

Abstract
C-methyltransferases (MTs) from modular polyketide synthase assembly lines are relatively rare and unexplored domains that are responsible for installing α-methyl groups into nascent polyketide backbones. The stage at which these synthase-embedded enzymes operate during polyketide biosynthesis has yet to be conclusively demonstrated. In this work we establish the activity and substrate preference for six MTs from the gephyronic acid polyketide synthase and demonstrate their ability to methylate both N-acetylcysteamine- and acyl carrier protein-linked β-ketoacylthioester substrates but not malonyl thioester equivalents. These data strongly indicate that MT-catalyzed methylation occurs immediately downstream of ketosynthase-mediated condensation during polyketide assembly. This work represents the first successful report of MT-catalyzed mono- and dimethylation of simple thioester substrates and provides the groundwork for future mechanistic and engineering studies on this important but poorly understood enzymatic domain.
Introduction
S-Adenosylmethionine (SAM)-dependent methyltransferases (MTs) constitute a broad class of enzymes that find frequent use in primary and secondary metabolism.1,2 Some MTs embedded within Type I polyketide synthases (PKSs) are able to add α-methyl branches to polyketide chains.2 The action of such MTs within PKS modules provides both an alternative route to the α-methyl branched intermediates typically introduced by methylmalonyl-CoA specific acyltransferase (AT) domains as well as the primary route for generating gem-dimethyl functionality.3,4
Polyketides comprise a major fraction of all known natural products, and the molecular factories responsible for synthesizing them represent long-standing targets of protein engineering efforts.5 The diversity of polyketides is remarkable, particularly in light of the few types of enzymatic domains and the simple building blocks employed in their assembly. A series of PKS modules, minimally comprised of a ketosynthase (KS) and an acyl carrier protein (ACP) [as well as an acyltransferase (AT) in cis-AT PKSs)], orchestrate two-carbon extensions of polyketide chains via decarboxylative condensations. Each module typically contains one or more processing domains, such as a ketoreductase (KR), dehydratase (DH), enoylreductase (ER), and C-methyltransferase (MT) that act on the α-/β-carbons of intermediates to confer complexity to the often large, stereo-dense final products.6 Though considerable effort has been dedicated to characterizing the structure and function of PKS biosynthetic pathways, limited work has been performed on embedded MT domains.7,8 Consequently, their substrate specificities and timing within the catalytic cycle of PKS modules have remained mysterious.9 Two routes for MT-catalyzed methyl incorporation into polyketides are plausible (Figure 1).9 In the first and traditionally-accepted route, methylation immediately follows KS-mediated condensation (aided by the decreased pKa of the β-ketoacylthioester intermediate). Conversely, in the second route, mono- or dimethylation occurs on the ACP-bound malonyl extender unit prior to condensation. Recent experiments on modules containing MTs catalyzing gem-dimethylation within the yersiniabactin and epothilone PKS pathways detected the formation of dimethylmalonyl-ACP via tandem MS and showed them to be suitable substrates for the subsequent condensation reaction indicating that the second route may be operative within these pathways.9
Figure 1.
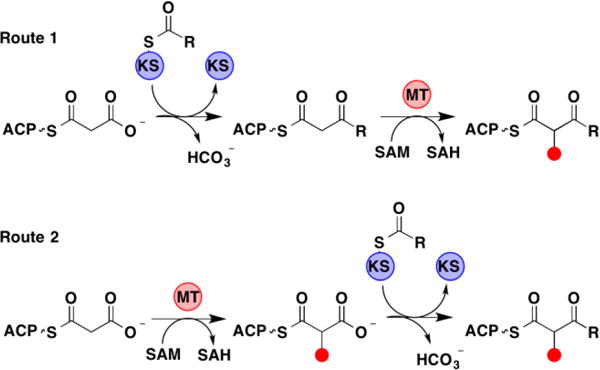
Potential routes for PKS MT-catalyzed α-methylation.
