

Abstract
Ion-pumping rhodopsins transfer ions across the microbial cell membrane in a light-dependent fashion. As the rate of biochemical characterization of microbial rhodopsins begins to catch up to the rate of identification in environmental and genomic sequence data sets, in vitro analysis of their light-absorbing properties and in vivo analysis of ion pumping will remain critical to characterizing these proteins. As we learn more about the variety of physiological roles performed by microbial rhodopsins in different cell types and environments, observing the localization patterns of the rhodopsins and/or quantifying the number of rhodopsin-bearing cells in natural environments will become more important. Here, we provide protocols for purification of rhodopsin-containing membranes, detection of ion pumping, and observation of functional rhodopsins in laboratory and environmental samples using total internal reflection fluorescence microscopy.
Keywords: rhodopsin, retinal, spectroscopy, proton pumping, TIRF microscopy
Introduction
Microbial rhodopsins are membrane-embedded pigment-protein complexes composed of a single polypeptide with seven transmembrane helices and one retinal chromophore (Brown, 2014). Sensory rhodopsins sense light and transmit a signal to the cell (Spudich, 2006), while ion-pumping rhodopsins transfer ions across the cell membrane in a light-dependent manner (Brown, 2014). If the ion translocated is H+, then the resulting ion gradient can contribute to the proton motive force (PMF) used to synthesize ATP or power flagellar rotation (Martinez et al., 2007; Walter et al., 2007). Microbial rhodopsins are generally assayed for their absorption and action spectra and ability to pump protons or other ions.
A variety of ion-pumping microbial rhodopsins have been characterized, including bacteriorhodopsin and halorhodopsin from the haloarchaea (proton and chloride pumps, respectively; Oesterhelt and Stoeckenius, 1973; Oesterhelt, 1995)), proteorhodopsin from marine Proteobacteria (proton pumps; Martinez et al., 2007; Steindler et al., 2011; Walter et al., 2007; Beja et al., 2001), several rhodopsins from the Flavobacteria (proton, chloride, and sodium pumps, respectively; Inoue et al., 2013, 2015; Yoshizawa et al., 2012, 2014), and actinorhodopsin from Actinobacteria (proton pump; Keffer et al., 2015a). Although the bacteriorhodopsins and halorhodopsins were initially identified and characterized biochemically in the 1970s, the other rhodopsins were first identified in metagenomic data sets or genome sequences, followed by biochemical analysis often in heterologous expression systems. Comparison of the sequences of this diverse family of proteins, combined with targeted mutagenesis in some, has identified the amino acids necessary for proton pumping, those necessary for spectral tuning, the conserved lysine residue required for covalent attachment of the cofactor (retinal), and amino acids involved in interactions with carotenoid antennae. This wealth of biochemical data allows us to predict the functions and substrates of novel rhodopsins – to a degree.
Despite the quantity of biochemical information available, novel rhodopsins are continuously being identified in cultivars (Sharma et al., 2009; Inoue et al., 2015; Harris et al., 2015; García-Martínez et al., 2015) and in metagenomic surveys (Atamna-Ismaeel et al., 2012; Sharma et al., 2008; Yutin and Koonin, 2012; Ugalde et al., 2011). The number of novel rhodopsins in uncultivated organisms vastly outnumbers those in cultivars (Brown, 2014; Boeuf et al., 2015). In the absence of cultivable organisms encoding these genes, they can be characterized biochemically in heterologous expression systems such as E. coli. However, E. coli cannot synthesize retinal or any of its precursors, so the retinal must be provided exogenously in the medium (Martinez et al., 2007; Beja et al., 2001), or the entire carotenoid biosynthesis pathway must be supplied in trans (Keffer et al., 2015a, 2015b).
Predictions of function cannot be made based solely on sequence data (Brown, 2014; Ugalde et al., 2011). A change in a single amino acid can shift the proteorhodopsin absorption spectrum (Man et al., 2003; Man-Aharonovich et al., 2004) or convert an outward-directed ion pump to an inward-directed ion pump (Kawanabe et al., 2009; Hasemi et al., 2015). Additionally, biochemical function may not predict physiological role(s). Although the proteorhodopsin in marine Dokdonia spp. pumps protons in response to light, growth in the light does not result in a higher ATP/ADP ratio, indicating limited contribution of the rhodopsin to the ATP generated by the PMF (Gomez-Consarnau et al., 2015). Instead, growth in the light is enhanced because the PMF-dependent TonB-type vitamin B1 transporters are more active in the light (Gomez-Consarnau et al., 2015). Additionally, the freshwater actinorhodopsin was predicted to be a proton pump (Sharma et al., 2009). When actinorhodopsin is expressed in E. coli that also synthesizes retinal, E. coli pumps protons in the light, indicating that actinorhodopsin is indeed a light-activated proton pump (Keffer et al., 2015a). However, spectroscopic analysis and proton-pumping assays in a native actinorhodopsin host, Rhodoluna lacicola, demonstrate that despite high levels of expression and protein production, R. lacicola does not pump protons in the light because it does not synthesize retinal (Keffer et al., 2015a). Without in vivo analyses, both of these rhodopsins would have been predicted to be proton-pumping rhodopsins that contribute to ATP synthesis in their respective hosts.
In this unit, we describe methods for purification of rhodopsin-containing membranes, detection of light-activated ion pumping, and use of total internal reflection fluorescence (TIRF) microscopy to visualize rhodopsins in vivo.
Basic Protocol 1
Membrane purification to enrich rhodopsin fraction
The most visible, fundamental property of microbial rhodopsins is their color, due to the light-absorbing properties of the pigment-protein complex. However, the molar absorptivity constant of rhodopsin is relatively low, and rhodopsin absorption maxima are in the same general range as those of carotenoid pigments (450-550 nm), which often co-occur with rhodopsins (McCarren and DeLong, 2007). Additionally, scattering by intact cells decreases the accuracy of the absorption spectrum, especially since the absorption of rhodopsin may be low relative to scattering (Fig. 1A). To clarify the solution and concentrate the rhodopsin, it is necessary to purify the membrane fraction (Fig. 1B). If possible, use a similar membrane preparation of uninduced cells, or cells not expressing the rhodopsin, to subtract as a baseline for absorption (Fig. 1C).
Figure 1. Absorption spectra of rhodopsin-expressing E. coli cells.
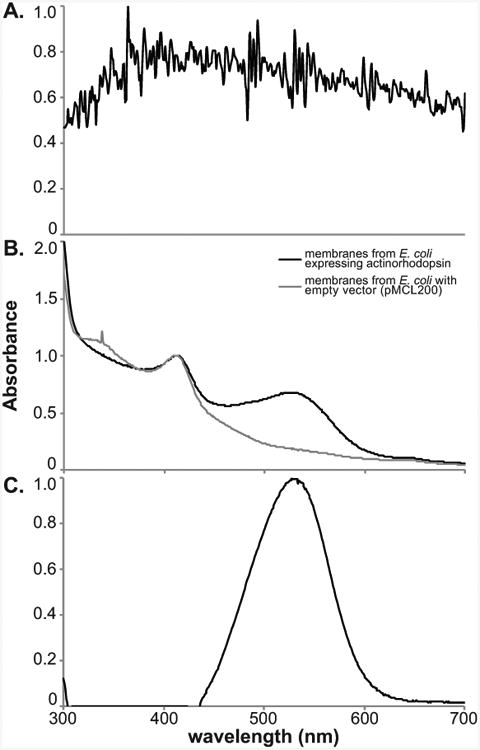
A Absorption spectrum of whole cells (spectrum normalized to highest peak). B. Absorption spectra of E. coli expressing retinal biosynthetic genes and actinorhodopsin (black line) and E. coli with the equivalent empty vectors (gray line). Both spectra are normalized to the peak at 412 nm. C. Difference spectrum of the two spectra in (B), with the rhodopsin peak now clearly visible. Spectrum is normalized to the highest peak, at 528 nm.
In this protocol, cells are lysed in a high-osmotic-strength sucrose buffer with lysozyme, then sonicated in a high-salt buffer. Hydrophobic components, including membrane proteins, are pelleted by centrifugation and resuspended in detergent.
