon the life cycle stage of Plasmodium, both T‐cell and antibody‐mediated mechanisms have been shown to play important roles in controlling infection and further development of parasite stages. When it comes to specific antigenic targets in the sexual stages, it is imperative that the vaccines are able to elicit antibodies that recognize disulphide‐bond‐constrained conformational epitopes as previously described. Moreover, an ideal vaccine candidate will induce potent and long‐lasting immune responses. Using Pfs48/45 as a model target antigen, we aimed to investigate whether GILT‐mediated processing of epitopes will influence recognition of epitopes, and kinetics and magnitude of immune responses against conformationally intact (non‐reduced) and unfolded (denatured, reduced and alkylated) forms of recombinant Pfs48/45 antigen. We also examined the establishment and maintenance of long‐lived plasma cells and the memory B‐cell pool.
Materials and methods
Pfs48/45 protein production and reduction/alkylation
Expression and purification of the codon‐harmonized, recombinant Pfs48/45 protein (CH‐rPfs48/45) from P. falciparum was performed according to previously established protocol.4 Purified CH‐rPfs48/45 (500 μg/ml) was reduced using 100 μm of dithiothreitol (DTT) in the presence of 6 m urea, for 1 hr at 37°, followed by treatment with 1 mM iodoacetamide (IAA; freshly dissolved in 100 mm Tris–HCl, pH 8·5) in the dark at room temperature for 30 min. Afterwards, the IAA was quenched with an equal molar amount of DTT, followed by extensive dialysis to remove excess DTT and IAA. Proteins [non‐reduced (NR)‐Pfs48/45 and reduced/alkylated (RA)‐Pfs48/45] were characterized using murine polyclonal anti‐Pfs48/45 antibodies by Western blot analysis, and protein concentration was determined using a BCA Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA) and tested for endotoxin (Pierce LAL Chromogenic Endotoxin Quantitation Kit).
Overlapping sub‐fragments spanning full‐length Pfs48/45
Full‐length Pfs48/45 sequence divided into five overlapping fragments ~ 100 amino acids long with ~ 20‐amino‐acid overlap (amino acid boundaries depicted in Fig. 1c), were cloned into pRSET‐A vector (Invitrogen, Carlsbad, CA, USA) and expressed in E. coli BL21 (DE3) after induction with 1·0 mm Isopropyl β‐D‐1‐thiogalactopyranoside. Induced bacteria were lysed by microfluidization. Expressed protein found in the inclusion bodies was solubilized using 2% sarcosyl and purified using nickel affinity chromatography. Bound protein was eluted using 400 mm imidazole and dialysed using PBS + 10% glycerol + 350 mm NaCl + 50 mm NaH2PO4 (pH 7·4). Protein fragments were characterized by Western blot analysis under non‐reduced and reduced conditions (see Supplementary material, Fig. S1b), and protein concentration was determined using a BCA Protein Assay kit (Thermo Fisher Scientific, Waltham, Ma, USA) and tested for endotoxin (Pierce LAL Chromogenic Endotoxin Quantitation Kit). A panel of 39 peptides (20 amino acids long with 10‐amino‐acid overlap) was synthesized by GenScript (sequences and relevant characteristics described in the Supplementary material, Table S1). Peptides were initially dissolved in 100 μl DMSO + 10% H2O + 1 mm DTT and then immediately diluted in Dulbecco's modified Eagle's medium (DMEM; 100 μg/ml), filter sterilized and stored at −20°.
Figure 1.
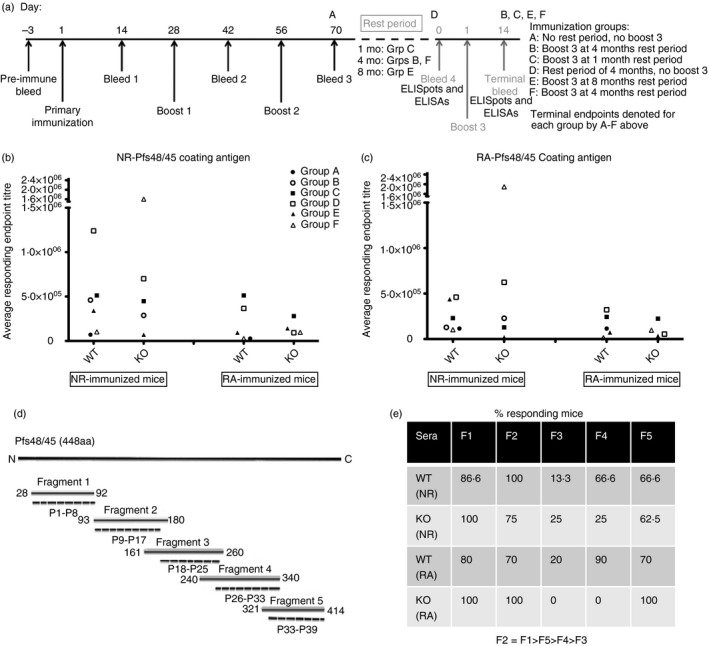
Serum antibody endpoint titres and sub‐fragment recognition. Immunizations, rest periods, blood collection and terminal end‐points are depicted in (a). Each independent immunization group (A to F) consisted of wild‐type (WT) and knockout (KO) mice immunized with non‐reduced (NR) and reduced/alkylated (RA) Pfs48/45 proteins, n = 5 mice for each form of immunogen. While B and F groups followed a very similar immunization schedule, they were totally independent of each other and used for different assays. Individual mouse serum collected after each immunization was tested by ELISA method for recognition of (b) NR‐Pfs48/45 coating antigen and (c) RA‐Pfs48/45 coating antigen. End‐point titres were defined as the last serum dilution found positive above pre‐immune sera mean + 3SD, and were averaged for responding mice in each group. Symbols represent average end‐point for each immunization group, for bleeds collected after the second booster immunization. Statistical analysis was performed using one‐way analysis of variance to compare end‐point titres across groups for each coating antigen and each immunogen. (d) Schematic representation of the amino acid boundaries for Pfs48/45 fragments and synthetic peptides (P1–P38), corresponding to the full‐length Pfs48/45 sequence. (e) Individual mice sera after second booster immunization were analysed (dilution 1 : 500) by Western blotting analysis. The recognition of individual Pfs48/45 fragments was tallied for each group, reported as percent responding mice, and presented according to immunogen; non‐reduced (NR) and reduced/alkylated (RA) Pfs48/45 protein.
Animals and immunization
Female WT C57BL/6 mice were purchased from the National Cancer Institute at 6–8 weeks of age. GILT−/− mice were originally generated by Dr Peter Cresswell and colleagues (Yale University). GILT−/− mouse line used in the present study was re‐derived using the plasmid generated by Dr Peter Cresswell, at Charles River before relocation to Tulane University. Mice were maintained and bred under specific pathogen‐free conditions in accordance with guidelines and regulations determined by the Institutional Animal Care and Use Committee of Tulane University.
For primary immunization, WT and KO mice (five mice per immunogen) received 10 μg of CH‐rPfs48/45 antigen (NR‐Pfs48/45 or RA‐Pfs48/45) formulated in complete Freund's adjuvant (Sigma, St Louis, MO, USA), administered through the intraperitoneal route. For various studies, immunizations were repeated in a series of six independent immunization groups (A–F) of WT and KO mice as described in Fig. 1(a). All mice were boosted twice via the intraperitoneal route at 4‐week intervals with 10 mg protein emulsified in incomplete Freund's adjuvant (Sigma). Mice in immunization groups (B, C, E and F) received a third boost after varying lengths of rest periods (1, 4 or 8 months). Blood was collected from the tail vein 1 week before primary immunization (pre‐immune sera), 2 weeks after each immunization, and at designated rest periods before boost 3 (1, 4 or 8 months). Terminal bleeds were collected at the time of sacrifice (denoted in Fig. 1a for each immunization group) using cardiac puncture. All blood samples were processed to collect serum and stored at −20° for further analysis.