Results and Discussion
To further elucidate the timing of embedded mono- and dimethylating MT domains, we purified and assayed the five monomethylating MTs and the one gem-dimethylating MT from the gephyronic acid biosynthetic pathway of Cystobacter violaceous (Figure 2).10 All six of these MTs display activity towards β-ketoacylthioester substrates but not towards equivalent malonyl thioester substrates, providing evidence that the first route is operative for both mono- and dimethylation within the gephyronic acid synthase.
Figure 2.
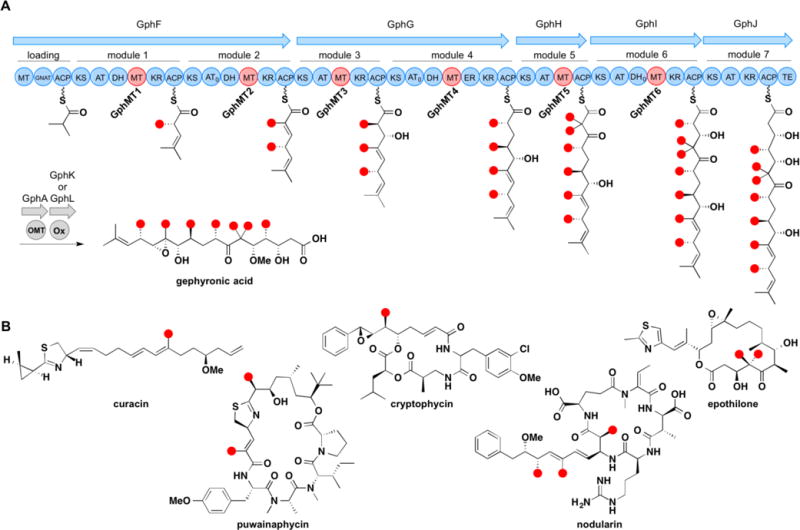
(A) Model of the gephyronic acid synthase and structure of gephyronic acid. (B) Polyketides that contain α-methyl branches installed by MT domains embedded within cis-AT PKSs.
To obtain soluble protein for the five monomethylating MT domains in the gephyronic acid synthase, four MTs were cloned and purified as MT+KR didomains (labeled as GphMT1, GphMT2, GphMT3, and GphMT6 for simplicity) and one as an MT+ER+KR tridomain (GphMT4) (Figure S1). A sequence alignment of cis-AT MTs reveals that MT domains are typically embedded within the KR structural subdomain, and our studies indicate that the inclusion of this region may be necessary for MT expression (Figure S2). Despite its location within a non-reducing module, the gem-dimethylating GphMT5 harbors a C-terminal region with striking sequence similarity to the KR structural subdomain (Figure S2). GphMT5 was assayed both as a discrete domain and in the context of its complete module (GphH). All six excised MT constructs and GphH were expressed in E. coli BL21(DE3) and E. coli K207-3 strains, respectively, and purified by Ni-NTA (nickel nitrilotriacetate) chromatography.
All monomethylating MTs were observed to catalyze the methylation of 3-oxopentanoyl-S-NAC, 1, to afford 2-methyl-3-oxopentanoyl-S-NAC, 2, with varying degrees of conversion, ranging from 2–24% after 72 h (Figure 3). Notably, after 72 h, no methylation of malonyl-S-NAC or malonyl-CoA was observed by any of the five monomethylating MTs. Taken together, these data suggest that all MT-catalyzed monomethylation occurs exclusively via route 1 in the gephyronic acid biosynthetic pathway. Dimethylation of 1 to afford 2,2-dimethyl-3-oxopentanoyl-S-NAC, 3, was not detected. With the exception of the MT+ER+KR tridomain, GphMT4, the KRs in each of the didomain constructs were capable of reducing 1 and 2 (Figures S4 and S5).
Figure 3.