Materials
“Sweet” buffer (see recipe)
Lysozyme: 50 mg mL-1 lysozyme in sddH2O, freshly prepared
“Salt” buffer (see recipe)
BOG buffer: 3% beta-octylglucopyranoside (BOG) in 10 mM HEPES, pH 7.1
Probe sonicator
Orbital shaker
Centrifuge and microcentrifuge
Vortexer
-
Grow cells. Grow the cells of interest to mid- to late-exponential phase.
Note that volumes here are optimized for a pellet from ∼500 mL Actinobacterial culture or ∼50 mL E. coli culture. For E. coli expressing rhodopsin from the pMCL200 vector and the retinal biosynthetic pathway cloned into pBAD-TOPO vector (Keffer et al., 2015b), grow cells overnight at 37°C with shaking at ∼200 rpm, in medium amended with arabinose (0.02 to 0.2%), chloramphenicol (34 μg mL-1) and ampicillin (100 μg mL-1). For R. lacicola, grow for several days at 28°C in 3 g L-1 NSY medium (Hahn et al., 2003) in the light. Conditions for optimal growth of other organisms need to be determined empirically.
-
Harvest cells by centrifugation.
For E. coli, centrifugation at room temperature for 10 min. at 5000 rpm is sufficient. For Actinobacteria or other small cells, longer centrifugation times will likely be necessary.
Wash cells. Resuspend cells in an equal volume of sterile, double-distilled water, then centrifuge again (2×).
Osmotic lysis. Resuspend cell pellet in 5 mL “sweet” buffer, add 1 mL lysozyme and incubate for 1h at 37°C with shaking.
Centrifuge cells (5000 rpm, 20 min., 4°C) and discard supernatant.
Sonication. Add 5 mL “salt” buffer to cell pellet. On ice, sonicate with a probe sonicator, [10s ON, 20s OFF] at 60% for 5 min.
-
Collect membrane fraction. Transfer solution to multiple 1.5 mL microcentrifuge tubes and centrifuge at 15,000 rpm, 30 min., 4°C.
If available, a higher speed centrifuge can be used.
-
Remove supernatant and discard.
Supernatant should be clear. Pellet will likely be lighter at the bottom, and darker at the top. The darker “film” has the highest concentration of rhodopsins. Remove film by carefully pipetting with a P200, and transfer to a clean microcentrifuge tube.
If you are purifying membranes from heterologous expression of a rhodopsin in E. coli, the pellet will be light pink at the bottom and darker pink at the top, where rhodopsins are enriched. Pelleted membranes from R. lacicola will appear light red at the bottom and dark red at the top. If your cells synthesize other carotenoids, the pellets will likely be a different color. However, the rhodopsin-enriched membrane fraction should be at the top of the pellet. In addition, although the darker “film” at the top of the pellet has the most rhodopsins, the lighter-colored pellet may also contain some rhodopsins, though usually less concentrated, as well as other membrane proteins.
-
Resuspend membrane film in BOG buffer.
A good rule of thumb is to use 50 μL BOG buffer in each microcentrifuge tube to resuspend the membrane film.
Combine all resuspended membrane fractions. Place on a vortexer and vortex overnight in the dark at 4°C.
-
Centrifuge membrane fraction at 10,000 rpm, 10 min., 4°C. Retain the colored supernatant and discard any pellet.
The colored supernatant contains solubilized rhodopsin. Any pellet that forms should not be colored and can be discarded. Solubilized rhodopsins should be stable for several weeks to months if kept at low temperature (4°C) in the dark.
-
Important note: if using SDS-PAGE to monitor protein recovery at different stages of the purification process, do not boil protein samples prior to loading in the gel.
Mix protein samples with loading buffer containing denaturing agents and incubate for 1 hour at room temperature. If the sample is boiled, rhodopsins will denature, then aggregate, and will remain in the stacking gel.
Basic Protocol 2
Monitoring proton pumping in rhodopsin-containing cells
If the rhodopsin contributes to the proton gradient across the cell membrane, its activity will lower the pH of an unbuffered solution in the light (Fig. 2). Similarly, changes in Na+ or Cl- concentration result in changes in pH (Inoue et al., 2015), though the assay buffer may need a different composition in this case. This method has been used to demonstrate that microbial rhodopsins pump protons since the 1970s (Oesterhelt and Stoeckenius, 1973; Beja et al., 2000; Martinez et al., 2007; Man et al., 2003; Wang et al., 2003; Walter et al., 2007; Keffer et al., 2015a; Gomez-Consarnau et al., 2015) and can also be used to demonstrate that they pump Na+ or Cl- ions (Inoue et al., 2013; Yoshizawa et al., 2014; Lanyi and Oesterhelt, 1982). Negative controls include assaying ion pumping in the presence of proton ionophores such as carbonyl cyanide m-chlorophenylhydrazone (CCCP), which disrupt the transmembrane proton gradient, or tetraphenylphosphonium bromide (TPP+), for Na+ or Cl- pumps (Oesterhelt and Stoeckenius, 1973; Schobert and Lanyi, 1982; Inoue et al., 2013; Yoshizawa et al., 2014).
Figure 2. Proton pumping by E. coli producing retinal and actinorhodopsin.
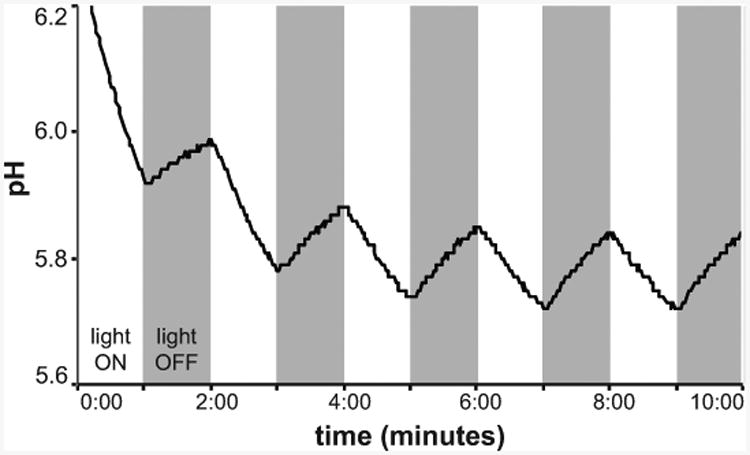
The pH decreases when the light is on, indicating outward transport of protons by the rhodopsin.
Materials
Medium appropriate for growing rhodopsin-expressing cells
Pumping buffer, freshly prepared (see recipe)
10 mM NaCl
10 mM MgCl2
100 μM CaCl2
Programmable timer (e.g. Leviton LT112 Digital Plug-In Timer)
250W halogen lamp (e.g. Utilitech 250-Watt Portable Work Light)
magnetic stir plate and small magnetic stir bars (5-8 mm × 1.5 mm)
micro pH electrode (for sample sizes < 1 mL, e.g. Mettler Toledo inLab Micro pH electrode)
data logger (must be able to connect to pH electrode and store time and pH data, e.g. Sper Scientific pH SD card data logger)
foil and transparent plastic shield
“floater” for microcentrifuge tubes (e.g. Ted Pella floating tube rack, item no. 20831-8, or USA Scientific floating foam rack, item no. 9138-7540)
-
Grow microbial cells (∼50 mL for E. coli) under rhodopsin-expressing conditions until mid-exponential growth phase.
These conditions will vary depending on the strain. For E. coli heterologously expressing a rhodopsin and retinal biosynthetic genes, use the conditions for induction of expression. For strains that express rhodopsins in the light, grow in continuous light. For strains that express rhodopsins under starvation conditions, it may be necessary to first grow cells to a high density in rich medium, then transfer to low-nutrient conditions to induce rhodopsin expression. If cells do not synthesize retinal, retinal (Sigma-Aldrich catalog no. R2500) should be added to a final concentration of 10 μg mL-1. Prepare a stock solution of 50 mg mL-1 retinal in ethanol, and store at -20°C in the dark. Add retinal to concentrated cultures (step 3) and incubate for 2-3 h, then harvest cells by centrifugation and resuspend in fresh pumping buffer.
