

Abstract
The affinity of therapeutic antibodies for Fcγ-receptors (FcγRs) strongly influences their antitumor potency. To generate antibodies with optimal binding and immunologic efficacy, we compared the affinities of different versions of an IgG1 Fc region that had an altered peptide backbone, altered glycans, or both. To produce IgG1 with glycans that lacked α1,6-fucose, we used CHO cells that were deficient in the enzyme UDP-N-acetylglucosamine (GnT1): α-3-D-mannoside-β-1,2-N-acetylglucosaminyltransferase I, encoded by the MGAT1 gene. Mature N-linked glycans require this enzyme, and without it, CHO cells synthesize antibodies carrying only Man5-GlcNAc2, which were more effective in antibody-dependent cell-mediated cytotoxicity (ADCC). Our engineered IgG1, hu3F8-IgG1, is specific for GD2, a neuroendocrine tumor ganglioside. Its peptide mutant is IgG1-DEL (S239D/I332E/A330L), both produced in wild-type CHO cells. When produced in GnT1-deficient CHO cells, we referred to them as IgG1n and IgG1n-DEL, respectively. Affinities for human FcγRs were measured using Biacore T-100 (on CD16 and CD32 polymorphic alleles), their immunologic properties compared for ADCC and complement-mediated cytotoxicity (CMC) in vitro, and pharmacokinetics and antitumor effects were compared in vivo in humanized mice. IgG1n and IgG1n-DEL contained only mannose and acetylglucosamine and had preferential affinity for activating CD16s, over inhibitory CD32B, receptors. In vivo, the antitumor effects of IgG1, IgG1-DEL, and IgG1n-DEL were similar but modest, whereas IgG1n was significantly more effective (P < 0.05). Thus, IgG1n antibodies produced in GnT1-deficient CHO cells may have potential as improved anticancer therapeutics.
Keywords: hu3F8, GD2, GnT1, glyco-enhanced, mannose, fucose
Introduction
Antibody based immunotherapy is an accepted modality in cancer treatment. In the presence of IgG antibodies, many tumor cells are susceptible to Fc dependent killing, which include NK (natural killer) cell-mediated antibody-dependent cytotoxicity (ADCC), granulocyte-mediated ADCC, monocyte-macrophage mediated cytotoxicity, and complement mediated cytotoxicity (CMC) (Reviewed in Ref (1)). All the IgG1 antibodies approved by the US FDA to date for cancer immunotherapy rely partly or solely on their Fc effector functions. The affinity of their Fc for the FcγRIIIA (CD16A) receptor on NK cells or macrophages, and the affinity for FcγRIIA (CD32A) receptor on myeloid cells (2) are critical determinants of the cytotoxic potential. The affinity for the neonatal Fc-receptor, FcR(n), can influence the serum half-life, thereby changing the cumulative drug exposure as measured by the area under the concentration curve (AUC) over time. Because of the importance of Fc affinity to therapeutic effects, we sought to improve the affinity of an antibody to GD2, a ganglioside prevalent on neuroendocrine tumors, by modifying either their amino acid sequence or their carbohydrate moieties.
In humans, FcγR polymorphism stratifies patients into those with higher affinity for human IgG1, FCGR3A(158V) or FCGR2A(131H), and those with lower affinity FCGR3A(158F) or FCGR2A(131R). Because of this affinity difference, patients benefit differentially from human IgG1 antibody treatment. Among lymphoma patients treated with rituximab, both higher affinity FCGR2A and FCGR3A alleles were shown to have better response with survival advantage in most but not all studies (3–6). When the receptor CD16A was the higher affinity allele, in vitro cytotoxicity could increase several fold (7), which translated into a 2-fold advantage in clinical response and in time to progression (3). When metastatic breast cancer was treated with Herceptin, patients with higher affinity FCGR3A allele had better overall response (83% versus 35%, P = 0.03), and longer progression-free survival (P = 0.005) (8), although in other studies no significant advantage was found (6, 9, 10). For metastatic colorectal cancer treated with cetuximab, patients with lower affinity FCGR2A and FCGR3A alleles had worse hazard ratios comparable to those with mutated KRAS (11).
Site directed mutagenesis has been successfully applied to the Fc region to enhance affinity for FcγR, creating antibodies with enhanced clinical potential (12). The triple mutation (S239D/I332E/A330L, DEL) is one example which increases FcγR affinity by 10 to 100 fold (13). However, mutations can increase the immunogenicity of IgGs. An alternative approach to enhancing FcγR affinity is by modifying Fc glycosylation (14). A key enzyme for the N-linked glycans is UDP-N-acetylglucosamine: α-3-D-mannoside-β-1,2-N-acetylglucosaminyltransferase I (GnT1) encoded by the MGAT1 gene. GnT1−/− CHO cells synthesize antibodies carrying Man5-GlcNAc2 without α1,6-fucose, exhibiting increased ADCC (15). Although antibodies carrying Man5 are susceptible to clearance through the mannose receptors on scavenger cells, serum half-lives of these IgGs have not been substantially affected (16). Recently a WT1-TCR mimic antibody produced in GnT1−/− CHO cells, specific for the WT1 RMF peptide/HLA-A*02:01 complex, was shown to have enhanced ADCC both in vitro and in vivo (17).
Anti-GD2 antibody immunotherapy has proven efficacy (18, 19). Besides NK-ADCC, granulocyte-ADCC appears to be important for clinical benefit (20, 21). Using the humanized version of 3F8 (hu3F8) (22) currently in clinical trials, we tested the effect of GnT1−/− and triple DEL mutations, alone and together, on FcR affinity, in vitro tumor cytotoxicity, and in vivo antitumor effects.