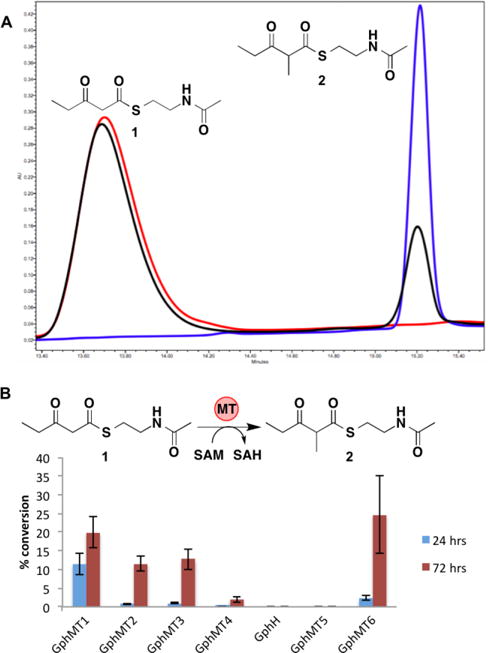
(A) HPLC traces monitoring 235 nm; red, synthetic standard of 1; blue, synthetic standard of 2; black, GphMT1 reaction after 72 h. All HPLC peaks collected and confirmed by HRMS. (B) Conversion of 1 to 2 catalyzed by 50 μM MT constructs and GphH with 10 mM 1 and 15 mM SAM. Error bars represent standard deviations of triplicate data.
Both the GphH module and the MT excised from it, GphMT5, are capable of dimethylating 1 to afford 3, albeit with low efficiency (Figure 4). Slightly higher conversion was observed for the GphH module, implying that the gem-dimethylating MT is more active in the context of its surrounding domains. Notably, 2 was not detected from reactions with either GphH or GphMT5, suggesting that the second methylation event occurs quickly after the first. Neither GphH nor GphMT5 catalyzed a detectable amount of methylation of malonyl-S-NAC or malonyl-CoA, suggesting that dimethylation, like monomethylation, occurs exclusively via route 1 in the gephyronic acid PKS pathway.
Figure 4.
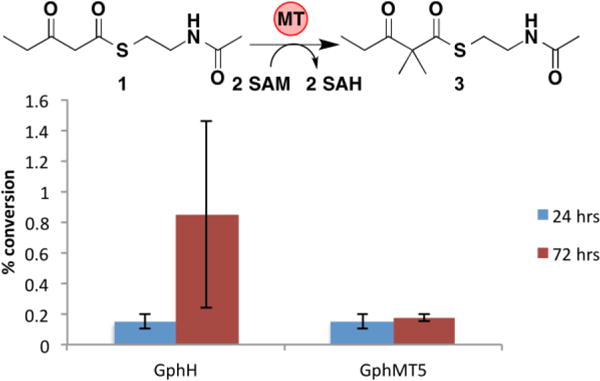
Conversion of 1 to 3 catalyzed by 50 μM GphH and GphMT5 with 10 mM 1 and 30 mM SAM. Error bars represent standard deviations of triplicate data.
To remove the generated S-adenosylhomocysteine (SAH) product, a potential inhibitor of MTs, we assayed the activity of each of the GphMTs in the presence of the E. coli SAH nucleosidase Pfs.2,11 Co-incubation of GphMTs with Pfs greatly increased conversion of 1 to 2 by GphMT1 and GphMT6, with 95% and 85% conversion, respectively, after 72 h (Figure 5). Scaled-up reactions with GphMT1 provided milligram quantities of 2 with isolated yields of 78%. The inclusion of Pfs in GphH and GphMT5 dimethylating reactions did not provide a significant increase in conversion, suggesting that the concentration of SAH generated from the lower conversion of substrate is at sub-inhibitory levels (Figure S3).
Figure 5.
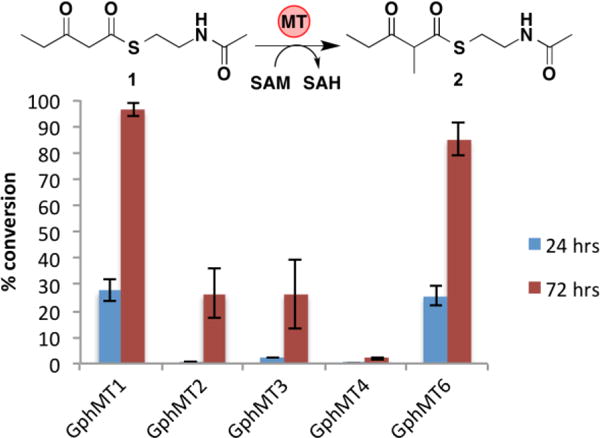
Conversion of 1 to 2 catalyzed by 50 μM monomethylating GphMT constructs in the presence of 25 μM Pfs with 10 mM 1 and 15 mM SAM. Error bars represent standard deviations of triplicate data.