Wash cells. Centrifuge cells gently (∼3000 rpm). Decant supernatant and resuspend pellet in 5 mL freshly-prepared pumping buffer.
Wash cells. Centrifuge again; remove supernatant by decanting or pipetting, and resuspend pellet in 2-3 mL pumping buffer.
- Program timer (Leviton LT112) for duration of experiment:
-
Choose start timeCells should equilibrate in the buffer for ∼30 min., so start time should be at 30 min. or later.
- Set 4 cycles of two minutes ON, two minutes OFF
- Set 1 cycle of two minutes ON, 10 minutes OFF
- Repeat for length of experiment
-
- Set up pumping apparatus. (See Fig. 3)
- Plug 250 watt halogen lamp into timer
- Set up lamp so that it shines directly into a 400 mL beaker of water set on a stir plate (Fig. 3).
-
Use a piece of clear, hard plastic approximately 8″ × 10″ as a heat shield. Wrap the plastic shield in aluminum foil, leaving a space in the center uncovered so that light can shine through. Set the plastic shield between lamp and beaker, with the uncovered space oriented so that the sample is illuminated directly.This will prevent the water bath and pH electrode from heating too much.
- Transfer 1 mL of cells into a microcentrifuge tube with a small stir bar.
- Place microcentrifuge tube of cells in a floating rack in the beaker so that tube is in the center of the light from the lamp (see Fig. 3).
- Turn on stir plate so that the magnetic stir bar gently rotates in the bottom of the microcentrifuge tube.
- Plug Mettler Toledo inLab Micro pH electrode into Sper Scientific pH SD card data logger. Calibrate pH electrode and set temperature if necessary, per the manufacturer's instructions.
- Submerge pH electrode in the cell suspension.
- Wrap pH electrode and upper portion of beaker, if necessary, in foil so that light and heat does not reach the electrode (not pictured in Fig. 3)
- Sync time in data logger to the timer. Program data logger to record data every 2 seconds.
- Begin recording. Light should turn on for the first time after 30 min.
Figure 3. Schematic of setup for measuring proton pumping.
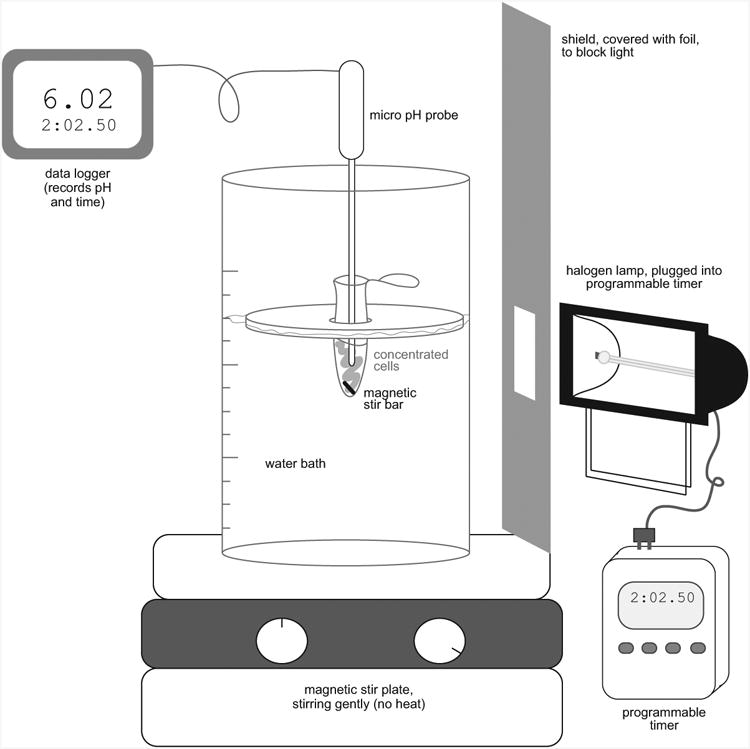
Necessary equipment and accessories for monitoring proton pumping in small (∼0.5 mL) volumes. This apparatus should be set up in a room that can be completely darkened. The foil-covered shield helps to prevent heating of the water bath; if necessary, the top of the water bath and pH probe can also be wrapped in foil to prevent heating (not pictured).
Basic Protocol 3
TIRF microscopy of rhodopsin-containing cells
Microbial rhodopsins have been visualized using atomic force microscopy, but the fluorescent yield of rhodopsins was thought to be too low to allow microscopy based on autofluorescence. However, because TIRF microscopy only excites molecules within 100-200 nm of the coverglass-sample interface, background fluorescence is low enough to allow visualization of rhodopsin fluorescence (Keffer et al., 2015b). This technique allows rhodopsin-containing cells in natural bacterial assemblages to be differentiated from other cells based on the wavelengths of light used to excite the fluorophores. TIRF microscopy may additionally enable fluorescence- based quantification of rhodopsins in individual cells, and more detailed analysis of subcellular localization.
We have successfully used this method to visualize rhodopsins both in pure culture and in environmental samples (Keffer et al., 2015b). Samples may be fixed with paraformaldehyde and stored at 4°C until processing, if necessary. Environmental samples should be counterstained with 4′,6-diamidino-2-phenylindole (DAPI), a DNA stain, to differentiate between autofluorescent particles and real cells. All cells are adhered to a gelatin-coated coverglass prior to microscopy.
This protocol includes instructions for all steps, including preparation of gelatin-coated coverslips, adhering cells to gelatin, microscopy, and image analysis.
Materials
Slide prepared as described in Support Protocol 1 and sample(s) prepared as described in Support Protocols 2-4.
Microscope: Fluorescence microscopy system capable of TIRF. Our system is a lab-built Zeiss Observer. A1 microscope. For imaging of bacteria, we use a 100/1.46-numerical-aperture (NA) oil immersion lens, with an additional 2 magnification after the tube lens.
Lasers:
405 nm (Coherent Cube), for imaging DAPI-stained cells
488 nm (Coherent Sapphire), for imaging carotenoid- and rhodopsin-containing cells
561 nm (Coherent Sapphire), for imaging rhodopsin-containing cells
641 nm (Coherent Cube), for imaging chlorophyll a-containing cells
Peltier cooled (-75°C) charge-coupled device (CCD) camera: for example, Andor iXON DU897 electron-multiplying CCD (eMCCD) camera, or Princeton Instruments Excelon ProEM512 CCD camera
Emission filter: 446/523/600/677 nm BrightLine® quad-band bandpass filter (Semrock, Inc., Part Number: FF01-446/523/600/677-25)
Computer equipped with NIH ImageJ
TIRF Microscopy
1. Focus lasers. For each laser, expand laser light to approximately 1-in. diameter, then focus onto the back aperture of the objective using a 500-mm achromatic doublet lens.
2. Focus on sample. Add a drop of immersion oil to the objective and mount slide on microscope stage with the coverslip facing down. If cells were stained with DAPI, use the 405-nm laser to illuminate the sample and focus. If cells were not stained, utilize the laser that will excite fluorophores in the greatest number of cells.
-
3. Image collection. Move slide to a new field of view. Set the number and frequency of image collection for a stack (e.g. 49 sequential images, collected every 30 ms). Illuminate with relevant lasers in order of decreasing wavelength and collect 49* sequential images for each excitation wavelength. Each image stack should be saved as an individual TIFF file in the same folder.
*The signal to noise ratio in the image, after averaging all of the images, will improve as a function of the square of the number of frames. A series of 49 frames will improve the signal to noise ratio by a factor of 7.
4. Analyze images of control samples. In this step, set the minimum background values for environmental samples for each wavelength imaged.
Average the sequential images
-
5. Using NIH ImageJ, open the folder of TIFF images of control samples.
Click on Image > Stacks > Z-project. In the drop-down “Projection Type” menu, choose Average Intensity, then click “OK”. This will generate a TIFF image that averages all 49 individual images. Save this image.
When choosing the number of frames to collect and the frequency of image collection, consider how immobile the cells are and whether anything is floating through the field of view. If one or more of the 49 individual images is significantly different from the others, it can be removed from the sequence prior to averaging. However, removing an image from the sequence will affect the signal to noise ratio.