Materials and Methods
Cell lines and antibodies
Neuroblastoma cell line LAN-1 and melanoma line M14 were obtained from University of California, Los Angeles in 1983 and 1984, respectively. The following cell lines were established at Memorial Sloan Kettering Cancer Center (MSK), New York: Neuroblastoma line SKNCM in 2007, and Ewing’s sarcoma line SKELP in 2004. The following cell lines were purchased from ATCC: Neuroblastoma IMR-32 in 1983; Melanoma SKMEL1 in 1984; Small cell lung cancer (SCLC) NCI-H524, NCI-H69 and NCI-H82 all in 2005. Ewing’s sarcoma line TC71 was from DSMZ in 2005. All cell lines have been tested and authenticated within 6 months of experiments by short tandem repeat profiling using PowerPlex 1.2 System (Promega), by comparison with either a public database, our original tumor samples, or the original batch of cells from outside sources. All cell lines were periodically tested for mycoplasma using a commercial kit (Lonza). The luciferase-labeled tumor cell line IMR-32-Luc was generated by stably expressing a SFG-GFLuc vector into the parental cells. All cell lines were grown in F10 medium (RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 ug/mL streptomycin) at 37°C in a 5% CO2 incubator.
The antibody hu3F8-IgG1 was produced as previously described (22). Nucleotide sequence encoding the parental hu3F8 (22) and the mutant form hu3F8-IgG1-DEL (S239D/I332E/A330L in the human IgG1-Fc region) was synthesized by GenScript, and was subcloned into a mammalian expression vector. The antibodies hu3F8-IgG1 and hu3F8-IgG1-DEL were produced in wild-type CHO-S cells, whereas hu3F8-IgG1n and hu3F8-IgG1n-DEL in GnT1-deficient CHO cells (Eureka Therapeutics, Inc.). Antibodies were purified by protein A affinity column chromatography. The purity of these antibodies was evaluated by both SDS-PAGE, and size-exclusion high-performance liquid chromatography (SE-HPLC) as previously described (22, 23).
Flow cytometry analysis
Lectin PHA binding to CHO cells was performed as previously described (24), using FITC-conjugated L-PHA (EY Laboratories, Inc.) at 2 μg per 106 cells.
In vitro binding kinetics studies by surface plasmon resonance (SPR)
Biacore T-100 Biosensor, CM5 sensor chip, and related reagents were purchased from GE Healthcare. Human recombinant CD16A-158V, CD16B and CD32B were purchased from R&D System; CD16A-158F was from Sino Biological. CD32A-131R and CD32A-131H were produced in E Coli as previously described (25). Human FcR(n) was purchased from R&D System, while mouse FcR(n) was purchased from ACRO Biosystems. All recombinant proteins as the active surface and blank as the reference were immobilized using the Amino Coupling kit (GE Healthcare).
The antibodies were diluted in standard HBS-EP buffer (0.01 M HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.05% v/v Surfactant P20) for CD16/CD32 study, and in FcR(n) running buffer (50 mM sodium phosphate pH 6.0, 0.15 M NaCl) for FcR(n) study, at varying concentrations (42 ~ 667 nM) prior to analysis. Samples were injected over the sensor surface at a flow rate of 30 uL/min over 1–2 min. Following completion of the association phase, dissociation was monitored for 3 min at the same flow rate. At the end of each cycle, the surface was regenerated using 10 mM NaOH for CD16/CD32 study, and FcR(n) regeneration buffer (0.1 M Tris pH 8.0, 0.2 M NaCl) for FcR(n) study, at a flow rate of 50 uL/min over 1–2 min. Prior to kinetics analysis, the control curves obtained with samples injected over reference surfaces were subtracted from the biosensor curves obtained after injection of the samples over active surfaces. The results were analyzed using Biacore T-100 evaluation software. The data for CD16 and CD32 were fitted either with two-state reaction model: A + B = AB = AB*, KD = (kd1/ka1)/(1 + ka2/kd2), or with steady-state (SS) equilibrium analysis as noted in the table footnotes; FcR(n) data were fitted with steady-state (SS) equilibrium analysis.
Cell cytotoxicity (51Cr release assay)
Cell cytotoxicity was assayed by 51Cr release as previously described (22), and EC50 was calculated using GraphPad Prism software. ADCC assays were performed using NK92MI cells stably transfected with the human CD16A-158V or human CD32A-131H Fc receptors, and effector to target ratio was kept at 20:1. CMC assays were performed using human serum from healthy donors.
Pharmacokinetics studies
Blood was collected from tail vein of nude mice at time 0.5, 2, 4, 8, 24, 48, 72, 96, and 168 hr after a bolus injection of 100 μg of antibodies. Quantitation of serum Hu3F8 was carried out by ELISA. In brief, antibody A1G4, specific for the 3F8 idiotype was used to capture serum hu3F8 (human IgG1 antibody) on 96-well microtiter plates, and it was quantified using a hu3F8-IgG1 standard curve ranging from 1.25 to 40 ng/ml. Diluted serum samples were allowed to bind to the ELISA plates, followed by reaction with peroxidase conjugated goat antibody to human IgG Fc. Color reactions were developed using hydrogen peroxide and chromogen o-phenylenediamine. Optical density was read with ELISA plate reader at 490 nm. Based on the standard curve, serum hu3F8 for each sample was calculated in ng/mL. Pharmacokinetic analysis was carried out by non-compartmental analysis of the serum concentration-time data using WinNonlin software program (Pharsight Corp., Mountain View, CA).
Antitumor effect in human tumor xenografts
All animal procedures were performed in compliance with Institutional Animal Care and Use Committee (IACUC) guidelines. The immunodeficient mice colony BALB-Rag2−/−IL2R-γc-KO (DKO) was maintained at MSK under sterile conditions, and provided with Sulfatrim food. In vivo experiments were performed with 6–10 week old mice, and each treatment group had 5 mice. Human peripheral blood mononuclear cells (PBMC) were prepared as previously described (23), and they had ~ 15% CD56+ NK cell subpopulations.
Fresh PBMC (4 × 106 cells) were mixed with IMR-32-Luc cells (6 × 106 cells) in Matrigel (BD Biosciences) and implanted subcutaneously (sc) into the right flank of DKO mice. Treatment schedules were detailed in the Results section. Tumor size was measured weekly by calipers, and tumor volumes calculated using the formula V = 0.5 (length × width × width). % tumor growth was the change in growth from pretreatment size.