Serum antibody analysis by ELISA
Sera collected from immunized animals were used to quantify Pfs48/45‐specific antibody titres as described previously.4 Pre‐immune sera were used as a negative control, and any sample absorbance reading found above the negative control mean + 3 standard deviations was considered positive. The lowest serum concentrations (highest dilutions), which tested positive, are reported as end‐point titres. End‐point titres determined for each individual mouse were averaged for each group. Data passed the D'Agostino–Pearson normality test and were subsequently compared using Student's t‐tests and one‐way analysis of variance (anova; reported as arithmetic means).
For isotype determination, ELISA was performed as described above with the following modifications: primary antibody was tested at a single dilution (one which gave an absorbance between 0·5 and 0·75 optical density at 405 nm for total IgGs) in duplicate wells, and secondary antibodies (horseradish peroxidase‐conjugated goat anti‐mouse IgG1, IgG2a, IgG2c and IgG3 from Southern Biotech, Birmingham, AL, USA) were used at a 1 : 5000 dilutions. Results for duplicates were averaged and are reported as optical density at 405 nm readings as well as IgG1 : IgG2c absorbance ratios.
Western blot analysis
Murine sera were analysed by Western blotting using recombinant sub‐fragments and purified gametocytes of P. falciparum (NF54). Recombinant sub‐fragments after SDS–PAGE (reducing 12·5%) were transferred to nitrocellulose membranes and probed with individual mouse sera and developed using horseradish peroxidase‐conjugated anti‐mouse IgG antibody (1 : 10 000 dilution) and ECL Prime Western blotting detection reagent (GE Healthcare Bio‐Sciences, Pittsburgh, PA, USA). Gametocytes were purified (> 80% enriched) using a Miltenyi LD paramagnetic column (Miltenyi Biotec, San Diego, CA, USA). Purified gametocytes (approximately 1 million gametocytes per lane) were lysed in SDS–PAGE sample buffer (non‐reducing) and fractionated SDS–PAGE using BioRad precast TGX gels (BioRad, Hercules, CA, USA) and probed as above at 1 : 5000 sera dilutions.
Antibodies and flow cytometry
Antibodies against indicated cell surface markers for flow analysis were purchased from Biolegend (San Diego, CA, USA) and included Fc Block (CD16/32, for blocking Fc receptors), CD19 (FITC, for B cells), CD138 (biotinylated, for plasma cells), streptavidin (phycoerythrin), CD4 (Alexafluor 647, for T helper cells), and CXCR5 (blue violet 421, for follicular helper T cells), CD3 (allophycocyanin, for T cells), IgM (blue violet 421, used with CD69‐ status for naive B cells), CD27 (phycoerythrin‐Cy7 for memory B cells) and CD69 (blue violet 605, for activation marker). Spleens were harvested and processed at the time of terminal bleeds from immunized mice. Cells were resuspended in DMEM and a portion was pooled from each mouse for compensation controls (single stain) and an unstained control. Cells were washed and incubated with anti‐mouse CD16/CD32 (clone 93) to block Fc binding. Subsequently, the cells were stained with primary antibody cocktail overnight, at 4°, in the dark. The following day cells were washed with 5 ml staining buffer and resuspended in staining buffer for incubation with streptavidin‐phycoerythrin on ice, in the dark for 30 min. Cells were washed once more and resuspended in staining buffer, filtered through 100‐μm mesh into BD tubes for flow cytometry using nine‐colour BD LSR II. Results were obtained using FlowJo software (Ashland, OR, USA), and analysed using GraphPad Prism. Data were determined to be Gaussian using the KS normality test and analysed using two‐way anova and Student's t‐tests. Results are reported as arithmetic mean and standard deviation.
T‐cell proliferation assays
Splenocytes were plated using DMEM + 10% fetal bovine serum (cDMEM) at 1 × 106 cells/ml and 200 μl each well in round‐bottomed, 96‐well cell culture‐treated plates (BD Biosciences, San Jose, CA, USA). Addition of various antigens varied between 2·5 and 10 μg/ml (final concentration), depending on the antigen. Various controls included mitogen phytohaemagglutinin and concanavalin A used at 5 μg/ml and unstimulated cells (medium only). Cells were incubated at 37°, 5% CO2 and pulsed at 72 hr (mitogen stimulation) and 120 hr (antigen stimulation) with 1 μCi/well of tritiated thymidine (American Radiolabeled Chemicals, Inc., St Louis, MO; specific activity of 20 Ci/mmol). Eighteen hours after pulsing, cells were harvested onto filtermats using a multi‐well cell harvester. Filter spots were dried and collected into scintillation vials containing scintillation fluid (National Diagnostics, Atlanta, GA, USA) and radioactivity (counts per minute, CPM) determined using a Packard Tri‐Carb 1600CA Liquid Scintillation Analyzer. The stimulation index was calculated as the ratio of CPM of stimulated/CPM of medium‐only cells and stimulation index values > 3 were considered positive. Data was determined to be Gaussian using the D'Agostino–Pearson normality test and then analysed using one‐way anova and Student's t‐tests to determine if groups were responding differently based on statistical comparison of ΔCPM values (ΔCPM = CPM in stimulated cells – CPM in medium‐only cells). Results are reported as mean ΔCPM with standard deviation.
Measurement of cytokine secretion by ELISA
Splenocytes (5 × 106 cells/ml in complete DMEM) were plated (750 μl per well) on 48‐well plates (BD Biosciences, San Jose, CA, USA) pre‐coated with 100 μl of anti‐mouse CD3 (BD Biosciences, San Jose, CA, USA) in PBS pH 7·4 at a concentration of 10 μg/ml at 4°. Cells were stimulated with 10 μg/ml Pfs48/45 in combination with purified anti‐mouse CD28 (Biolegend) or media alone and incubated at 37°, 5% CO2 for 48 hr. Cell supernatants were harvested and stored at −20° until further use. For determination of cytokines by ELISA, capture antibody was diluted in sterile 0·1 m carbonate coating buffer, pH 9·6. Interferon‐γ capture antibody (BD Biosciences, clone R4‐682) and interleukin‐4 (IL‐4) capture antibody (BD Biosciences, clone IIBII) were used at 2 μg/ml. Plates were blocked with assay diluent (sterile PBS + 10% fetal bovine serum) at room temperature for at least 1 hr, followed by addition of 100 μl culture supernatant (directly or diluted) and twofold serial dilutions of standards (murine recombinant IFN‐γ or IL‐4). Plates were incubated overnight at 4° and then reacted (1 hr at room temperature) with detection antibodies (biotinylated IFN‐γ detection antibody: clone XMG1.2 from BD Biosciences and biotinylated IL‐4 detection antibody: clone BVD6‐2462 from BD Biosciences). Plates were then incubated with extravidin–horseradish peroxidase (Sigma) at room temperature for 30 min, followed by addition of ABTS (2,2′‐Azinobis [3‐ethylbenzothiazoline‐6‐sulfonic acid]‐diammonium salt) substrate (KPL, Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD, USA) in the dark. Absorbance (405 nm) was read every 5 min, up to 45 min using a VersaMax ELISA reader (Molecular Devices, Sunnyvale, CA, USA) and SoftMax Pro software (Molecular Devices, Sunnyvale, CA, USA). Absorbance values of standards were used to determine the concentration of cytokine in culture supernatants.
Memory B‐cell expansion and ELISpot assays
Memory B cells were selectively expanded from spleens of immunized mice using a previously described protocol15 and plated in duplicate wells at various cell densities in ELISpot plates (EMD, Millipore, Billerica, MA, USA; Fisher Cat#:MAIPS4510). Total IgG‐secreting and Pfs48/45‐specific antibody‐secreting cells were quantified using a modification of the ELISpot method described previously.16 Wells were scanned and spots were counted, both using CTL‐Immunospot Analyzer. Background spots (from antibody‐uncoated wells) were subtracted from individual mouse groups, and expressed as number of spots/million spleen cells and compared between and within groups using prism software. Data were normalized for total IgG‐secreting cells and analysed using one‐way and two‐way anova. Results are reported as arithmetic mean and standard deviation.