-
6. Set minimum values for the background. Using the image generated in (b), click on Analyze > Measure. This will open a table that includes the minimum and maximum fluorescence values in the image. Under the Process > Math menu, choose “MIN” and set the minimum value to a value close to the minimum in the table. Vary this number until the sharpest image of the control cells is observed. Record this value as “MIN.” Save each image and repeat for control images for each excitation wavelength.
This step ensures that all samples have the same background levels and that those levels will maximize visibility of cells of interest at each wavelength. The minimum value will likely be different for each wavelength.
Analyze images of laboratory or field samples
-
7. Using NIH ImageJ, open the folder of TIFF images of control samples.
Average the sequential images. Click on Image > Stacks > Z-project. In the drop-down “Projection Type” menu, choose Average Intensity, then click “OK”. This will generate a TIFF image that averages all 49 individual images. Save this image.
- 8. Set minimum values for the background. Under the Process > Math menu, choose “MIN” and set the minimum value to the value recorded in step (1c) for each wavelength. Save the image.
- This step further reduces background noise.
Support Protocol 1
Preparation of gelatin-coated coverslips
Prior to TIRF microscopy, cells should be adhered to gelatin-coated cover glasses, so they cannot move during imaging. This support protocol describes how to coat cover glasses with gelatin.
Materials
Sterile, double-distilled water (sddH2O)
0.1N hydrochloric acid
95% ethanol
gelatin (Sigma C-6144)
chromium sulfate hydrate
Coverslips (Nunc 155409)
slide chamber (e.g. SPI slide mailer, SPI Item Number 01253-FA)
parafilm
Forceps
Sonicator
Orbital shaker
Laminar flow hood
Wash with water. Place coverslips in the slide chamber and cover with 25 mL sddH2O and gently invert the slide chamber for 2 min. Decant water and add 25 mL fresh sddH2O.
Sonicate the coverslips for 15 min (30 sec on, 10 sec off; amp 20%). Decant water, add 25 mL fresh sddH2O, and sonicate again.
Acid-wash. Decant the water and add 25 mL of 0.1 N HCl. Seal the top of the slide chamber with parafilm and incubate for 1 hour on the orbital shaker (∼30 rpm). Decant HCl into appropriate waste container.
Wash with water. Repeat step 1, 3×.
Wash with ethanol. Add 25 mL 95% ethanol to the slide chamber. Seal the top of the slide chamber with parafilm and incubate for 1 hour on the orbital shaker (∼30 rpm). Decant ethanol into appropriate waste container.
Wash with water. Repeat step 1, 2×.
Dry and sterilize. Remove coverslips from chamber and rinse with ethanol. Place at an angle in a rack. Air-dry, then sterilize under a UV light for 15 min.
-
Prepare gelatin. Boil 100 mL sddH2O. Add 0.5 g gelatin (Sigma C-6144) to the boiling water and stir until dissolved completely. Cool slightly (to ∼95°C), then add 10 mg chromium sulfate hydrate and stir to dissolve.
Make sure that gelatin is completely dissolved prior to adding the chromium sulfate. If the gelatin is not completely dissolved, the chromium sulfate will stick to the gelatin.
-
Coat the coverslips with gelatin. Cool gelatin solution to ∼55°C. Using forceps, dip coverslips into heated gelatin. Place coated slides in a rack and dry at an angle overnight in a laminar-flow hood or for ∼1 hour in a vacuum chamber.
Gelatin solution can be reused by loosening the lid and heating to 95°C. Allow to cool to ∼ 55°C and prepare coverslips as in Step 9.
Support Protocol 2
Preparation of unfixed, unstained bacterial cells for TIRF microscopy
If cells have been grown in an axenic culture and can be analyzed immediately, neither counter-staining with DAPI (a DNA stain) nor fixation is necessary. In this case, cells can merely be washed, concentrated, and placed on coverslips prior to microscopy.
Materials
sterile ddH2O
gelatin-coated cover glasses as described above
clean glass slides
nail polish or other sealant
Centrifuge
Forceps
Grow cells to be analyzed until mid-exponential growth phase under appropriate conditions.
-
Harvest cells. For E. coli, centrifuge at 5000 rpm, for 10 min.
Modify as appropriate for different cell types.
Wash and concentrate cells. Decant supernatant and resuspend pellet in sddH2O. Repeat 2×. After the last wash, resuspend the cells in a smaller volume of sddH2O to concentrate.
-
Adhere cells to gelatin. Place 50 μL concentrated cells onto a cleaned and gelatin-coated coverslip and incubate 10 min at RT.
Adhering cells to gelatin keeps them immobilized during microscopy. Cells must be resuspended in sddH2O, not medium or buffer, prior to additional to gelatin.
-
Prepare slide. Tilt the coverslip and remove as much liquid as possible from the coverslip with a pipette. Place the coverslip cell-side down on a clean glass slide and seal the coverslip to a slide for TIRF.
Nail polish of any color can be used as the sealant. Use different colors to help label different samples.
Support Protocol 3
Preparation of fixed cells from an axenic culture for TIRF microscopy
If cells have been grown in an axenic culture and cannot be analyzed immediately, counter-staining with DAPI (a DNA stain) is not necessary, but cells should be fixed to arrest growth and preserve fluorescence properties. In this case, cells can be washed, fixed in 2-4% paraformaldehyde, and stored at 4°C. They should then be washed, collected on a filter, and placed on coverslips prior to microscopy.
Materials
sterile ddH2O
Phosphate-buffered saline (PBS; see recipe)
20% paraformaldehyde in PBS
NucBlue Fixed Cell ReadyProbes Reagent (Life Technologies, catalog R37606)
gelatin-coated cover glasses as described above
25 mm 0.2 μm polycarbonate filters (EMD Millipore)
clean glass slides
nail polish or other sealant
Centrifuge
Filter holder with stainless steel support for 25 mm filters (e.g. EMD Millipore, item no. XX1002540)
Vacuum pump
Forceps
Grow cells to be analyzed until mid-exponential growth phase under appropriate conditions.
-
Harvest cells. For E. coli, centrifuge at 5000 rpm, for 10 min.
Modify as appropriate for different cell types.
Wash cells. Decant supernatant and resuspend pellet in 0.5 volumes 1× PBS.
Fix cells. Add 20% paraformaldehyde (PFA) to a final concentration of ∼4%. Incubate overnight at 4°C.
-
Stain cells. Dilute 10 μl fixed cells into 10 mL sddH2O, then filter the solution through a 25-mm 0.2 μm polycarbonate filter (EMD Milipore) until volume is ∼3 mL. Place 1 drop of NucBlue Fixed Cell ReadyProbes Reagent (Life Technologies, catalog R37606) per mL of sample into the solution and incubate for 10 min at room temperature.
NucBlue Fixed Cell ReadyProbes is a purified DAPI stain for nucleic acids.
Adhere cells to gelatin. Filter the remaining 3 mL onto the 0.2 μm polycarbonate filter. Carefully transfer filter to a gelatin-coated coverslip, sample side touching the gelatin. Incubate at room temperature for 10 min. Peel off the polycarbonate filter and discard.
-
Prepare slide. Add a 10 μL drop of sddH2O to a clean glass slide. Place the coverslip cell-side down on the drop of water and seal the coverslip to the slide
Coverslip should be placed on water, and not the reverse, so that water does not move when coverslip is inverted.
Nail polish of any color can be used as the sealant. Use different colors to help label different samples.
Support Protocol 4
Preparation of environmental sample for TIRF
For analysis of environmental samples that cannot be analyzed immediately, cells should be fixed to arrest growth and preserve fluorescence properties. Further, counter-staining with DAPI (a DNA stain) is necessary to ensure that all fluorescent objects have cellular material, since autofluorescent, abiotic particles are common in natural samples. In this case, samples should be fixed in paraformaldehyde and stored at 4°C until the return to the lab. They should then be collected on a filter, stained, and placed on coverslips prior to microscopy.