Statistics
Differences between samples indicated in the figures and tables were tested for statistical significance (P < 0.05) by Student t-test.
Results
Biochemical purity and stability of the 4 constructs
The antibodies hu3F8-IgG1 and hu3F8-IgG1-DEL were produced in CHO-S cells, whereas hu3F8-IgG1n and hu3F8-IgG1n-DEL were produced in GnT1-deficient CHO cells. Antibodies were purified by protein A affinity chromatography, and tested by SEC-HPLC with 99%, 90%, 92%, and 98% purity, respectively. On reducing SDS-PAGE, heavy chain and light chain of all four antibodies migrated to the same expected molecular size (data not shown). All four antibodies remained stable after multiple freeze and thaw cycles when assayed by SDS-PAGE and SEC-HPLC (data not shown).
GnT1-deficient CHO lines lost reactivity with PHA
Loss of PHA binding could demonstrate that these GnT1-deficient CHO cells had the GnT1-deficient glycosylation phenotype (24). Thus, wild-type CHO-S cells or GnT1-deficient CHO cells were analyzed by flow cytometry with FITC-conjugated L-PHA. As shown in Fig. 1, wild-type parental and hu3F8-IgG1 stably transfected CHO-S cells had PHA binding (MFI at 50–60), whereas hu3F8-IgG1n- and hu3F8-IgG1n-DEL-producing GnT1-deficient CHO cells lost most of PHA binding (MFI at 8–10, control baseline MFI set at 5). Polymerase chain reaction (PCR) of genomic DNA from these GnT1-deficient CHO cells with primers flanking the single exon encoding MAGT1 gene produced no detectable PCR band, whereas a single band at the right size (~1.4 kb) was detected from wild-type CHO-S cells (24) (data not shown), suggesting GnT1-deficient CHO cells either totally or partially lost MAGT1 gene segment.
Figure 1. GnT1-deficient CHO lines lost reactivity with PHA.
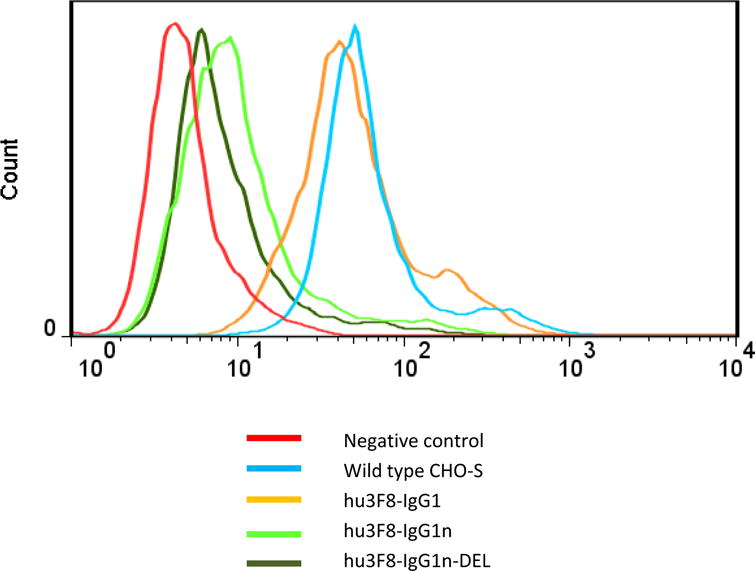
FACS histograms of L-PHA binding to CHO cells, with wild-type CHO-S cells, and CHO cells expressing hu3F8-IgG1, hu3F8-IgG1n and hu3F8-IgG1n-DEL were shown. Cells without adding FITC-PHA were used as negative control for each cell lines (baseline MFI set at 5). At least two independent experiments were performed, with one representative set of data was presented.
Binding kinetics by SPR
Binding kinetics of the four antibodies to human recombinant FcRs were compared by SPR using Biacore T-100, with different FcRs coated onto CM5 chips. One primary goal of our study was to compare kinetics that seemed critical for clinical efficacy. The method that gave the best fit was chosen. The two-state reaction model fit medium-ranged affinities best (as shown in Suppl Fig S1). In these studies, after the initial fast-association phase, a slow increase in binding continued, suggestive of a two-state mechanism, instead of a flat plateau (typical for 1:1 model), presumably due to the potential multivalent binding and/or conformational changes on the chip surface. However, when the overall affinity was too low (KD ≥ 1 uM, see footnote of Table 1), the two-state kinetics analysis started losing accuracy. In these cases, the data were also fit using the steady-state (SS) equilibrium method. As expected, hu3F8-IgG1 had higher overall affinity KD for CD16A(158V) than CD16A(158F), and higher KD for CD32A(131H) than CD32A(131R) (Table 1). The inhibitory CD32B had lower affinity than their activating counterparts, translating into a more favorable activating: inhibitory (A:I) ratio.
Table 1.
Hu3F8-IgG1: Affinities for FcRs*
| ka1 (1/Ms) | kd1 (1/s) | ka2 (1/s) | kd2 (1/s) | KD (M) | |
|---|---|---|---|---|---|
| CD16A(158V) | 1.34E+05 | 2.12E-01 | 1.05E-02 | 8.97E-03 | 7.26E-07* |
| CD16A(158F) | – | – | – | – | 2.09E-06** |
| CD16B | – | – | – | – | 2.03E-06** |
| CD32A(131R) | – | – | – | – | 7.86E-07** |
| CD32A(131H) | – | – | – | – | 5.41E-07** |
| CD32B | – | – | – | – | 2.40E-06** |
The results were fitted either with Two-State Reaction: A+B=AB=AB*, KD = (kd1/ka1)/(1 + ka2/kd2),
or with steady-state equilibrium analysis if the affinity was too low (KD ≥ 1 uM) when determined by Two-State Reaction method. For CD32A(131H), where the KDs were near identical or identical when analyzed by either steady-state equilibrium or Two-State Reaction analysis, only the steady-state equilibrium result was shown for comparability with that of CD32A(131R).