Immunofluorescence assay
Plasmodium falciparum (NF54) gametes and gametocytes (purified using Nycodenz gradient centrifugation) were spotted onto multi‐well immunofluorescnece assay (IFA) slides, fixed in pre‐chilled acetone for 20 min at −20° and washed with chilled PBS. Slides were incubated with serial dilutions (PBS containing 1% non‐fat milk) of mouse sera (immune and pre‐immune) for 1 hr in a humid chamber at room temperature. Slides were washed with PBS, and incubated with FITC‐conjugated goat anti‐mouse IgG antibody (Southern Biotech, 1 : 100 dilution, 60 min at room temperature) to detect binding of antibodies to parasites. Parasite nuclei were stained using NucBlue® Fixed Cell ReadyProbes® Reagent (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA) as per product manual. Slides were washed with PBS and mounted using Citifluor AF‐3 anti‐fade solution (Ted Pella, Inc., Redding, CA, USA). Slides with all samples blinded for microscopy were examined using the Olympus BX41 fluorescent microscope, photographed using a QIClick Imaging CCD camera and analysed using the qcapture pro, QCapture Pro 7 Software.
For live IFA,4 gametes of P. falciparum were purified by Nycodenz gradient centrifugation and incubated for 45 min with 1 : 100 dilution (RPMI‐1640 medium containing 1% milk) of various mouse sera (immune and pre‐immune) on ice with intermittent gentle mixing. Cells were washed three times with chilled RPMI (1 min at 2095 × g, ‘Immufuge II’, Baxter) and incubated with FITC‐conjugated goat anti‐mouse IgG antibody (Southern Biotech., 1 : 100 dilution, 45 min on ice) and washed as above. NucBlue® Fixed Cell ReadyProbes® Reagent (Life Technologies) was used as per product manual and no DAPI staining was seen indicating viability of gametes through purification and staining procedure. Washed parasites were examined at 1000 × magnification (100 × oil immersion) using an Olympus BX41 Fluorescence microscope, photographed using a QIClick Imaging CCD camera and monochrome images were tinted using the ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
All statistical analyses were performed using both excel (Microsoft Office Software) and GraphPad Prism statistical software package (GraphPad Software Inc., CA). Normality tests were performed for each data set, and parametric tests were used accordingly. anova were performed for multiple group comparisons and multiple variable analyses. Follow‐up tests were performed in conjunction with anova, including Tukey–Kramer testing for comparison of all columns. Student's t‐tests were used for direct comparisons and anytime variances were considered significantly different by the F test, Welch's correction was applied.
Results
Serum IgG responses do not differ significantly between WT and KO mice
We hypothesized that the GILT‐mediated processing of S‐S bonds in Pfs48/45 will impact the T‐cell epitopes presented on APCs, which in turn will guide the activated T cells’ ability to help antigen‐specific B cells for the production of antibodies. We also speculated that such proposed altered T helper cell responses in turn will influence the ability of Pfs48/45‐specific B cells to produce qualitative and quantitative differences in antibody production, including overall repertoire of antibody epitopes generated. Although it is unlikely that GILT can influence the specificity of a given antigen‐specific B cell, downstream B‐cell functions such as expansion, isotype switch, affinity maturation and development of memory B cells are expected to be affected because of dependence on T‐cell help. We aimed to assess quantitative differences in antibody titres as well as differences in terms of recognition of antigenic epitopes.
The ELISAs were performed to determine antigen‐specific (NR‐Pfs48/45 and RA‐Pfs48/45) serum IgG antibodies for each bleed. Individual mouse serum was serially diluted twofold and the end‐point titres were defined as the last serum dilution found positive above pre‐immune sera mean + 3 SD, and were averaged for responding mice in each group (Fig. 1b,c). Immunization resulted in boosting of antibody responses after each immunization dose in mice immunized with NR‐ and RA‐Pfs48/45 proteins (data not shown). Mice immunized with RA‐Pfs48/45 protein consistently showed lower antigen‐specific IgG responses to both NR‐ and RA‐Pfs48/45 antigens used to coat ELISA plates (Fig. 1b,c). The KO mice showed a trend for lower antibody titres; however, the differences were not statistically significant because of large animal‐to‐animal variations in different immunization groups.
Western blotting analysis was used to further characterize antibody reactivity patterns using whole protein and various overlapping fragments of Pfs48/45 (Fig. 1d). Sera collected for all bleeds were diluted 1 : 500 and tested from individual mice, for recognition of the five sub‐fragments. Mice within groups (n ~ 10) were tallied for the presence or absence of bands visualized for each fragment and results are reported as % responding mice for each group. Results for bleed 3 are summarized in Fig. 1(e). Most of the mice in all groups responded well throughout various blood collection time‐points to fragments 1, 2 and 5 in the NR‐immunized group, whereas those immunized with RA‐Pfs48/45 responded mostly to fragments 2 and 5. Mice immunized with NR‐ and RA‐Pfs48/45 showed weak or no recognition of fragment 3, suggesting overall reactivity patterns as F2 = F1 > F5 > F4 > F3. The fragment recognition pattern did not differ between WT and KO mice.
Unique T‐cell specificities are found in GILT KO mice
We reasoned that the availability of GILT within APCs would impact the repertoire of potential T‐cell epitopes for MHC class II‐mediated activation of T helper cells. If GILT‐mediated reduction of Pfs48/45 antigen is required for antigen processing, we expected to find observable differences in the T‐cell epitope specificities, as well as a skewed cytokine profile between NR‐immunized WT and GILT KO mice.
Flow cytometry was used to enumerate lymphocyte populations from the spleens of immunized (NR‐ or RA‐Pfs48/45 immunogens) WT and KO mice (Table 1). Differences in total B and T lymphocyte populations in immunized WT and KO mice were not statistically significant. We then compared T‐cell proliferation responses in the presence of whole Pfs48/45 protein antigens (NR‐ and RA‐Pfs48/45), overlapping fragments (immunization group B), pools of synthetic peptides (immunization group C) and individual synthetic peptides spanning Pfs48/45 (immunization group F). T‐cell proliferation studies using whole antigens (NR‐ and RA‐Pfs48/45) revealed that splenocytes from RA‐immunized mice were stimulated to a lesser extent than those from the NR‐immunized groups (Fig. 2a). Likewise, splenocytes from GILT KO mice (immunized with NR protein) also showed a trend of lower stimulation compared with WT mice (Fig. 2a).
Table 1.
Immunophenotyping with flow cytometry
| Flow cytometry data (% of parent population) | NR‐immunized | RA‐immunized | |||
|---|---|---|---|---|---|
| Cell type | Cell surface markers | WT | KO | WT | KO |
| Lymphocytes | SSC‐A × FSC‐A gate (singlets) | 48·1 (5·6) | 52·7 (2·2) | 45·9 (4·8) | 55·1 (2·5) |
| Total T cells | CD19– CD3+ | 23·7 (4·0) | 19·8 (3·0) | 16·9 (2·0) | 16·8 (3·2) |
| CD4+ T cells | CD19– CD3+ CD4+ CD8– | 11·4 (1·6) | 15·5 (2·7) | 11·7 (1·6) | 16·0 (2·9) |
| Follicular T cells | CD19– CD3+ CXCR5+ | 1·2 (0·2) | 3·4 (0·5) | 1·8 (0·2) | 2·8 (0·7) |
| Total B cells | CD3– CD19+ CD45+ | 60·6 (4·7) | 64·6 (0·6) | 61·8 (6·5) | 65·2 (1·4) |
| Naive B cells | CD3– CD19+ CD45+ IgM+ CD69– | 3·2 (1·7) | 1·3 (1·1) | 0·3 (0·2) | 0·5 (0·2) |
| Activated B cells | CD3– CD19+ CD45+ CD69+ | 1·5 (0·3) | 0·9 (0·1) | 1·4 (0·5) | 0·8 (0·1) |
| Memory B cells | CD3– CD19+ CD45+ CD138– CD27+ | 19·3 (2·3) | 18·6 (7·5) | 21·0 (6·8) | 15·7 (1·4) |
| Plasma cells | CD3– CD19+ CD45+ CD138+ | 0·7 (0·1) | 1·0 (0·4) | 0·4 (0·1) | 0·5 (0·2) |
| Long‐lived plasma cells | CD3– CD19+ CD45+ CD138+CD27+ | 0·1 (0) | 0·1 (0) | 0·1 (0) | 0·1 (0) |
Lymphocyte subpopulations in the spleens at necropsy were selected for by size, and single cells were used for further gating (excluding doublets). Total B‐cell and T‐cell populations were mutually exclusive and were defined as either CD19+ or CD3+, respectively. Additional gating was used to differentiate CD4+ T cells, follicular CXCR5+ T cells, CD69+ activated B cells from IgM+ naive B cells. Further gating on activated B cells, was used to enumerate CD138+ plasma cells compared with CD27+ B memory cells. Statistics generated in flowjo software were used for percentages of cell types found in the individual spleens of wild‐type (WT) and knockout (KO) mice, immunized with either non‐reduced (NR) ‐Pfs48/45 or reduced/alkylated (RA) ‐Pfs48/45 antigen (groups E and F). One‐way analysis of variance (anova) was used to compare across immunization groups and comparisons for WT and KO for specific lymphocyte populations were carried out using two‐way anova and Student's t‐tests. No statistically significant differences were found. Mean with (SD) are depicted above.