Materials
sterile ddH2O
Phosphate-buffered saline (PBS; see recipe)
20% paraformaldehyde in PBS
NucBlue Fixed Cell ReadyProbes Reagent (Life Technologies, catalog R37606)
gelatin-coated cover glasses as described above
25 mm 0.2 μm polycarbonate filters
clean glass slides
nail polish or other sealant
Filter holder with stainless steel support for 25 mm filters (e.g. EMD Millipore, item no. XX1002540)
Vacuum pump
Forceps
Collect and stain cells. Pre-filter environmental water sample if desired with a larger-pore size filter to remove eukaryotic cells and large debris. Add 20% PFA to sample to a final concentration of 1-4% PFA and store at 4°C overnight or until needed.
-
Stain cells. Filter fixed sample on to a 25-mm diameter 0.2 μm polycarbonate filter (EMD Milipore) until volume is ∼3 mL. Place 1 drop of NucBlue Fixed Cell ReadyProbes Reagent (Life Technologies, catalog R37606) per mL of sample into the solution and incubate for 10 min at room temperature.
NucBlue Fixed Cell ReadyProbes is a purified DAPI stain for nucleic acids.
Adhere cells to gelatin. Filter the remaining 3 mL onto the 0.2 μm polycarbonate filter. Carefully transfer filter to a gelatin-coated coverslip, sample side touching the gelatin. Incubate at room temperature for 10 min. Peel off the polycarbonate filter and discard.
-
Prepare slide. Place a 10 μL drop of sddH2O on a clean glass slide. Place the coverslip cell-side down on the water and seal the coverslip to the slide.
Coverslip should be placed on water, and not the reverse, so that water does not move when coverslip is inverted.
Nail polish of any color can be used as the sealant. Use different colors to help label different samples.
Reagents and Solutions
Phosphate-buffered saline (PBS)
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
“Sweet” buffer
0.4 M sucrose
0.075 M Tris pH 8.0
2.0 mM MgSO4
“Salt” buffer
50 mM Tris pH 7.6
10 mM MgSO4
0.8 M NaCl
Pumping buffer, freshly prepared
10 mM NaCl
10 mM MgCl2
100 μM CaCl2
Commentary
Background Information
Microbial rhodopsins (bacteriorhodopsins) were originally isolated from Haloarchaea as the “purple membrane” (Oesterhelt and Stoeckenius, 1974, 1973; Racker and Stoeckenius, 1974), followed by chloride-pumping halorhodopsins isolated from the same organisms (Lanyi and Oesterhelt, 1982). Following the discovery of rhodopsins in marine Proteobacteria (Beja et al., 2000), proteorhodopsins were extensively characterized by heterologously expressing them in E. coli (Beja et al., 2001; Man et al., 2003; Martinez et al., 2007; Walter et al., 2007). More recently, novel classes of rhodopsins have been discovered and characterized in the halophilic bacterium Salinibacter ruber (Balashov et al., 2005), the cyanobacteria Gloeobacter violaceus, Anabaena, and Mastigocladopsis repens (Balashov et al., 2010; Irieda et al., 2012; Hasemi et al., 2015), Gram-positive Actinobacteria (Sharma et al., 2008; Keffer et al., 2015a), and marine Flavobacteria (Inoue et al., 2015).
Characterization of the spectroscopic properties of rhodopsins is critical to understanding what wavelengths of light are utilized by individual rhodopsins. Further, as we learn more about the absorption spectra of novel rhodopsins, our ability to predict these properties will improve. However, because rhodopsins have a relatively low molar absorptivity constant and often co-occur with other light-absorbing molecules, absorption due to rhodopsins can only rarely be identified in whole cells. Instead, the membrane fraction must be purified and concentrated, reducing scattering and increasing the rhodopsin signal. Purifying membranes from native hosts, rather than (or in addition to) generating a tagged protein that can be heterologously expressed and easily purified, has two advantages. First, the rhodopsin signal can be analyzed in the context of other light-absorbing compounds and complexes in the in the native host membrane, and the relative intensity of the rhodopsin determined. This should also allow for identification and characterization of rhodopsins that have bound carotenoid antennae, similar to xanthorhodopsin (Lanyi and Balashov, 2008). Second, bulky protein tags may interfere with formation of rhodopsin multimers in the membrane.
Most microbial rhodopsins translocate ions across cell membranes, and light-induced ion-pumping by microbial rhodopsins has been measured in both whole cells and reconstituted vesicles (Racker and Stoeckenius, 1974). Outwardly-directed proton-pumping rhodopsins decrease the solution pH in the light, and their activity is abolished by proton ionophores or inhibitors that disrupt the transmembrane proton gradient (Martinez et al., 2007; Keffer et al., 2015a; Inoue et al., 2015). In contrast, Cl- or Na+ pumping rhodopsins increase the solution pH in the light. CCCP alone will not affect their activity, but CCCP in conjunction with TPP+ will abolish it (Inoue et al., 2015).
The negative controls for in vivo ion-pumping experiments should include a closely related strain that does not have a rhodopsin, or an empty-vector control, in the case of heterologous expression, to demonstrate that in the absence of a rhodopsin, no light-induced pumping is observed. A dark control (never exposed to light) may also be included, to quantify drift in the pH meter.
To better understand rhodopsins and their quantities and localizations in vivo, a method utilizing TIRF microscopy to analyze both laboratory and environmental samples was recently developed (Keffer et al., 2015b). Fluorescence microscopy of rhodopsins in both laboratory and natural samples has been rarely used due to the very low fluorescent yield of rhodopsins (Cheminal et al., 2013; Kochendoerfer and Mathies, 1996; Kralj et al., 2012). However, TIRF microscopy is not only capable of detecting rhodopsin fluorescence, but also of differentiating between unpigmented bacteria, carotenoid-producing bacteria, and rhodopsin-producing bacteria (Keffer et al., 2015b). Further, the subcellular localization of the rhodopsins can be observed in the relatively large E. coli cells, and presumably in other similarly-sized cells. Other techniques for fluorescence analysis of rhodopsins are reviewed in Alexiev and Farrens (2013).
TIRF microscopy only illuminates the 100-200 nm closest to the coverglass-sample interface. For this reason, it typically yields higher signal-to-noise ratios than confocal laser scanning microscopy (CSLM), because the imaging volume in a TIRF image is ∼1/10 that of a CLSM image, and less background scattering is visible. For a comparison of superresolution microscopy techniques, including both TIRF and CLSM, see the recent review by Schermelleh et al. (Schermelleh et al., 2010). For an introduction to TIRF microscopy, see (Axelrod, 2008).
Because cyanobacterial phycobiliproteins absorb and fluoresce in the same range as microbial rhodopsins, these pigments cannot be distinguished with this technique. To prevent false positives due to phycobiliprotein fluorescence, the 641-nm laser can be used to identify chlorophyll a (Chl a)-containing cells. The 561-nm laser should selectively excite rhodopsins and phycobiliproteins; fluorescence from phycobiliproteins can be excluded by excluding cells with Chl a, which fluoresces when excited with the 641-nm laser. The 488-nm laser will excite both rhodopsins and carotenoids, and is not critical unless identifying carotenoid-containing cells is relevant. The 405-nm laser will excite DAPI-stained cells (Keffer et al., 2015b).
In sum, the methods described here provide guidelines for concentrating rhodopsins from either native or heterologous hosts, observing proton pumping in vivo, and using TIRF microscopy to identify rhodopsin-hosting cells in natural or laboratory samples. These methods can be used to characterize microbial rhodopsins both in their native hosts and in heterologous hosts. Critical parameters to consider, expected results, and suggestions for troubleshooting are outlined below.
Critical Parameters
Protocol 1: Membrane purification
Please note that other membrane proteins and hydrophobic carotenoid compounds are likely to be purified along with the rhodopsins. However, this procedure concentrates rhodopsins up to 1000-fold (from ∼500 mL culture to 0.5 mL in the membrane preparation), strongly increasing the signal from the rhodopsin. Having a more concentrated protein in solution, without scattering due to whole cells, improves the resolution of the absorption spectrum measurably (see Fig.1A and Fig. 1B).