The comparison between the four antibodies was shown in Table 2 (the data were fitted with either two-state reaction or steady-state as noted), in which we used low-affinity polymorphic alleles (CD16A-158F and CD32A-131R, respectively) as reference. GnT1−/− CHO cell-produced antibodies (IgG1n and IgG1n-DEL) contained only mannose and acetylglucosamine, gaining affinity preferentially for CD16, but not for CD32B, resulting in higher A:I FcγR affinity ratios than parental IgG1. In contrast, IgG1-DEL mutant gained affinity for both CD16 and CD32B, resulting in A:I ratio comparable to parental. IgG1n-DEL mutant gained more affinity for CD16 but not for CD32B, resulting in highest A:I ratio. In Table 2, only the A:I ratio of CD16A-158V/CD32B was calculated, although the trends were similar irrespective of any of the three activating CD16 receptors (CD16A-158V, CD16A-158F or CD16B). The data were also fit with the two-state reaction only, and shown in Supplemental Table S1. Although the numbers were different, the overall trends were still the same, especially for A:I ratio.
Table 2.
Affinities relative to hu3F8-IgG1*
| Affinity for CD16 | Affinity for CD32 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Antibody | FcR | n | Relative to hu3F8-IgG1 on CD16A(158F) | SD | P value | FcR | n | Relative to hu3F8-IgG1 on CD32A(131R) | SD | P value | A:I Ratioˆ |
| hu3F8-IgG1 | CD16A(158V) | 3 | 3 | 0.2 | 0.001 | CD32A(131R) | 3 | 1.0** | 0.1 | 9 | |
| CD16A(158F) | 3 | 1** | 0.1 | CD32A(131H) | 3 | 1.3** | 0.1 | 0.01 | |||
| CD16B | 3 | 1** | 0.2 | CD32B | 3 | 0.3** | 0.0 | ||||
| hu3F8-IgG1n | CD16A(158V) | 4 | 11 | 1.7 | 0.002 | CD32A(131R) | 4 | 0.5** | 0.1 | 0.01 | 35 |
| CD16A(158F) | 4 | 4 | 0.7 | 0.002 | CD32A(131H) | 4 | 0.6** | 0.1 | 0.003 | ||
| CD16B | 4 | 2** | 0.1 | 0.01 | CD32B | 4 | 0.3** | 0.1 | NS | ||
| hu3F8-IgG1-DEL | CD16A(158V) | 3 | 25 | 5 | 0.01 | CD32A(131R) | 3 | 1.4** | 0.1 | 0.01 | 18 |
| CD16A(158F) | 3 | 16 | 3 | 0.01 | CD32A(131H) | 3 | 1.3** | 0.2 | NS | ||
| CD16B | 3 | 15 | 1 | 0.002 | CD32B | 3 | 1.4** | 0.3 | 0.02 | ||
| hu3F8-IgG1n-DEL | CD16A(158V) | 6 | 63 | 16 | 0.0002 | CD32A(131R) | 4 | 0.3** | 0.1 | 0.003 | 165 |
| CD16A(158F) | 6 | 37 | 9 | 0.0002 | CD32A(131H) | 4 | 0.3** | 0.2 | 0.0002 | ||
| CD16B | 6 | 14 | 4 | 0.0004 | CD32B | 4 | 0.4** | 0.1 | NS | ||
Affinity reference highlighted in grey.
These results were fitted with steady-state equilibrium analysis as noted in the footnote of Table 1. All others were fitted with Two-State Reaction.
A:I ratio = Activating: inhibitory = (relative KA on CD16-158V)/(relative KA on CD32B).
Tumor cytotoxicity in vitro
To examine the cytotoxic potency of these four antibodies for individual activating FcγR (CD16A-158V or CD32A-131H) in the absence of inhibitory FcγR (CD32B), NK92MI cells that do not carry human FcRs were stably transfected with either of these human high affinity FcγRs, and ADCC was measured using a standard 4-hr 51Cr release assay. A representative panel of human tumor cell lines, including neuroblastoma (LAN1, IMR-32, and SKNCM), Ewing’s sarcoma (TC71 and SKELP), SCLC (NCI-H524, NCI-H69, and NCI-H82), and melanoma (M14 and SKMEL1), was tested for cytotoxicity, and the potencies relative to hu3F8-IgG1 (EC50 for IgG1/EC50 for IgG1 variants) were summarized in Fig, 2. Both IgG1n and DEL mutants had significantly enhanced killing potency when engaging CD16A-158V compared to parental IgG1 (Fig. 2A). In contrast, DEL mutant maintained potency, while IgG1n lost potency, when engaging CD32A-131H (Fig. 2B). All the ADCC results were consistent with the affinity data summarized in Table 2. Interestingly, both IgG1n and DEL mutants had decreased CMC activity, whereas IgG1n-DEL lost almost all of its CMC activity (Fig. 2C).
Figure 2. Potency of ADCC and CMC relative to hu3F8-IgG1.