Figure 2.
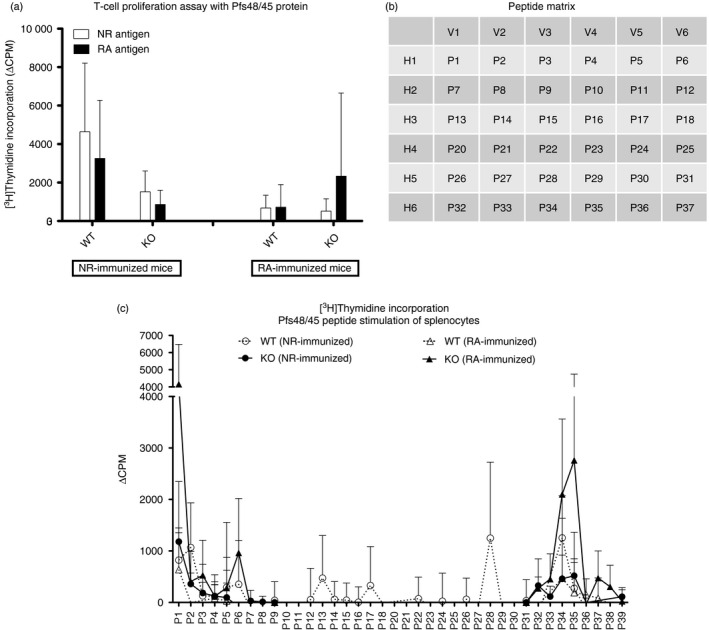
T‐cell proliferation. Splenocytes collected at terminal end‐points from individual wild‐type (WT) and knockout (KO) mice (immunized with either non‐reduced (NR) or reduced/alkylated (RA) ‐Pfs48/45 antigens) in groups B, C and F were stimulated with final concentrations of 2·5 μg/ml of NR‐ or RA‐Pfs48/45 antigens (a). Results from all three immunization groups (B, C and F) were pooled for statistical comparisons. Bars show arithmetic mean and SD (n = 14 total after pooling data from individual mouse in the three inmmunization groups). (b) shows peptide matrix for pooling peptides used for stimulation of splenocytes. These pools consisted of vertical and horizontal pool designation that allowed testing of several peptides simultaneously, while deducing individual peptides resulting in stimulation. Splenocytes from individual mice (immunization group C, n = 5 per immunogen) were tested against each peptide pool (10 μg/ml) and individual peptides 38 and 39. (c) In a separate experiment, splenocytes from WT and KO mice in the immunization group F (n = 5 per immunogen) were assayed for stimulation by individual peptides. Spleen cells from WT mice immunized with NR‐Pfs48/45 were tested against all peptides corresponding to full‐length Pfs48/45. Peptides corresponding to only fragments 1 and 5 of Pfs48/45 were used for in vitro stimulation of cells from NR‐Pfs48/45 immunized KO mice and RA‐Pfs48/45 immunized WT and KO mice, Cells were harvested after 18 hr of pulsing with [3H]thymidine and radioactivity was recorded as counts per minute (CPM). ΔCPM was derived after subtraction of CPM of control cells (unstimulated) from stimulated cells. Graphs depict mean and SD within immunization groups.
We then evaluated overlapping fragments in T‐cell proliferation assays. WT mice immunized with NR‐Pfs48/45 protein compared with KO‐immunized mice had the largest responses, with both WT and GILT KO mice responding particularly well to fragment 5, indicating the presence of T‐cell‐stimulating epitopes toward the C‐terminus of the protein (data not shown). Further T‐cell proliferation studies employed overlapping synthetic peptides to further identify epitopes. Initially, we carried out proliferation experiments using peptide pools before testing individual peptides. A peptide matrix was designed using six horizontal pools (denoted H1–H6) and six vertical pools (denoted V1–V6) (Fig. 2b). We had hoped that this method would allow us to scan stimulation responses of multiple peptides simultaneously, while also allowing us to infer proliferative responses to individual peptides and compare results to previous fragment data. Results revealed that the pools H1, V1 and V3 showed the largest proliferation (data not shown). When the peptide pools were mapped back to the matrix, it suggested peptides 1 and 3 to be probably responsible for the proliferative responses found. These peptides are contained within fragment 1, and these results contrast with previous data obtained from T‐cell proliferation assays using the fragments as stimulatory antigens. In light of these ambiguous data, we decided to repeat proliferation studies using individual peptides spanning the Pfs48/45 sequence. Peptides P1–P8 and P32–P39 correspond to fragment 1 and fragment 5, respectively.
As shown in Fig. 2(c), peptides 1, 2, 34 and 35 gave consistent T‐cell stimulation in most NR‐Pfs48/45 immunized mice, regardless of the availability of GILT. These shared immune responses to peptides correspond to fragments 1 and 5, corroborating previous T‐cell stimulation experiments with fragments and peptide pools (above). Some differences in stimulatory epitopes were also found between WT and KO mice, with WT mice uniquely responding to peptide 5, whereas the KO mice showed unique responses to peptides 7, 8, 32, 33 and 39.
T cells in GILT KO mice are skewed for IL‐4 production
Initial investigations into cytokine production of stimulated T cells showed detectable antigen‐specific secretion of IL‐4 in only NR‐Pfs48/45 immunized groups, and IFN‐γ secretion most prominent in groups stimulated by the NR antigen (data not shown). These studies also revealed differences among various immunogen groups in the overall stimulation by anti‐CD3, leading us to normalize antigen‐specific responses as a ratio of anti‐CD3 responses for comparing across various immunogen groups. Focusing on antigen‐specific responses alone, we found an apparent trend that NR‐Pfs48/45 was a more potent stimulant of IFN‐γ responses than RA‐Pfs48/45 (Fig. 3a). Further analysis also revealed that KO mice immunized with NR‐Pfs48/45 produced barely detectable to low levels of IFN‐γ in response to both NR‐ and RA‐Pfs48/45 antigens. In contrast, a similar analysis of IL‐4 secretion did not reveal any such difference between NR‐ and RA‐Pfs48/45 antigens used as in vitro stimulants (Fig. 3b). These findings prompted us to further analyse cytokine secretion data by expressing as a ratio of IL‐4 : IFN‐γ. Results in Fig. 3(c) demonstrate a sharp skew for IL‐4 secretion in the KO mice immunized with NR‐Pfs48/45 and stimulated with either NR‐ or RA‐Pfs48/45 in vitro.
Figure 3.