Lysis conditions may vary with cell type. Although the lysozyme treatment and osmotic lysis make this protocol broadly applicable to different cell types, difficult-to-lyse cells will likely need harsher lysis conditions.
Choice of detergent is critical. Some detergents bleach certain rhodopsins; for example, the beta-octyl glucopyranoside BOG) used in this protocol bleaches bacteriorhodopsins. Other detergent solutions that have been used to solubilize purified rhodopsins or membranes include dodecyl maltoside (0.1% dodecyl maltoside, 300 mM NaCl, 50 mM Tris-HCl, 150 mM imidazole, pH 7.0 (Kawanabe et al., 2009), or 0.03% dodecyl maltoside in HEPES, pH 7.4 (Luck et al., 2015)), sodium cholate (3% in 4M NaCl and 25 mM Tris, pH 8.0, for halophiles (Duschl et al., 1990)), octyl glucoside (1% in 300 mM NaCl, 10 mM imidazole, and 50 mMTris, pH 6.8 (Sineshchekov et al., 2005)), lauryl maltoside (1% in 300 mM NaCl, 10% glycerol, and 50 mM Tris, pH 8.0 (Jung et al., 2003)). Buffer composition varies depending on salt tolerance and optimal pH of the bacterial strain; imidazole is typically added for elution of His-tagged proteins from a nickel column.
Choice of buffer for the detergent solution can be important: the pH of the buffer may affect absorption spectrum of the rhodopsin.
Protocol 2: Proton pumping
This protocol works best if set up in a room or chamber that can be closed and left dark, such as a microscope room, so that light or temperature fluctuations as doors open or close will not affect the results.
This procedure may require some adjustment with regards to equilibration. We have had our best results after allowing the cells to equilibrate in the buffer for 30 min. in the dark prior to starting the assay.
Pumping solution should be freshly prepared so that pH is between 6-7. Different starting pH values may be necessary for different rhodopsin types.
The programmable automatic timer and data recorder eliminate human error and/or variability related to data acquisition: the light turns on and off in a precise sequence, and pH is recorded every 2 seconds so no small changes are missed. However, it is important to coordinate the timer and data recorder, so that the pH and time measurements are in sync.
Protecting the electrode from direct light is essential so that the electrode does not heat up during the course of the experiment.
A negative control that does not pump protons in response to light is necessary to demonstrate that any recorded change in pH from the rhodopsin expressing strain is due to changes in the light and not changes in temperature, pH or other factors.
Including experiments utilizing an ionophore for suspected proton pumping rhodopsins is essential for demonstrating that protons are being transported across the membranes.
Protocol 3: TIRF Microscopy
Positive controls for axenic cultures. In the case of laboratory samples known to express rhodopsins, these are less critical. However, when analyzing laboratory samples not known to produce rhodopsins, a positive control is important. We have been using a strain of E. coli expressing retinal biosynthetic genes along with an actinorhodopsin (Keffer et al., 2015b) for this purpose.
Positive controls for environmental samples. When analyzing environmental samples, we use a suite of 4 control cultures: non-pigmented E. coli stained with DAPI, E. coli producing lycopene or beta-carotene, E. coli producing retinal and actinorhodopsin, and an algal isolate as a chlorophyll a control (Keffer et al., 2015b). These controls are used to confirm that each cell type can be visualized under the conditions present and to set the background levels in the images during processing.
Troubleshooting
Protocol 1: Membrane purification
If the membrane fraction cannot be solubilized, confirm that the cell lysis was successful. If cells were successfully lysed, try a different detergent or buffer solution.
Protocol 2: Proton pumping
If pumping is not observed, first try another light source. The intensity of the light must be bright enough to affect cells after passing though the shield and water bath.
If pumping is not observed after changing the light source, confirm that buffer has the correct pH. Buffer pH may change during storage, so it is best to use freshly-prepared buffer.
If pumping is still not observed, confirm that the rhodopsin is being expressed and that retinal is present.
If pH is drifting, check temperature of sample.
Protocol 3: TIRF Microscopy
If no cells are seen in the microscope images, check the focus and alignment of lasers, try increasing the number of cells in a sample, and test the focus on a sample with high cell density.
Anticipated Results
Protocol 1: Membrane purification
When starting with a 50-mL culture of E. coli expressing a rhodopsin, this procedure will typically result in ∼1400 μg mL-1 total protein. Total protein can be quantified using a detergent-compatible protein assay (such as the Pierce™ BCA Protein Assay Kit, ThermoFisher catalog no. 23225). Because this is a membrane purification protocol, the solution will also contain hydrophobic carotenoids, lipids, and other membrane proteins (Keffer et al., 2015a). The ratio of total protein to rhodopsin and degree of rhodopsin purification can be estimated by measuring absorbance at 280 nm and absorbance at the maximum absorption wavelength of the rhodopsin (empirically determined; see Fig. 1C) of the cell lysate and the concentrated, purified membranes.
Protocol 2: Proton pumping
If rhodopsin is a proton pump, pH should decrease when the light is on, and drift upward or demonstrate no additional change when the light is off. Proton pumping should be abolished in the presence of the ionophore CCCP. No pH change should occur when no rhodopsin is present.
If rhodopsin is a sodium or chloride pump, pH should increase when the light is on, and drift downward when the light is off. CCCP alone will not affect pumping, but CCCP and TPP+ together will abolish pumping activity (Inoue et al., 2013; Yoshizawa et al., 2014).
Since the pH will increase if the pump is either a Na+ or Cl- pump, components of the pumping buffer can be altered to differentiate between these two activities (Yoshizawa et al., 2014; Inoue et al., 2013). If the rhodopsin is a sodium pump, no change in pH will be observed if the pumping buffer is 100 mM KCl. Similarly, if the rhodopsin is a chloride pump, activity will still be observed in a KCl solution, but not in a solution of sodium sulfate (Na2SO4, 100 mM).
Protocol 3: TIRF Microscopy
In cells >1 μm, rhodopsin fluorescence should be localized to the cell membrane, though this level of resolution may not be possible for smaller cells.
Time Considerations
Protocol 1: Membrane purification
Excluding time to grow cells, lysis and solubilization takes 3-4 hours, followed by an overnight incubation.
Protocol 2: Proton pumping
Although pumping can be observed immediately (at the first onset of bright light), most experiments are set up to be recorded for 2-2.5 hours for one experiment.
Protocol 3: TIRF Microscopy
Coverslip coating
this process will take a total of 2.5-3 h, though with several long incubation times.
Sample preparation
If the cells are fixed, filtering and slide/coverslip sealing can take place the day before the microscope is to be used, so that the sealant has enough time to dry. With live cells, coverslip/slide preparation should take place ∼1-2 hr before the microscope time so that the sealant can dry.
TIRF microscopy
Generally, a half day should be set aside to use the microscope at one time, and samples are imaged in batches. As proficiency increases, 10 different samples can be viewed and imaged with all 4 lasers during a 3-4 hour session.
Image processing
Processing 30-36 fields of view, with each one exposed to all four lasers (a total of 120-144 image sequences) will take ∼2 hours.
Acknowledgments
Development of the TIRF microscopy method was supported by an Institutional Development Award (IDeA) award from the National Institute of General Medical Sciences of the National Institutes of Health under grant number 5 P30 GM103519. Dr. Chandran Sabanayagam and the University of Delaware BioImaging Center provided access to the microscopy system.