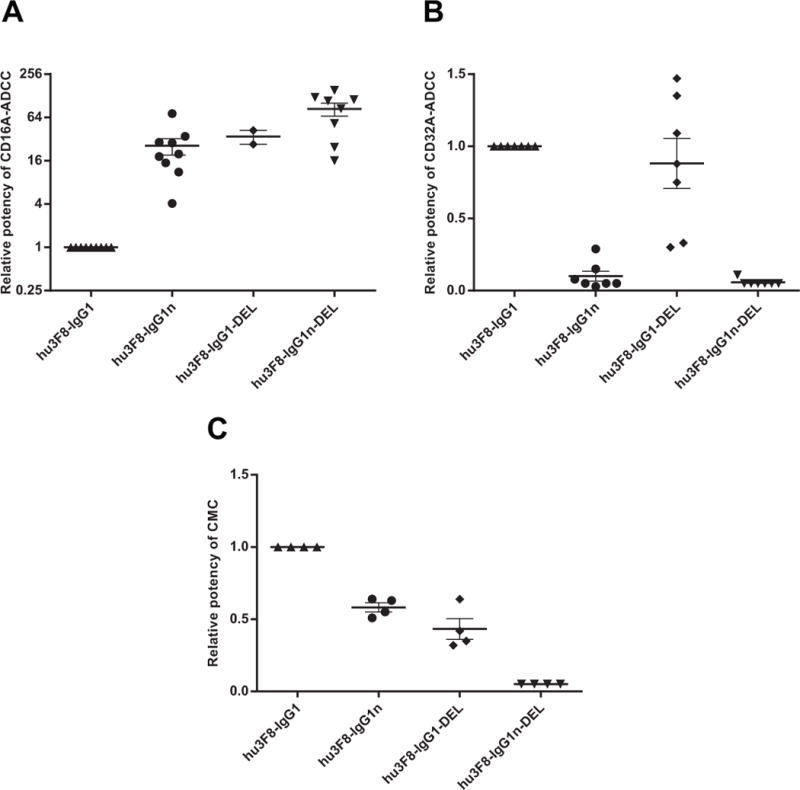
Dot plots shown the relative potency (EC50 for IgG1/EC50 for IgG1 variants for each cell line) of ADCC for CD16A(158V) (A) and CD32A(131H) (B); and CMC for human serum (C). E:T = 20:1 for ADCC. Each dot represents one cell line. At least two different cell lines were assayed for each IgG1 variants. Bars denote Mean ± SEM. Relative potency below detection level was assigned as ≤ 0.05.
Pharmacokinetics (PK) of antibodies in mice
Serum pharmacokinetic analysis was performed in nude mice after intravenous injections of these antibodies, where serum antibody levels were quantified by ELISA assay. As shown in Table 3 and Supplemental Fig. S2, while IgG1, IgG1n, and IgG1-DEL had comparable serum half-life (t1/2), Cmax, AUC, and clearance (Cl); IgG1n-DEL had markedly shorter serum half-life, lower AUC, and faster clearance. In contrast, all four antibodies had similar binding affinity to both human FcR(n) and mouse FcR(n) (Supplemental Table S2), suggesting other mechanisms might be involved in the inferior pharmacokinetics of IgG1n-DEL in mice.
Table 3.
Pharmacokinetics of antibodies in nude mice*
| Antibody | t1/2 (hr) | Cmax (ug/mL) | Cmin (ug/mL) | AUC (hr.ug/ml) | Vz (ml) | Cl (mL/hr) |
|---|---|---|---|---|---|---|
| hu3F8-IgG1 | 17 | 55 | 0.0 | 1171 | 2.1 | 0.1 |
| hu3F8-IgG1n | 22 | 69 | 0.1 | 902 | 3.5 | 0.1 |
| hu3F8-IgG1-DEL | 31 | 84 | 1.5 | 2029 | 2.1 | 0.0 |
| hu3F8-IgG1n-DEL | 2 | 103 | 0.0 | 293 | 0.9 | 0.3 |
100 ug iv per mouse at time 0. Cmax measured at 30 minutes post injection.
Antitumor effect in xenografted mice
For in vivo therapy studies, BALB-Rag2−/−IL2R-γc-KO (DKO) mice were used for xenografting human tumors (26). Using a humanized mouse xenograft models (23)(sc effector cells plus sc tumors to simulate effector cells residing within tumor), iv antibodies showed high activity against established tumors. No clinical toxicities were observed. IMR-32-Luc cells were mixed with fresh PBMC (unactivated, from buffy coat) and planted subcutaneously. On day 7, treatment with antibodies (50 ug iv, twice per week for 4 wk) was started and tumor size was measured. As shown in Fig. 3, human IgG control antibody palivizumab failed to shrink tumors. Although the antitumor effect of the IgG1, IgG1-DEL, and IgG1n-DEL forms of hu3F8 were similar, but modest, hu3F8-IgG1n was significantly more effective (p < 0.05). The experiment was repeated once with similar settings, and similar results were observed.
Figure 3. Efficacy of the antibodies in humanized DKO mice.
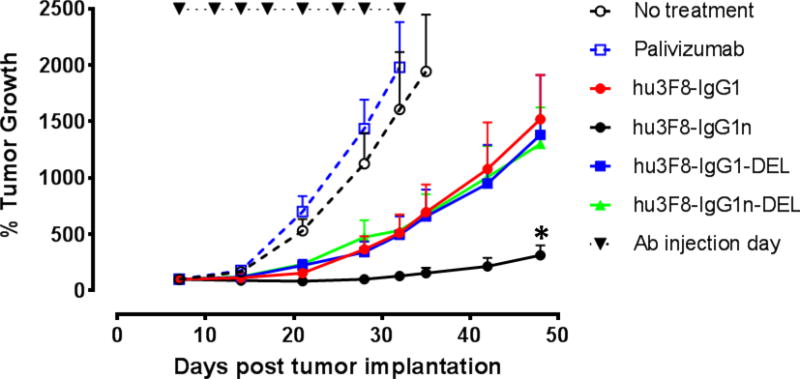
% tumor growth of IMR-32 neuroblastoma was shown. Treatment schedules, doses of antibodies were detailed in the Methods and Results. Data shown as mean + SEM (n = 5); * P < 0.05 determined by Student t test when IgG1n group was compared with other three treatment groups, respectively. Two independent experiments were performed, with one representative set of data was presented.