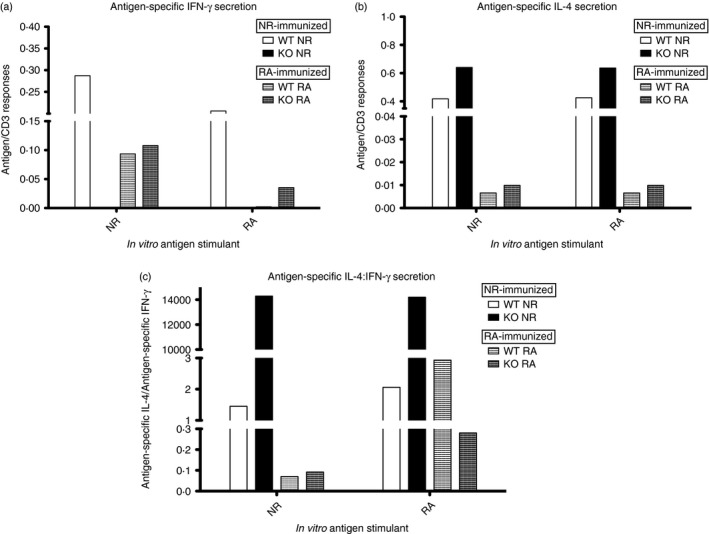
Analysis of antigen‐specific interferon‐γ (IFN‐γ) and interleukin‐4 (IL‐4) responses. Spleen cells from mice after third boost (immunization group F mice, n = 5 per immunogen) were stimulated individually in vitro with Pfs48/45 proteins or anti‐CD3, in the presence of an anti‐CD28 co‐stimulator. Supernatants of stimulated cells were used to determine cytokines by ELISA. Absorbance values of standards (murine recombinant IFN‐γ and IL‐4) were used to determine the concentration of IFN‐γ and IL‐4 cytokine secretion in culture supernatants. Given the differences in stimulation of cells from wild‐type (WT) and knockout (KO) mice by anti‐CD3, cytokine results were normalized by expressing antigen‐specific responses as a ratio of anti‐CD3 responses for both IFN‐γ (a) and IL‐4 (b) secretion. (c) Further comparison of antigen‐specific cytokine responses expressed as a ratio of IL‐4 to IFN‐γ.
Long‐lived serum antibody levels wane faster in GILT KO mice
In view of the results on the lack of any significant differences in the antibody levels, assayed directly after booster immunizations, between Pfs48/45‐immunized WT and GILT KO mice; and T‐cell responses, indicated by differential epitope recognition and cytokine bias, we investigated the possibility of altered B‐cell and T‐cell interactions. Long‐lived plasma cells (LLPCs) and memory B cells (MBCs) require sustained T‐cell help to differentiate and survive for long‐lived antibody production and antigen recall responses. Wild‐type and GILT KO mice immunized with NR‐ or RA‐Pfs48/45 were rested for different time periods, followed by evaluation of various B‐cell populations and antigen recall responses with additional boosting.
Figure 4 shows Pfs48/45‐specific antibody levels after rest periods of 4 and 8 months and during an antigen recall response after further boosting with Pfs48/45. The end‐point titres after rest periods were significantly lower (statistically) in KO mice compared with WT mice immunized with NR‐Pfs48/45; however, such differences in antibody titres were not found when RA‐Pfs48/45 was used as an immunogen. To compare differences in the persistence of antibody titres after the rest period across groups, we expressed differences as fold reduction of titres after resting time‐points (Fig. 4a–d). Fold reduction was found to be significantly different at 4 months of resting, with KO mice exhibiting significantly reduced IgG titres (Fig.4a). However, these differences were equalized between WT and KO mice analysed after 8 months of rest (Fig.4b). These results indicated that although all mice lost a substantial amount of antigen‐specific IgG after 8 months, the KO mice immunized with NR‐antigen did so much faster (significantly lower by the 4 month rest time‐point). These kinetic differences appeared to be influenced by GILT‐mediated processing of the NR‐Pfs48/45 antigen, as RA‐immunized mice showed no significant differences in antibody titre at either rest periods (Fig. 4c,d). We do not know whether these differences are the result of faster loss of antigen‐specific antibody in KO mice immunized with NR‐Pfs48/45 or whether WT mice continue to produce antibodies for longer periods of time.
Figure 4.
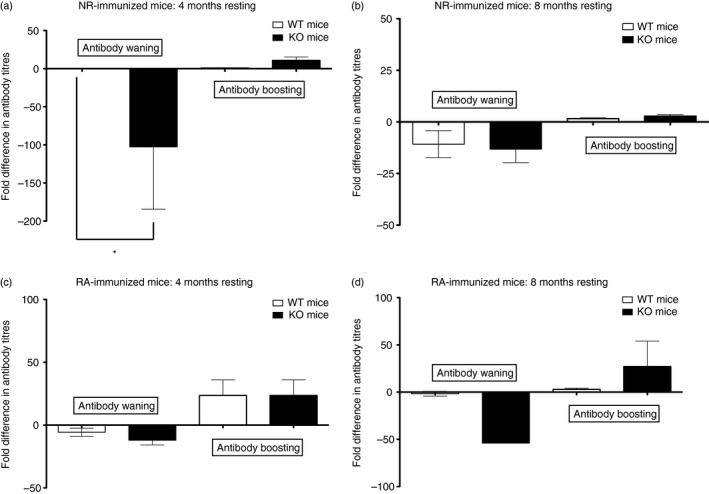
Antibody waning and recall responses. The ELISA analysis for end‐point titres in the sera collected after rest period and third booster immunization (see immunization and blood collection schedule in Fig. 1a) was carried out for individual mice and antibody end‐point titres were expressed as fold difference, Antibody waning was calculated by comparing bleed 3 and bleed 4. Comparison of bleed 4 and terminal bleed provided data on antibody boosting. The fold reduction or increase between bleeds calculated for each mouse were averaged and also used to calculate standard error of the mean pertaining to waning and boosting titres for each immunogen group. Results were graphed separately above, according to immunogen and rest period: (b) non‐reduced (NR) ‐Pfs48/45 immunized mice (group F), rested 4 months (c) NR‐Pfs48/45 immunized mice (group E), rested 8 months (d) reduced/alkylated (RA) ‐Pfs48/45 immunized mice (group F), rested 4 months (e) RA‐Pfs48/45 immunized mice (group E), rested 8 months. Comparisons of wild‐type (WT) and knockout (KO) groups were performed using Student's t‐test. Mean fold differences and SEM are shown.
We next evaluated the antigen recall response after additional boosting (after resting for 4 and 8 months) with Pfs48/45 antigen. As shown (Fig. 4b,e), although KO mice lost anti‐Pfs48/45 IgG faster, their boosting responses were intact, and able to reach levels found in WT counterparts. Similar to waning results, this kinetic difference found between WT and KO mice for boosting of antibody titres was limited to NR‐immunized mice, as it was no longer apparent in RA‐immunized mice – once again implicating the influence of GILT in these responses. Seeking an explanation for differences in the kinetics of antibody waning and recall responses, we analysed antibody isotypes and found that KO mice were biased toward IgG2c isotype but only after 8 months of resting, which became similar to WT after the antigen recall phase (Fig. 5a,b).
Figure 5.