To gain insight into the substrate tolerance of embedded PKS MT domains, we next assayed the most active domains, GphMT1 and GphMT6, with 2 additional substrates of varying chain length, 3-oxohexanoyl-S-NAC, 4, and 3-oxobutanoyl-S-NAC, 6 (Figure 6). Since 1 most closely mimics the natural substrate of GphMT1, the decrease in activity observed for both the larger substrate 4 and the smaller substrate 6 suggests that GphMT1 exerts a moderate degree of selectivity for molecules most closely resembling the natively encountered substrate. Similarly, the decreased conversion of 6 by GphMT6 relative to the larger substrates 1 and 4 suggests that mild selectivity is again arising from protein-substrate interactions beyond the β-keto group. The preference of GphMT6 for larger hydrophobic substrates can be rationalized by its presence within the penultimate module. In aggregate, these results are similar to the recent kinetic characterization of the MT domain from the lovastatin synthase LovB in that despite showing a preference for compounds closely resembling the natural substrates they are promiscuous enough to act on a variety of β-ketoacyl compounds12.
Figure 6.
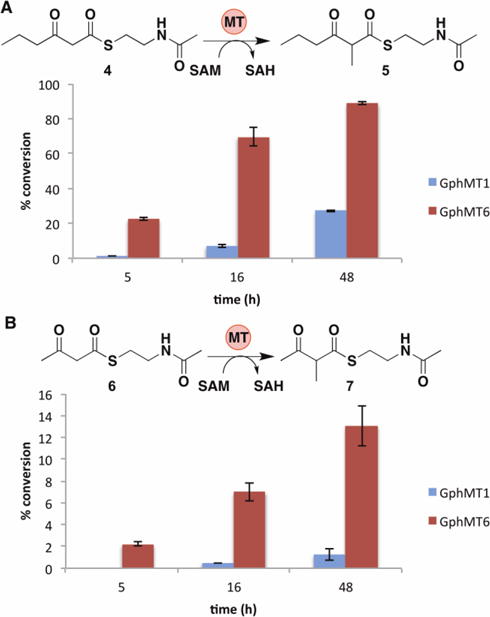
Impact of chain length on GphMT1 and GphMT6 activity using substrates 4 (A) and 6 (B) (10 mM) with 15 mM SAM, 50 μM MT, and 25 μM Pfs (Note scale change on y-axis). Error bars represent standard deviations of triplicate data.
It is conceivable that the methylation of small molecule substrate mimics may not accurately reflect the methylation of the natural substrates of MTs. Therefore, we examined the activities of two MTs towards their cognate holo-ACPs bound either to a β-ketopentanoyl moiety or a malonyl extender unit. GphACP1 and GphACP6 (the cognate ACPs of GphMT1 and GphMT6, respectively) were expressed in E. coli K207-3 to afford holo-ACP domains that were then thioesterified with either substrate 1 or malonyl-CoA. For both GphMT constructs a 14-Da mass increase was observed by HRMS analysis (Figure 7). MS/MS spectra confirm MT-catalyzed methylation occurred at the α-position of the diketide attached to the pantetheinyl arm on the conserved ACP serine. Consistent with previous results, no mass increase was observed during methylation attempts of either malonyl-S-ACP. The selective methylation of β-ketoacyl-S-ACPs provides further evidence that MT-catalyzed α-branching occurs via route 1 in gephyronic acid biosynthesis.
Figure 7.