Discussion
Cancer immunotherapy using human IgG relies on its Fc to mediate ADCC. Affinity of Fc for CD16A (FcγRIIIA) receptor on white cells can be substantially enhanced through either site-directed mutagenesis of the Fc or expression of IgG in GnT1−/− CHO cell lines. However, they have different effects on the affinity for CD32, especially on the inhibitory FcγR allele CD32B, translating into a superior A:I ratio when using antibodies generated with the GnT1−/− approach, which gives rise to a loss of fucose and a gain of Man5-GlcNAc2 (27). Yet, in vitro FcR affinity and in vitro cytotoxicity assays cannot accurately predict the in vivo antitumor effect in mice models, nor the varying influence of different Fc modifications on their in vivo pharmacokinetics. For hu3F8-IgG1n-DEL, despite its highest A:I FcγR affinity ratio, its fast clearance, and low AUC (Table 3), presumably because of lower stability of the antibody in vivo, and higher affinity for FcR-bearing auxiliary cells circulating in the blood, appeared to negate all the advantages of enhanced FcγR affinities. The fact that hu3F8-IgG1n-DEL lost nearly all of its CMC capability (Fig. 2C) might also partially contribute to its inferior in vivo antitumor effect.
Besides the GnT1−/− phenotype, defucosylation alone (FUT8−/−) (28) and bisected oligosaccharides (29) are other examples where changes in the Fc oligosaccharides have led to substantial changes in the affinity of IgG1 for the FcγR. Removing fucose from asparagine 297 of the heavy chain results in a 5-fold increase in the affinity of Fc for CD16A, and a 2- to 11-fold improvement in the EC50 in ADCC assays (30). Affinities for CD32B and for the 131R allele of the CD32A also increase slightly, but not for the 131H allele (31). Binding to CD64(FcγRI), C1q and FcR(n) are unaffected (31). Human NK cells bearing CD16A stimulated with a defucosylated antibody to CD20 (obinutuzumab; Gazyva™) exhibit downstream signaling through the Vav-1, MAPK, and PI3K pathways, resulting in an increase in actin rearrangement and degranulation (32). The cytotoxic potential of each individual NK cell is improved resulting in better antitumor effects in mouse models (30, 33). Macrophages also express CD16A, and their ADCC may also benefit from defucosylated antibodies (2, 34). Because serum antibodies compete with fucosylated therapeutic antibodies for FcγRs (35), antibodies with enhanced affinity for CD16A have a definite affinity advantage in dwell time and productive signaling (36), especially when antigen density is low (37, 38). Based on superior clinical responses, obinutuzumab became FDA approved for CLL in 2013 (39, 40). Bisected carbohydrates can also be introduced into IgGs using the enzyme β1,4-N-acetylglucosaminyltransferase III to enhance affinity and ADCC activity (29). Bisected, afucosylated carbohydrates have been made with the additional Golgi α-mannosidase II enzyme in CHO cells (41), thereby enhancing their affinity for CD16A, as well as their in vitro and in vivo antitumor effects (42). Since the IgG produced in GnT1−/− CHO cells also lacks the core fucose residues, one might question the additional value of Man5-GlcNAc2 over defucosylation alone. Here we showed that antibodies from GnT1−/− CHO cells gained affinity for activating CD16, but not for inhibitory CD32B, thereby enhancing the A:I ratio, which translated into superior antitumor effects in vivo. In contrast, IgG-DEL affinities for both CD16 and CD32B were increased, resulting in an unfavorable A:I ratio. Glycoproteins containing high mannose structures bind the mannose receptor during clearance, resulting in a shorter serum half-life, and an adverse effect on antibody PK (43, 44). Unexpectedly in mouse studies, these high mannose IgG preparations had similar half-lives as wild-type IgGs (45), which was confirmed in our current study. High mannose containing IgGs bind to CD16A with increased affinity, enhancing ADCC functions while losing CMC capacity (46).
Besides CD16A and CD16B, CD32A is another key receptor on granulocytes and myeloid effectors in performing their ADCC functions (47) and its high affinity allele FCGR2A(131R) for murine mAb 3F8 was associated with a more favorable patient outcome (48–50). In our studies of mice models humanized with human PBMC, human granulocytes were removed during Ficoll gradient separation, and only monocyte-macrophages were recovered; hence the antitumor effect of granulocyte-ADCC could not be ascertained. Although the IgG1n antibodies showed decreased affinity for CD32A, the enhanced affinity by 2-fold for CD16B on granulocytes should compensate for the net ADCC ability of these IgG1n antibodies.
In summary, this study systematically compared two common ways to enhance affinity of Fc for CD16A (FcγRIIIA), through either GnT1-deficient glyco-engineering or site-directed mutagenesis of the Fc. Hu3F8-IgG1n produced in GnT1-deficient CHO cells turned out to have more promising potential as a therapeutic antibody than the parental hu3F8-IgG1 or its other Fc enhanced variants (summarized in Table 4), in terms of both in vitro and in vivo antitumor effects. This platform may be useful for enhancing antitumor potential of other IgG antibodies, and may have broad implications for therapeutic antibodies in cancer therapy.
Table 4.
Summary of Fc enhanced hu3F8
| hu3F8-IgG1 | hu3F8-IgG1n | hu3F8-IgG1-DEL | hu3F8-IgG1n-DEL | |
|---|---|---|---|---|
| KD (CD16A-158F)* | 1 | 4 | 16 | 37 |
| KD (CD16A-158V)* | 3 | 11 | 25 | 63 |
| KD (CD32A-131R)** | 1.0 | 0.5 | 1.4 | 0.3 |
| KD (CD32A-131H)** | 1.3 | 0.6 | 1.3 | 0.3 |
| A:I FcR ratioˆ | 9 | 35 | 18 | 165 |
| ADCC(CD16A-158V) | 1 | 26 | 35 | 84 |
| ADCC(CD32A-131H) | 1.0 | 0.1 | 0.9 | 0.1 |
| CMC | 1.0 | 0.6 | 0.4 | ≤0.05 |
| t1/2 | 17 | 22 | 31 | 2 |
| Cmax | 55 | 69 | 84 | 103 |
| AUC | 1171 | 902 | 2029 | 293 |
| In vivo antitumor effect | + | +++ | + | + |
Normalized to KD of hu3F8-IgG1 on CD16A-158F.