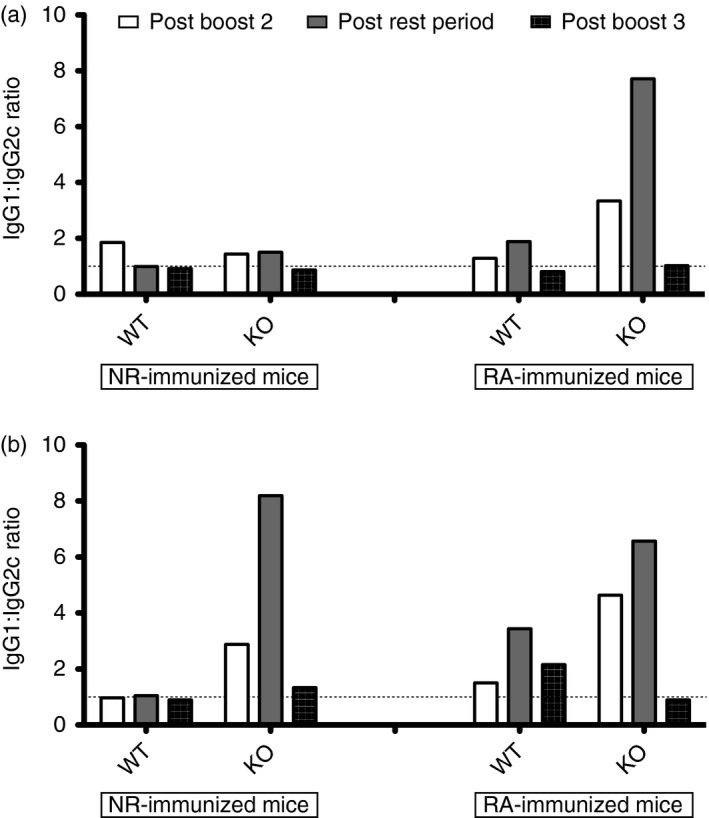
Antibody isotyping data. All the isotyping data were obtained using non‐reducing (NR) ‐Pfs48/45 as coating antigen. Antibody isotype ratios for IgG1 : IgG2c were determined by ELISA, for sera collected after the second boost (bleed 3) against the NR coating antigen, with defined rest periods (bleed 4) and post third boost (terminal bleed). Results were graphed separately above, according to rest period: (a) mice rested 4 months (group F) after boost 2; (b) mice rested 8 months (group E) after boost 2.
Recognition of Pfs48/45 by antibodies in both WT and KO mice immunized with NR‐ and RA‐Pfs48/45
We also tested antibodies generated in WT and KO mice immunized with NR‐Pfs48/45 antigen (rested for 4 or 8 months and subsequently boosted) for recognition of antigen in the sexual stages of P. falciparum. Sera from four to five mice for each group were pooled and tested by Western blotting and IFA (Fig. 6a–c). Antibodies in sera from WT and KO mice showed strong recognition of antigen on the surface of live P. falciparum gametes (Fig. 6a). To compare antibody titres by IFA and ELISA, pooled sera were tested by IFA using acetone‐fixed gametes and gametocytes. Figure 6(b) shows results demonstrating a comparable trend for antibody titres by IFA and ELISA. Recognition of Pfs48/45 in the parasite was further confirmed by Western blot analysis using purified gametocyte lysate under non‐reducing conditions. Pooled sera from WT and KO mice showed strong recognition of Pfs48/45 protein in the parasite lysate (Fig. 6c).
Figure 6.
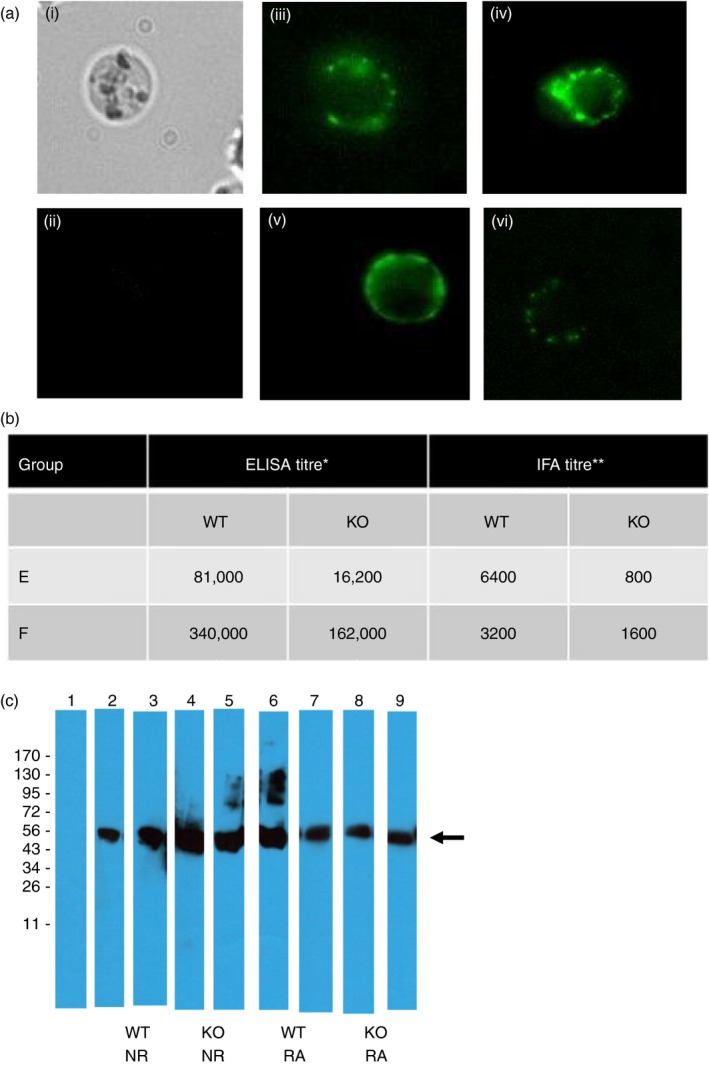
Recognition of Pfs48/45 by antibodies in wildtype and GILT knockout (KO) mice immunized with non‐reducing (NR) and reduced/alkylated (RA) ‐Pfs48/45 antigens. Terminal bleed sera from individual immunized mice were pooled and tested by live immunofluorescence assay (IFA) (a), fixed IFA (b) and Western blotting (c). Sera were tested at 1 : 100 dilution in live IFA (a): panel (i) shows a representative bright‐field image of live gamete, (ii) shows IFA using pre‐immune C57BL/6 mouse sera at 1 : 100 dilution, (iii) and (iv) show two representative IFA patterns obtained using sera from wild‐type (WT) mice immunized with NR‐Pfs48/45. (v) and (vi) show two representative IFA patterns using sera from KO mice immunized with NR‐Pfs48/45. For titration by fixed IFA (b), immune and pre‐immune mouse sera were serially diluted (1 : 200 to 1 : 6400) for incubation with parasites, Slides were blinded before examination by fluorescence microscopy. IFA titres are defined as the last serum dilution found to be visually positive compared with pre‐immune mouse serum used as a negative control at corresponding dilutions. End‐point ELISA titres in (b) are defined as the last serum dilution found positive above pre‐immune sera mean + 3SD. For Western blotting (c) immune sera were tested at 1 : 5000 and 1 : 1000 dilutions of NR‐Pfs48/45 and RA‐Pfs48/45, respectively, immunized WT and KO mice. Lanes show pre‐immune mouse serum negative control (lane 1), two different groups of WT mice immunized with NR‐Pfs48/45 (lanes 2, 3) and RA‐Pfs48/45 (lanes 6, 7), and two different groups of KO mice immunized with NR‐Pfs48/45 (lanes 4, 5) and RA‐Pfs48/45 (lanes 8, 9). An arrow indicates the position of Pfs48/45 in the gametocyte lysates under non‐reducing SDS–PAGE.
Frequency of antigen‐specific LLPCs and MBCs
Finally, we compared frequency and specificity of total MBCs and LLPCs in the spleens of immunized mice using a modification of the ELISpot method previously described.16 Initially we performed in vitro re‐stimulation ELISpots, wherein antibody‐secreting cells were collected from spleens after resting mice for 4 months, and then tested for response to Pfs48/45 antigens. MBCs from the same spleens were selectively expanded in culture then tested by ELISpot analysis using wells coated with Pfs48/45 and sub‐fragments. Based on previous Western data we decided to test the specificity of LLPCs and MBCs for reactivity to whole proteins and Pfs48/45 fragments 2 and 5. We estimated total number of IgG‐secreting cells and antigen‐specific B cells and normalized ELISpot data by expressing data as a ratio of antigen‐specific cells to total IgG‐secreting cells for comparison across all groups. After normalizing for total IgG‐secreting cells, similar numbers of LLPCS and MBCs were found in all groups for reactivity to all antigens (Table 2). Within individual groups, however, an apparent general trend was found that the largest responses, or the larger numbers of B cells, were responsive to NR‐Pfs48/45 and fragment 5. Little to no response was found to RA‐Pfs48/45.