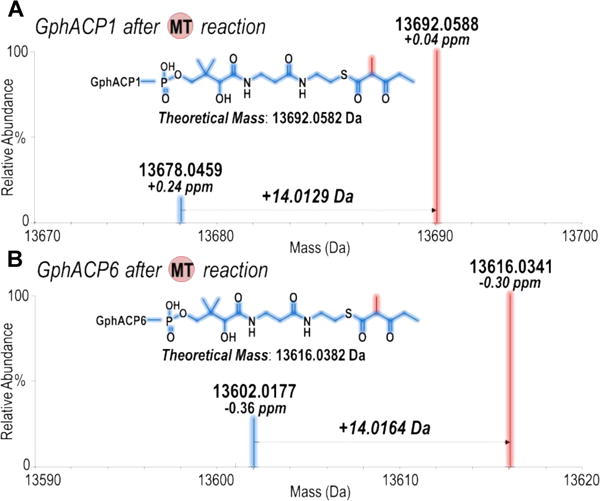
Deconvoluted ESI-MS (mass range 13590 – 13620 Da) of β-ketopentanoyl- (A) GphACP1 and (B) GphACP6, which show a 14-Da mass increase after the MT reaction.
Conclusions
In closing, our results provide significant evidence that MT-catalyzed methylation immediately follows KS-mediated condensation in the gephyronic acid PKS pathway. The data also represent the first reported activity of mono- and dimethylating MTs excised from a modular PKS and help to provide insight into the substrate promiscuity of embedded MTs as well as the order of chemistry performed by PKS processing domains. As it stands, the reported variability in the timing of α-branching presents MTs as unique, versatile domains within PKS pathways that require further exploration.9,10 Additionally, the robust activity of several of the GphMT domains when coupled with Pfs activity, make them potential tools for the incorporation of α-branches into previously reported PKS biocatalytic platforms to allow a greater access to polyketide chemical diversity.
Supplementary Material
Acknowledgments
The National Institutes of Health (GM106112), the National Science Foundation (CHE1402753), and the Welch Foundation (F-1712, AKC; F-1155, JSB) funded this work. We thank the University of Texas at Austin mass spectrometry facility for their help in obtaining high-resolution masses for 1–7.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Ansari MZ, Sharma J, Gokhale S, Mohanty D. BMC Bioinf. 2008;9:454. doi: 10.1186/1471-2105-9-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liscombe DK, Louie GV, Noel JP. Nat Prod Rep. 2012;29:1238. doi: 10.1039/c2np20029e. [DOI] [PubMed] [Google Scholar]
- 3.Ishida K, Fritzsche K, Hertweck C. J Am Chem Soc. 2007;42:129. doi: 10.1021/ja075524e. [DOI] [PubMed] [Google Scholar]
- 4.Schenk A, Xu Z, Pfeiffer C, Steinbeck C, Hertweck C. Angew Chem Int Ed. 2007;46:7035. doi: 10.1002/anie.200702033. [DOI] [PubMed] [Google Scholar]
- 5.Williams G. Curr Opin Struct Biol. 2013;23:603. doi: 10.1016/j.sbi.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keatinge-Clay AT. Nat Prod Rep. 2012;29:1050. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 7.Winter JM, Chiou G, Bothwell IR, Xu W, Garg KG, Luo KM, Tang Y. Org Lett. 2013;15:3775. doi: 10.1021/ol401723h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller AM, Luo L, Hillson N, Keating TA, Walsh CT. Chem Biol. 2002;9:333. doi: 10.1016/s1074-5521(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 9.Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, Keasling JD. Angew Chem Int Ed. 2015;54:2370. doi: 10.1002/anie.201410124. [DOI] [PubMed] [Google Scholar]
- 10.Young J, Stevens DC, Carmichael R, Tan J, Rachid S, Boddy CN, Müller R, Taylor RE. J Nat Prod. 2013;76:2269. doi: 10.1021/np400629v. [DOI] [PubMed] [Google Scholar]
- 11.Hendricks CL, Ross JR, Pichersky E, Noel JP, Zhou ZS. Anal Biochem. 2004;326:100. doi: 10.1016/j.ab.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Cacho RA, Thuss J, Xu W, Sanichar R, Gao Z, Nguyen A, Vederas JC, Tang Y. J Am Chem Soc. 2015;137:15688. doi: 10.1021/jacs.5b11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.