Normalized to KD of hu3F8-IgG1 on CD32A-131R.
A:I = Activating: inhibitory = (relative KA on CD16-158V)/(relative KA on CD32B).
Supplementary Material
Acknowledgments
We thank Dr. Mamoru Ito of Central Institute for Experimental Animals (Kawasaki, Japan) for providing the DKO mice, Dr. Gloria Koo, Hospital for Special Surgery (New York, NY) for her advice in handling DKO mice, and Dr. Cheng Liu of Eureka Therapeutics Inc., for his expert advice and service in providing the GnT1-deficient CHO cell line. We also thank Dr. Mahiuddin Ahmed for his expertise in antibody structural design.
Financial support: This study was supported, in part, by grants from the Band of Parents, Arms Wide Open Cancer Foundation, Kids Walk for Kids with Cancer NYC, Robert Steel Foundation, and Enid A. Haupt Chair Endowment Fund. Technical service provided by the MSK Small-Animal Imaging Core Facility was supported in part by the NIH Cancer Center Support Grant P30 CA008748.
Footnotes
Disclosure: NK Cheung and H Xu were named as co-inventors on a patent application for humanized anti-GD2 antibodies hu3F8 filed by Memorial Sloan Kettering Cancer Center. NK Cheung and MSKCC have financial interest in hu3F8 which is licensed to YmAbs Inc. The other authors declare they have no competing interests as defined by the Journal, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
- 1.Dahal LN, Roghanian A, Beers SA, Cragg MS. FcgammaR requirements leading to successful immunotherapy. Immunol Rev. 2015;268:104–22. doi: 10.1111/imr.12342. [DOI] [PubMed] [Google Scholar]
- 2.Braster R, O’Toole T, van Egmond M. Myeloid cells as effector cells for monoclonal antibody therapy of cancer. Methods. 2014;65:28–37. doi: 10.1016/j.ymeth.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 3.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–7. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 5.Kenkre V, Hong F, Cerhan JR, Lewis M, Sullivan L, Williams ME, et al. Fc gamma receptor 3A and 2A polymorphisms do not predict response to rituximab in follicular lymphoma. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. 2013;6:1. doi: 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niwa R, Hatanaka S, Shoji-Hosaka E, Sakurada M, Kobayashi Y, Uehara A, et al. Enhancement of the antibody-dependent cellular cytotoxicity of low-fucose IgG1 Is independent of FcgammaRIIIa functional polymorphism. Clin Cancer Res. 2004;10:6248–55. doi: 10.1158/1078-0432.CCR-04-0850. [DOI] [PubMed] [Google Scholar]
- 8.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 9.Hurvitz SA, Betting DJ, Stern HM, Quinaux E, Stinson J, Seshagiri S, et al. Analysis of Fcgamma receptor IIIa and IIa polymorphisms: lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin Cancer Res. 2012;18:3478–86. doi: 10.1158/1078-0432.CCR-11-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. FcgammaR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol. 2011;22:1302–7. doi: 10.1093/annonc/mdq585. [DOI] [PubMed] [Google Scholar]
- 11.Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 12.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 13.Oganesyan V, Damschroder MM, Leach W, Wu H, Dall’Acqua WF. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol Immunol. 2008;45:1872–82. doi: 10.1016/j.molimm.2007.10.042. [DOI] [PubMed] [Google Scholar]
- 14.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–73. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 15.Zhong X, Cooley C, Seth N, Juo ZS, Presman E, Resendes N, et al. Engineering novel Lec1 glycosylation mutants in CHO-DUKX cells: molecular insights and effector modulation of N-acetylglucosaminyltransferase I. Biotechnol Bioeng. 2012;109:1723–34. doi: 10.1002/bit.24448. [DOI] [PubMed] [Google Scholar]
- 16.Goetze AM, Liu YD, Zhang Z, Shah B, Lee E, Bondarenko PV, et al. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21:949–59. doi: 10.1093/glycob/cwr027. [DOI] [PubMed] [Google Scholar]
- 17.Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res. 2014;20:4036–46. doi: 10.1158/1078-0432.CCR-13-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobrenkov K, Cheung NK. GD2-targeted immunotherapy and radioimmunotherapy. Semin Oncol. 2014;41:589–612. doi: 10.1053/j.seminoncol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, et al. Murine Anti-GD2 Monoclonal Antibody 3F8 Combined With Granulocyte-Macrophage Colony-Stimulating Factor and 13-Cis-Retinoic Acid in High-Risk Patients With Stage 4 Neuroblastoma in First Remission. J Clin Oncol. 2012;30:3264–70. doi: 10.1200/JCO.2011.41.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metelitsa LS, Gillies SD, Super M, Shimada H, Reynolds CP, Seeger RC. Antidisialoganglioside/granulocyte macrophage-colony-stimulating factor fusion protein facilitates neutrophil antibody-dependent cellular cytotoxicity and depends on FcgammaRII (CD32) and Mac-1 (CD11b/CD18) for enhanced effector cell adhesion and azurophil granule exocytosis. Blood. 2002;99:4166–73. doi: 10.1182/blood.v99.11.4166. [DOI] [PubMed] [Google Scholar]
- 22.Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. OncoImmunology. 2012;1:477–86. doi: 10.4161/onci.19864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer immunology research. 2015;3:266–77. doi: 10.1158/2326-6066.CIR-14-0230-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Unligil UM, Rini JM, Stanley P. Independent Lec1A CHO glycosylation mutants arise from point mutations in N-acetylglucosaminyltransferase I that reduce affinity for both substrates. Molecular consequences based on the crystal structure of GlcNAc-TI. Biochemistry. 2001;40:8765–72. doi: 10.1021/bi015538b. [DOI] [PubMed] [Google Scholar]
- 25.Sondermann P, Jacob U. Human Fcgamma receptor IIb expressed in Escherichia coli reveals IgG binding capability. Biol Chem. 1999;380:717–21. doi: 10.1515/BC.1999.090. [DOI] [PubMed] [Google Scholar]