Table 2.
Long‐lived plasma cell and memory B‐cell ELISpots
| % Antigen‐specific ASC/Total ASCs | LLPCs Mean (SD) | MBCs Mean (SD) | ||
|---|---|---|---|---|
| Capture antigen | WT | KO | WT | KO |
| NR‐Pfs48/45 | 7·4 (5·5) | 10·8 (14·7) | 6·0 (3·3) | 34·0 (5·5) |
| RA‐Pfs48/45 | 0 | 0 | 2·1 (1·8) | 2·15 (1·0) |
| Fragment 2 | 3·0 (2·2) | 5·4 (16·0) | 25·7 (9·2) | 8·0 (1·7) |
| Fragment 5 | 12·7 (7·8) | 19·3 (10·3) | 58·3 (6·8) | 44·4 (7·5) |
ELISpot method was used to determine frequency and specificity of antibody‐secreting cells (ASCs) after mice were rested for 4 months (group D mice, n = 5 for each immunogen). Long‐lived plasma cells (LLPCs) were tested directly after mice were killed, whereas memory B cells (MBCs) were selectively expanded in culture and then tested. The analysis was performed on cells from each spleen individually. Background spots (from uncoated wells) were subtracted from individual mouse groups, averaged for duplicate wells and reported as average number of spots per million cells. ELISpot data were normalized for total IgG‐secreting cells and expressed as a percentage of antigen‐specific cells : total IgG‐secreting cells. Using one‐way analysis of variance, no significant differences were found for antigen‐specific spots produced between wild‐type (WT) and knockout (KO) mice or between immunization groups (reduced/alkylated‐immunized mice not shown).
Discussion
We initially reasoned that a lack of GILT in the antigen‐processing pathway would result in altered Pfs48/45‐specific T helper cell responses and this in turn would influence the ability of Pfs48/45‐specific B cells to produce qualitative and/or quantitative differences in antibody production, including overall repertoire of antibody epitopes generated. However, our results indicate that the NR form of recombinant Pfs48/45 antigen apparently does not require GILT‐mediated processing for generation of antibodies. Though we did find that immunization with the NR‐Pfs48/45 tended to produce higher titres of antigen‐specific IgG in both WT and KO mice than the RA‐Pfs48/45 antigen. In addition, antibodies produced in both WT and KO mice were largely specific for epitopes at the amino‐ and carboxyl‐termini of Pfs48/45. Although initial production of antibodies seemed not to differ between WT and KO mice throughout various time‐points of immunizations, the persistence of antigen‐specific IgG was different. Data from serum collected over time after resting indicate that although all mice showed antibody waning after 8 months, the KO mice immunized with NR‐antigen did so much faster, with significantly lower titres by the 4‐month time‐point. We conclude that these kinetic differences are due to influences of GILT‐mediated processing of the NR‐Pfs48/45 antigen, as RA‐immunized mice showed no significant differences in antibody titre at either rest period. This could be explained with either KO mice losing antigen‐specific antibody at a faster rate, or by WT mice producing a larger quantity of antibody for longer periods of time. With subsequent boosting after resting, antigen recall was intact for both WT and KO mice; however, WT showed a generally higher titre of antigen‐specific antibodies revealed by ELISA as well as IFA. Antibodies in both WT and KO mice immunized with NR‐Pfs48/45 antigen were capable of recognizing Pfs48/45 antigen in the parasite, as revealed by strong surface reactivity in live gametes, indirect IFA using fixed gametes and gametocytes, and by Western blot analysis using purified gametocytes. It is reasonable to suggest that the production of antigen‐specific antibodies is influenced by the availability of GILT, most likely through the interactions required by B cells with helper T cells.
It has been established that T‐cell epitopes can influence the production of antibodies against specific B‐cell epitopes.17, 18, 19 A recent study of CD4+ T‐cell responses to the malaria protein, MSP1‐33, found that specific T‐cell epitopes were capable of inducing higher titres and more protective antibodies to the larger precursor protein (MSP1‐42), and skewed immunity to a T helper type 2‐biased response.19 Other studies have provided evidence that interactions of CD4+ T helper cells and B cells are linked through cognate antigen‐specific epitopes.20, 21 This study was designed to correlate antibody responses with specific CD4+ T‐cell responses. We reasoned that GILT‐mediated processing within APCs would impact the repertoire of potential T‐cell epitopes for MHC class II‐mediated activation of T helper cells. It has been demonstrated that the hierarchy of immunodominant epitopes presented in MHC class II molecules is dependent on GILT, and it can change in its absence, to produce an alternative repertoire of epitopes presented by B cells, for T‐cell recognition.11, 14 Moreover, if the priming of the relevant CD4+ T cells by classical APCs is altered based on the protein conformation and availability of GILT, the possibility of altered B‐cell interactions with T helper cells would be apparent. This should lead to a skewed stimulation for B‐cell proliferation and antibody production.
In fact we did detect unique T‐cell epitopes in KO mice and IFN‐γ secretion was barely detectable with a skew for elevated IL‐4 production. We expected that there would be detectable differences in the recognition of peptides by T cells in GILT KO mice exposed to NR‐Pfs48/45 compared with WT mice. We also predicted that T‐cell epitopes in KO mice would be limited, as was previously demonstrated for hen egg lysozyme peptides.9 We theorized that in WT mice immunized with NR‐Pfs48/45, there would be dominant epitopes responsible for the activation of specific CD4+ T cells required for eliciting a potent antibody response. We thought it probable that the dominance patterns of T‐cell epitopes should be altered in GILT−/− mice immunized with the NR antigen. As anticipated, T‐cell proliferation studies showed impaired T‐cell responses to whole antigens in NR‐immunized KO mice. Results also showed that the RA‐Pfs48/45 is seemingly less immunogenic, and although responses were low we did find that RA‐immunized mice achieved comparable responses between WT and KO groups when stimulated with whole antigen. This is expected for the RA‐Pfs48/45 antigen as both WT and KO mice should be able to process and present RA‐Pfs48/45 with similar efficiency. Fragments 1 and 5, which correspond to the first and last 100 amino acids in the Pfs48/45 protein, seemed to be most stimulatory of antigen‐specific splenocytes, and when assayed for specific epitopes, we found that peptides 1–2 and 34–35 might represent putative helper T‐cell epitopes common to all WT and KO groups. These stimulatory regions may indicate major domains for T‐cell epitope mining, and should prompt additional studies using all peptides spanning the Pfs48/45 protein. As we saw large responses to these common epitopes in both WT and KO groups immunized with NR‐Pfs48/45, we conclude that these peptides are efficiently presented in a GILT‐independent manner. The stimulatory epitopes found in fragment 1 (peptides 1 and 2) are located at the N‐terminus of the native protein, which contains a reduction‐insensitive domain for recognition by monoclonal antibodies in both reducing and non‐reducing conditions. The stimulatory epitopes found in fragment 5 (peptides 34 and 35) are located at the C‐terminus, which contains a reduction‐sensitive domain, targeted by the most potent transmission‐blocking antibodies. This C‐terminal protein domain includes a highly conformational six‐cysteine domain common to several unique Plasmodium antigens. If the peptides corresponding to fragment 5 are efficiently presented to T cells without the help of GILT‐mediated processing, it may support the hypothetical structure proposed by Carter et al.,6 which suggested that this domain contains a large structural loop. This external loop would have areas accessible to proteases present in the antigen‐processing pathway and may account for the efficient display of epitopes in the absence of disulphide‐bond reduction.