Literature Cited
- Alexiev U, Farrens DL. Fluorescence spectroscopy of rhodopsins: Insights and approaches. BBA - Bioenergetics. 2013:1–16. doi: 10.1016/j.bbabio.2013.10.008. Available at: http://dx.doi.org/10.1016/j.bbabio.2013.10.008. [DOI] [PMC free article] [PubMed]
- Atamna-Ismaeel N, Finkel OM, Glaser F, Sharon I, Schneider R, Post AF, Spudich JL, von Mering C, Vorholt JA, Iluz D, et al. Microbial rhodopsins on leaf surfaces of terrestrial plants. [Accessed November 10, 2015];Environmental microbiology. 2012 14:140–6. doi: 10.1111/j.1462-2920.2011.02554.x. Available at: http://onlinelibrary.wiley.com/wol1/doi/10.1111/j.1462-2920.2011.02554.x/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D. Chapter 7 Total Internal Reflection Fluorescence Microscopy BT - Biophysical Tools for Biologists, Volume Two: In Vivo Techniques. In: Correia John J, III, Detrich H William., editors. Biophysical Tools for Biologists, Volume Two: In Vivo Techniques. Academic Press; 2008. pp. 169–221. Available at: http://www.sciencedirect.com/science/article/pii/S0091679X08006079. [Google Scholar]
- Balashov SP, Imasheva ES, Boichenko VA, Anton J, Wang JM, Lanyi JK. Xanthorhodopsin: A Proton Pump with a Light-Harvesting Carotenoid Antenna. Science (New York, NY) 2005;309:2061–2064. doi: 10.1126/science.1118046. Available at: http://www.sciencemag.org/cgi/doi/10.1126/science.1118046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashov SP, Imasheva ES, Choi AR, Jung KH, Liaaen-Jensen S, Lanyi JK. Reconstitution of Gloeobacter Rhodopsin with Echinenone: Role of the 4-Keto Group. Biochemistry. 2010;49:9792–9799. doi: 10.1021/bi1014166. Available at: http://pubs.acs.org/doi/abs/10.1021/bi1014166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beja O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science (New York, NY) 2000;289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- Beja O, Spudich EN, Spudich JL, Leclerc M, Delong EF. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411:786–789. doi: 10.1038/35081051. Available at: http://www.nature.com/nature/journal/v411/n6839/full/411786a0.html. [DOI] [PubMed] [Google Scholar]
- Boeuf D, Audic S, Brillet-Guéguen L, Caron C, Jeanthon C. MicRhoDE: a curated database for the analysis of microbial rhodopsin diversity and evolution. [Accessed January 30, 2016];Database : the journal of biological databases and curation. 2015 2015 doi: 10.1093/database/bav080. bav080–. Available at: http://database.oxfordjournals.org/content/2015/bav080.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LS. Eubacterial rhodopsins — Unique photosensors and diverse ion pumps. BBA - Bioenergetics. 2014;1837:553–561. doi: 10.1016/j.bbabio.2013.05.006. Available at: http://www.sciencedirect.com/science/article/pii/S0005272813000972. [DOI] [PubMed] [Google Scholar]
- Cheminal A, Léonard J, Kim SYY, Jung KHH, Kandori H, Haacke S. Steady state emission of the fluorescent intermediate of Anabaena Sensory Rhodopsin as a function of light adaptation conditions. Chemical Physics Letters. 2013;587:75–80. Available at: http://dx.doi.org/10.1016/j.cplett.2013.09.044. [Google Scholar]
- Duschl A, Lanyi JK, Zimanyi L. Properties and photochemistry of a halorhodopsin from the haloalkalophile, Natronobacterium pharaonis. J Biol Chem. 1990;265:1261–1267. Available at: http://www.nature.com/articles/srep07798. [PubMed] [Google Scholar]
- García-Martínez J, Brunk M, Avalos J, Terpitz U. The CarO rhodopsin of the fungus Fusarium fujikuroi is a light-driven proton pump that retards spore germination. [Accessed December 17, 2015];Scientific reports. 2015 5:7798. doi: 10.1038/srep07798. Available at: http://www.nature.com/srep/2015/150115/srep07798/full/srep07798.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Consarnau L, Gonzalez JM, Riedel T, Jaenicke S, Wagner-Dobler I, Sanudo-Wilhelmy SA, Fuhrman JA. Proteorhodopsin light-enhanced growth linked to vitamin-B1 acquisition in marine Flavobacteria. ISME J. 2015 doi: 10.1038/ismej.2015.196. Available at: http://dx.doi.org/10.1038/ismej.2015.196. [DOI] [PMC free article] [PubMed]
- Hahn MW, Lunsdorf H, Wu Q, Schauer M, Hofle MG, Boenigk J, Stadler P. Isolation of Novel Ultramicrobacteria Classified as Actinobacteria from Five Freshwater Habitats in Europe and Asia. Applied and Environmental Microbiology. 2003;69:1442–1451. doi: 10.1128/AEM.69.3.1442-1451.2003. Available at: http://aem.asm.org/content/69/3/1442.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Ljumovic M, Bondar AN, Shibata Y, Ito S, Inoue K, Kandori H, Brown LS. A new group of eubacterial light-driven retinal-binding proton pumps with an unusual cytoplasmic proton donor. [Accessed December 9, 2015];Biochimica et biophysica acta. 2015 1847:1518–29. doi: 10.1016/j.bbabio.2015.08.003. Available at: http://www.sciencedirect.com/science/article/pii/S0005272815001590. [DOI] [PubMed] [Google Scholar]
- Hasemi T, Kikukawa T, Kamo N, Demura M. Characterization of a Cyanobacterial Chloride-Pumping Rhodopsin and Its Conversion into a Proton Pump. [Accessed December 16, 2015];The Journal of biological chemistry. 2015 291 doi: 10.1074/jbc.M115.688614. M115.688614–. Available at: http://www.jbc.org/content/early/2015/11/17/jbc.M115.688614.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Kato Y, Kandori H. Light-driven ion-translocating rhodopsins in marine bacteria. [Accessed August 18, 2015];Trends in microbiology. 2015 23:91–8. doi: 10.1016/j.tim.2014.10.009. Available at: http://www.sciencedirect.com/science/article/pii/S0966842X14002248. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ono H, Abe-Yoshizumi R, Yoshizawa S, Ito H, Kogure K, Kandori H. A light-driven sodium ion pump in marine bacteria. 2013;4:1678. doi: 10.1038/ncomms2689. Available at: http://dx.doi.org/10.1038/ncomms2689. [DOI] [PubMed] [Google Scholar]
- Irieda H, Morita T, Maki K, Homma M, Aiba H, Sudo Y. Photo-induced Regulation of the Chromatic Adaptive Gene Expression by Anabaena Sensory Rhodopsin. Journal of Biological Chemistry. 2012;287:32485–32493. doi: 10.1074/jbc.M112.390864. Available at: http://www.jbc.org/content/287/39/32485.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KH, Trivedi VD, Spudich JL. Demonstration of a sensory rhodopsin in eubacteria. [Accessed December 17, 2015];Molecular Microbiology. 2003 47:1513–1522. doi: 10.1046/j.1365-2958.2003.03395.x. Available at: http://dx.doi.org/10.1046/j.1365-2958.2003.03395.x. [DOI] [PubMed] [Google Scholar]
- Kawanabe A, Furutani Y, Jung KH, Kandori H. Engineering an inward proton transport from a bacterial sensor rhodopsin. [Accessed December 16, 2015];Journal of the American Chemical Society. 2009 131:16439–44. doi: 10.1021/ja904855g. Available at: http://dx.doi.org/10.1021/ja904855g. [DOI] [PubMed] [Google Scholar]
- Keffer JL, Hahn MW, Maresca JA. Characterization of an unconventional rhodopsin from the freshwater Actinobacterium Rhodoluna lacicola. [Accessed June 22, 2015];Journal of bacteriology. 2015a 197:2704–2712. doi: 10.1128/JB.00386-15. Available at: http://jb.asm.org/content/197/16/2704.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keffer JL, Sabanayagam CR, Lee ME, DeLong EF, Hahn MW, Maresca JA. Using total internal reflection fluorescence microscopy to visualize rhodopsin-containing cells. [Accessed April 29, 2015];Applied and environmental microbiology. 2015b 81:3442–50. doi: 10.1128/AEM.00230-15. Available at: http://aem.asm.org/content/81/10/3442.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendoerfer GG, Mathies RA. Spontaneous emission study of the femtosecond isomerization dynamics of rhodopsin. Journal of Physical Chemistry. 1996;100:14526–14532. Available at: http://pubs.acs.org/doi/abs/10.1021/jp960509%2B?journalCode=jpchax. [Google Scholar]
- Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D, Cohen AE. Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nature Chemical Biology. 2012;9:90–95. doi: 10.1038/nmeth.1782. Available at: http://www.nature.com/nmeth/journal/v9/n1/full/nmeth.1782.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanyi JK, Balashov SP. Xanthorhodopsin: a bacteriorhodopsin-like proton pump with a carotenoid antenna. Biochimica et biophysica acta. 2008;1777:684–688. doi: 10.1016/j.bbabio.2008.05.005. Available at: papers2://publication/uuid/8C72B0AA-BEC5-44C5-8A71-E6A027E51A2F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanyi J, Oesterhelt D. Identification of the retinal-binding protein in halorhodopsin. J Biol Chem. 1982;257:2674–2677. Available at: http://www.jbc.org/content/257/5/2674.long. [PubMed] [Google Scholar]
- Luck M, Bruun S, Keidel A, Hegemann P, Hildebrandt P. Photochemical chromophore isomerization in histidine kinase rhodopsin HKR1. [Accessed December 17, 2015];FEBS letters. 2015 589:1067–71. doi: 10.1016/j.febslet.2015.03.024. Available at: http://www.sciencedirect.com/science/article/pii/S001457931500201X. [DOI] [PubMed] [Google Scholar]
- Man D, Wang W, Sabehi G, Aravind L, Post AF, Massana R, Spudich EN, Spudich JL, Beja O. Diversification and spectral tuning in marine proteorhodopsins. The EMBO journal. 2003;22:1725–1731. doi: 10.1093/emboj/cdg183. Available at: http://emboj.embopress.org/content/22/8/1725.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man-Aharonovich D, Sabehi G, Sineshchekov OA, Spudich EN, Spudich JL, Beja O. Characterization of RS29, a blue-green proteorhodopsin variant from the Red Sea. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology. 2004;3:459–462. doi: 10.1039/b316071h. Available at: http://pubs.rsc.org/en/content/articlelanding/2004/pp/b316071h#!divAbstract. [DOI] [PubMed] [Google Scholar]
- Martinez A, Bradley AS, Waldbauer JR, Summons RE, Delong EF. Proteorhodopsin photosystem gene expression enables photophosphorylation in a heterologous host. Proceedings of the National Academy of Sciences. 2007;104:5590–5595. doi: 10.1073/pnas.0611470104. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.0611470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarren J, DeLong EF. Proteorhodopsin photosystem gene clusters exhibit co-evolutionary trends and shared ancestry among diverse marine microbial phyla. Environmental Microbiology. 2007;9:846–858. doi: 10.1111/j.1462-2920.2006.01203.x. Available at: http://doi.wiley.com/10.1111/j.1462-2920.2006.01203.x. [DOI] [PubMed] [Google Scholar]
- Oesterhelt D. Structure and Function of Halorhodopsin. Israel Journal of Chemistry. 1995;35:475–494. Available at: http://dx.doi.org/10.1002/ijch.199500044. [Google Scholar]
- Oesterhelt D, Stoeckenius W. [69] Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane BT - Biomembranes Part A. In: Sidney Fleischer LP, editor. Biomembranes Part A. Academic Press; 1974. pp. 667–678. Available at: http://www.sciencedirect.com/science/article/pii/0076687974310725. [DOI] [PubMed] [Google Scholar]
- Oesterhelt D, Stoeckenius W. Functions of a New Photoreceptor Membrane. [Accessed May 28, 2015];Proceedings of the National Academy of Sciences. 1973 70:2853–2857. doi: 10.1073/pnas.70.10.2853. Available at: http://www.pnas.org/content/70/10/2853.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racker E, Stoeckenius W. Reconstitution of Purple Membrane Vesicles Catalyzing Light-driven Proton Uptake and Adenosine Triphosphate Formation. Journal of Biological Chemistry. 1974;249:662–663. Available at: http://www.jbc.org/content/249/2/662.abstract. [PubMed] [Google Scholar]
- Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. [Accessed July 10, 2014];The Journal of cell biology. 2010 190:165–75. doi: 10.1083/jcb.201002018. Available at: http://jcb.rupress.org/content/190/2/165.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert B, Lanyi JK. Halorhodopsin is a light-driven chloride pump. The Journal of biological chemistry. 1982;257:10306–10313. [PubMed] [Google Scholar]
- Sharma AK, Sommerfeld K, Bullerjahn GS, Matteson AR, Wilhelm SW, Jezbera J, Brandt U, Doolittle WF, Hahn MW. Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. The ISME Journal. 2009;3:726–737. doi: 10.1038/ismej.2009.13. Available at: http://dx.doi.org/10.1038/ismej.2009.13. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Zhaxybayeva O, Papke RT, Doolittle WF. Actinorhodopsins: proteorhodopsin-like gene sequences found predominantly in non-marine environments. Environmental Microbiology. 2008;10:1039–1056. doi: 10.1111/j.1462-2920.2007.01525.x. Available at: http://doi.wiley.com/10.1111/j.1462-2920.2007.01525.x. [DOI] [PubMed] [Google Scholar]
- Sineshchekov OA, Trivedi VD, Sasaki J, Spudich JL. Photochromicity of Anabaena Sensory Rhodopsin, an Atypical Microbial Receptor with a cis-Retinal Light-adapted Form. [Accessed December 17, 2015];Journal of Biological Chemistry. 2005 280:14663–14668. doi: 10.1074/jbc.M501416200. Available at: http://www.jbc.org/content/280/15/14663.abstract. [DOI] [PubMed] [Google Scholar]
- Spudich JL. The multitalented microbial sensory rhodopsins. [Accessed July 29, 2014];Trends in Microbiology. 2006 14:480–487. doi: 10.1016/j.tim.2006.09.005. Available at: http://www.sciencedirect.com/science/article/pii/S0966842X06002319. [DOI] [PubMed] [Google Scholar]
- Steindler L, Schwalbach MS, Smith DP, Chan F, Giovannoni SJ. Energy Starved Candidatus Pelagibacter Ubique Substitutes Light-Mediated ATP Production for Endogenous Carbon Respiration. PLoS ONE. 2011;6:e19725. doi: 10.1371/journal.pone.0019725. Available at: http://dx.plos.org/10.1371/journal.pone.0019725.s013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde JA, Podell S, Narasingarao P, Allen EE. Xenorhodopsins, an enigmatic new class of microbial rhodopsins horizontally transferred between archaea and bacteria. Biology Direct. 2011;6:1–8. doi: 10.1186/1745-6150-6-52. Available at: http://dx.doi.org/10.1186/1745-6150-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JM, Greenfield D, Bustamante C, Liphardt J. Light-powering Escherichia coli with proteorhodopsin. Proceedings of the National Academy of Sciences. 2007;104:2408–2412. doi: 10.1073/pnas.0611035104. Available at: http://www.pnas.org/content/104/7/2408.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WW, Sineshchekov OA, Spudich EN, Spudich JL. Spectroscopic and Photochemical Characterization of a Deep Ocean Proteorhodopsin. Journal of Biological Chemistry. 2003;278:33985–33991. doi: 10.1074/jbc.M305716200. Available at: http://www.jbc.org/cgi/doi/10.1074/jbc.M305716200. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Kawanabe A, Ito H, Kandori H, Kogure K. Diversity and functional analysis of proteorhodopsin in marine Flavobacteria. Environmental Microbiology. 2012;14:1240–1248. doi: 10.1111/j.1462-2920.2012.02702.x. Available at: http://onlinelibrary.wiley.com/doi/10.1111/j.1462-2920.2012.02702.x/abstract. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Kumagai Y, Kim H, Ogura Y, Hayashi T, Iwasaki W, DeLong EF, Kogure K. Functional characterization of flavobacteria rhodopsins reveals a unique class of light-driven chloride pump in bacteria. Proceedings of the National Academy of Sciences. 2014;111:6732–6737. doi: 10.1073/pnas.1403051111. Available at: http://www.pnas.org/content/111/18/6732.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N, Koonin EV. Proteorhodopsin genes in giant viruses. Biology direct. 2012;7:34. doi: 10.1186/1745-6150-7-34. Available at: http://www.biologydirect.com//content/7/1/34. [DOI] [PMC free article] [PubMed] [Google Scholar]