- 26.Andrade D, Redecha PB, Vukelic M, Qing X, Perino G, Salmon JE, et al. Engraftment of peripheral blood mononuclear cells from systemic lupus erythematosus and antiphospholipid syndrome patient donors into BALB-RAG-2−/− IL-2Rgamma−/− mice: a promising model for studying human disease. Arthritis Rheum. 2011;63:2764–73. doi: 10.1002/art.30424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–2. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 28.Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614–22. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- 29.Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol. 1999;17:176–80. doi: 10.1038/6179. [DOI] [PubMed] [Google Scholar]
- 30.Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70:4481–9. doi: 10.1158/0008-5472.CAN-09-3704. [DOI] [PubMed] [Google Scholar]
- 31.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–40. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 32.Liu SD, Chalouni C, Young JC, Junttila TT, Sliwkowski MX, Lowe JB. Afucosylated antibodies increase activation of FcgammaRIIIa-dependent signaling components to intensify processes promoting ADCC. Cancer immunology research. 2015;3:173–83. doi: 10.1158/2326-6066.CIR-14-0125. [DOI] [PubMed] [Google Scholar]
- 33.Gasdaska JR, Sherwood S, Regan JT, Dickey LF. An afucosylated anti-CD20 monoclonal antibody with greater antibody-dependent cellular cytotoxicity and B-cell depletion and lower complement-dependent cytotoxicity than rituximab. Mol Immunol. 2012;50:134–41. doi: 10.1016/j.molimm.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Herter S, Birk MC, Klein C, Gerdes C, Umana P, Bacac M. Glycoengineering of therapeutic antibodies enhances monocyte/macrophage-mediated phagocytosis and cytotoxicity. J Immunol. 2014;192:2252–60. doi: 10.4049/jimmunol.1301249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iida S, Kuni-Kamochi R, Mori K, Misaka H, Inoue M, Okazaki A, et al. Two mechanisms of the enhanced antibody-dependent cellular cytotoxicity (ADCC) efficacy of non-fucosylated therapeutic antibodies in human blood. BMC Cancer. 2009;9:58. doi: 10.1186/1471-2407-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nechansky A, Schuster M, Jost W, Siegl P, Wiederkum S, Gorr G, et al. Compensation of endogenous IgG mediated inhibition of antibody-dependent cellular cytotoxicity by glyco-engineering of therapeutic antibodies. Mol Immunol. 2007;44:1815–7. doi: 10.1016/j.molimm.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 37.Velders MP, van Rhijn CM, Oskam E, Fleuren GJ, Warnaar SO, Litvinov SV. The impact of antigen density and antibody affinity on antibody-dependent cellular cytotoxicity: relevance for immunotherapy of carcinomas. Br J Cancer. 1998;78:478–83. doi: 10.1038/bjc.1998.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 39.Cartron G, de Guibert S, Dilhuydy MS, Morschhauser F, Leblond V, Dupuis J, et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: final data from the phase 1/2 GAUGUIN study. Blood. 2014;124:2196–202. doi: 10.1182/blood-2014-07-586610. [DOI] [PubMed] [Google Scholar]
- 40.Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–10. doi: 10.1056/NEJMoa1313984. [DOI] [PubMed] [Google Scholar]
- 41.Ferrara C, Brunker P, Suter T, Moser S, Puntener U, Umana P. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol Bioeng. 2006;93:851–61. doi: 10.1002/bit.20777. [DOI] [PubMed] [Google Scholar]
- 42.Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res. 2013;19:1126–38. doi: 10.1158/1078-0432.CCR-12-0989. [DOI] [PubMed] [Google Scholar]
- 43.Jones AJ, Papac DI, Chin EH, Keck R, Baughman SA, Lin YS, et al. Selective clearance of glycoforms of a complex glycoprotein pharmaceutical caused by terminal N-acetylglucosamine is similar in humans and cynomolgus monkeys. Glycobiology. 2007;17:529–40. doi: 10.1093/glycob/cwm017. [DOI] [PubMed] [Google Scholar]
- 44.Wright A, Sato Y, Okada T, Chang K, Endo T, Morrison S. In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology. 2000;10:1347–55. doi: 10.1093/glycob/10.12.1347. [DOI] [PubMed] [Google Scholar]
- 45.Millward TA, Heitzmann M, Bill K, Langle U, Schumacher P, Forrer K. Effect of constant and variable domain glycosylation on pharmacokinetics of therapeutic antibodies in mice. Biologicals: journal of the International Association of Biological Standardization. 2008;36:41–7. doi: 10.1016/j.biologicals.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Q, Shankara S, Roy A, Qiu H, Estes S, McVie-Wylie A, et al. Development of a simple and rapid method for producing non-fucosylated oligomannose containing antibodies with increased effector function. Biotechnol Bioeng. 2008;99:652–65. doi: 10.1002/bit.21598. [DOI] [PubMed] [Google Scholar]
- 47.Kushner BH, Cheung NK. Absolute requirement of CD11/CD18 adhesion molecules, FcRII and the phosphatidylinositol-linked FcRIII for monoclonal antibody-mediated neutrophil antihuman tumor cytotoxicity. Blood. 1992;79:1484–90. [PubMed] [Google Scholar]
- 48.Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol. 2006;24:2885–90. doi: 10.1200/JCO.2005.04.6011. [DOI] [PubMed] [Google Scholar]
- 49.Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, et al. Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 2010;70:9554–61. doi: 10.1158/0008-5472.CAN-10-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheung NK, Cheung IY, Kramer K, Modak S, Kuk D, Pandit-Taskar N, et al. Key Role for Myeloid Cells: Phase II Results of Anti-G Antibody 3F8 Plus Granulocyte-Macrophage Colony-Stimulating Factor for Chemoresistant Osteomedullary Neuroblastoma. Int J Cancer. 2014 doi: 10.1002/ijc.28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.