We expected cytokine results to be indicative of helper responses, as they relate to isotyping data, and that this information would be valuable in understanding immunodominant T‐cell epitopes as they relate to mechanisms important for functional T‐cell–B‐cell interaction and resulting antibody production. We found that most WT mice immunized with either NR‐ or RA‐Pfs48/45 were skewed for IFN‐γ secretion. The IFN‐γ secretion at the T‐cell–B‐cell interface results in the production of IgG2c, with potent complement fixation and antigen‐binding functions.22 In contrast, a lack of IFN‐γ secretion was apparent in the NR‐immunized KO mice. Previous work has shown that cytokine profiles of T cells can be influenced at an epitope‐specific level.19, 23 Based on unique epitopes mapped in KO splenocytes, we speculate that these epitopes might be responsible for the skew of IL‐4 secretion in the same group. Interleukin‐4 secretion during the T‐cell–B‐cell interaction has been known to produce the IgG1 isotype, which is used for a more classical T helper type 2 response (primarily mast cell binding).22 Indeed it was determined that KO mice are largely biased for IgG1 production, which increases over longer rest periods. This would lead us to believe that the remaining persistent long‐lived Pfs48/45‐specific antibody‐secreting cells are producing IgG, probably in response to IL‐4 stimulation by T cells specific for non‐buried epitopes in the Pfs48/45 antigen.
Together, our results indicate a possible GILT‐dependent kinetic influence for antigen‐recall responses, but it requires further investigation. Results showed GILT‐dependent differences in immune responses with rest and antigen‐recall responses. These influences may indicate that antigen structure, and epitope specificity can select for specific memory cells and functions, and that this should be further validated. Overall, we aimed to gain a better understanding of the immunological mechanisms critical to generate effective and long‐lasting humoral immunity against the target antigen being pursued as a transmission‐blocking vaccine antigen. The results from these and future studies will contribute significantly to our understanding of Pfs48/45 for use as a transmission‐blocking vaccine. We believe that this investigation has begun to lay the necessary groundwork for consideration of the implications of S‐S bond‐containing epitopes and their presentation by APCs through processing by the GILT pathway for induction of functional B‐cell responses possibly leading to functional antibody responses.
Disclosure
The authors declare no conflicts of interest.
Supporting information
Figure S1. SDS–PAGE analysis of purified rPfs48/45.
Table S1. Synthetic peptide sequences and chemical properties.
Acknowledgements
We thank Dr Samuel Landry and Dr James Mclachlan and their lab members Dr Nam Nguyen, Dr Jonathan Kurtz and Dr Daniel Frederick for discussions and assistance with T‐cell proliferation studies and flow cytometry work. KM was supported by The Robert D. Watkins Fellowship provided by the American Society for Microbiology. We acknowledge additional financial support from NIH grant RO1‐AI47089.
References
- 1. WHO : World Malaria Report. 2015.
- 2. Kappe SHI, Vaughan AM, Boddey JA, Cowman AF. That was then but this is now: malaria research in the time of an eradication agenda. Science 2010; 328:862–6. [DOI] [PubMed] [Google Scholar]
- 3. van Dijk MR, Janse CJ, Thompson J, Waters AP, Braks JA, Dodemont HJ, et al A central role for P48/45 in malaria parasite male gamete fertility. Cell 2001; 104:153–64. [DOI] [PubMed] [Google Scholar]
- 4. Chowdhury DR, Angov E, Kariuki T, Kumar N. A potent malaria transmission blocking vaccine based on codon harmonized full length Pfs48/45 expressed in Escherichia coli . PLoS One 2009; 4:1–10. e6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milek RL. Pfs48/45, Major Transmission‐Blocking Vaccine Candidate of the Human Malaria Parasite Plasmodium falciparum. The Netherlands: University of Nijmegen, 1999: 1–156. [Google Scholar]
- 6. Carter R, Coulson A, Bhatti S, Taylor BJ, Elliott JF. Predicted disulfide‐bonded structures for three uniquely related proteins of Plasmodium falciparum, Pfs230, Pfs48/45 and Pf12. Mol Biochem Parasitol 1995; 71:203–10. [DOI] [PubMed] [Google Scholar]
- 7. Outchkourov NS, Roeffen W, Kaan A, Jansen J, Luty A, Schuiffel D, et al Correctly folded Pfs48/45 protein of Plasmodium falciparum elicits malaria transmission‐blocking immunity in mice. Proc Natl Acad Sci USA 2008; 105:4301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Outchkourov N, Vermunt A, Jansen J, Kaan A, Roeffen W, Teelen K, et al Epitope analysis of the malaria surface antigen pfs48/45 identifies a subdomain that elicits transmission blocking antibodies. J Biol Chem 2007; 282:17148–56. [DOI] [PubMed] [Google Scholar]
- 9. Maric M, Arunachalam B, Phan UT, Dong C, Garrett WS, Cannon KS, et al Defective antigen processing in GILT‐free mice. Science 2001; 294:1361–5. [DOI] [PubMed] [Google Scholar]
- 10. Li P, Gregg JL, Wang N, Zhou D, O'Donnell P, Blum JS, et al Compartmentalization of class II antigen presentation: contribution of cytoplasmic and endosomal processing. Immunol Rev 2005; 207:206–17. [DOI] [PubMed] [Google Scholar]
- 11. Li P, Haque MA, Blum JS. Role of disulfide bonds in regulating antigen processing and epitope selection. J Immunol 2002; 169:2444–50. [DOI] [PubMed] [Google Scholar]
- 12. Mirano‐Bascos D, Steede NK, Robinson JE, Landry SJ. Influence of disulfide‐stabilized structure on the specificity of helper T‐cell and antibody responses to HIV envelope glycoprotein gp120. J Virol 2010; 84:3303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hastings KT, Lackman RL, Cresswell P. Functional requirements for the lysosomal thiol reductase GILT in MHC class II‐restricted antigen processing. J Immunol 2006; 177:8569–77. [DOI] [PubMed] [Google Scholar]
- 14. Haque M, Li P, Jackson S, Zarour H, Hawes J, Phan U, et al Absence of γ‐interferon‐inducible lysosomal thiol reductase in melanomas disrupts T cell recognition of select immunodominant epitopes. J Exp Med 2002; 195:1267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ndungu FM, Cadman ET, Coulcher J, Nduati E, Couper E, Macdonald DW, et al Functional memory B cells and long‐lived plasma cells are generated after a single Plasmodium chabaudi infection in mice. PLoS Pathog 2009; 5:1–15. e1000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eriksson K, Nordström I, Horal P, Jeansson S, Svennerholm B, Vahlne A, et al Amplified ELISpot assay for the detection of HIV‐specific antibody‐secreting cells in subhuman primates. J Immunol Methods 1992; 153:107–13. [DOI] [PubMed] [Google Scholar]
- 17. Oseroff C, Sidney J, Kotturi MF, Kolla R, Alam R, Broide DH, et al Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol 2010; 185:943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scherle PA, Gerhard W. Functional analysis of influenza‐specific helper T cell clones in vivo. T cells specific for internal viral proteins provide cognate help for B cell responses to hemagglutinin. J Exp Med 1986; 164:1114–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pusic K, Hashimoto C, Lehrer A, Aniya C, Clements D, Hui G. T cell epitope regions of the P. falciparum MSP1‐33 critically influence immune responses and in vitro efficacy of MSP1‐42 vaccines. PLoS One 2011;6:1–13. e24782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sette A, Moutaftsi M, Moyron‐Quiroz J, McCausland M, Davies D, Johnston R, et al Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity 2008; 28:847–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C, Dow C, Wang P, Sidney J, Read A, Harmsen A, et al Identification of CD4+ T cell epitopes in C. burnetii antigens targeted by antibody responses. PLoS One 2011;6:1–10. e17712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hussain R, Dawood G, Abrar N, Toossi Z, Minai A, Dojki M, et al Selective increases in antibody isotypes and immunoglobulin G subclass responses to secreted antigens in tuberculosis patients and healthy household contacts of the patients. Clin Diagn Lab Immunol 1995; 2:726–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milich D, McLachlan A, Moriarty A, Thornton G. A single 10‐residue pre‐S(1) peptide can prime T cell help for antibody production to multiple epitopes within the pre‐S(1), pre‐S(2), and S regions of HBsAg. J Immunol 1987; 138:4457–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SDS–PAGE analysis of purified rPfs48/45.
Table S1. Synthetic peptide sequences and chemical properties.