

Abstract
Acute liver failure is a rare and devastating clinical condition. At present, emergency liver transplantation is the only life-saving therapy in advanced cases, yet the feasibility of transplantation is affected by the presence of systemic inflammation, infection and resultant multi-organ failure. The importance of immune dysregulation and acquisition of infection in the pathogenesis of acute liver failure and its associated complications is now recognised. In this review we discuss current thinking regarding the role of infection and inflammation in the pathogenesis of and outcome in human acute liver failure, the implications for the management of such patients and suggest directions for future research.
Keywords: Inflammation, Neuroinflammation, Acute liver failure, Biomarker, Infection
Core tip: Acute liver failure is a serious and rare condition, for which emergency liver transplantation is the only rescue therapy in advanced cases. The medical need for liver transplantation and feasibility of such an intervention are affected by the presence of systemic inflammation and infection. This review will discuss current thinking with regards to the role of infection and inflammation in the pathogenesis of human acute liver failure, and its effect on outcome. We also provide clinical guidance for the management of these patients and suggest directions for future research.
INTRODUCTION
Acute liver failure (ALF) is a rare and devastating clinical condition, resulting from massive loss of hepatic parenchyma and metabolic function. ALF is estimated to affect 2000 people per year in the United States, and is associated with a significant mortality rate[1,2]. The development of hepatic encephalopathy (HE) defines ALF, and confers a poor prognosis. With the use of emergency liver transplantation, up to 75% of patients should now be expected to survive[3,4]. Acetaminophen (APAP; paracetamol) is the most common cause of ALF in the West, compared with viral causes in the developing world.
Many of the extra-hepatic features of ALF-including hemodynamic disturbance and multi-organ failure- are now thought to be driven by the secondary immune response to hepatocyte cell death. Clinically ALF shares many features with severe sepsis, including a systemic inflammatory response and progression to multi-organ failure. The hemodynamic profile of ALF mirrors that of septic shock, suggesting that the systemic release of inflammatory mediators might be responsible for progression from the inflammatory response to multi-organ failure. Immune dysregulation is now recognised to be central to the pathogenesis of ALF[5,6] and it is likely that the clinical features and outcomes in ALF relate to an individual patients innate immune response to liver injury, as opposed to the liver injury and hepatocyte cell death itself. As a result of this immune dysregulation, patients with ALF demonstrate increased susceptibility to infection which is associated with the development of further complications. Currently, emergency liver transplantation is the only rescue therapy for patients with advanced ALF, yet the development of infection, sepsis and the resultant inflammatory response and multi-organ failure may preclude the opportunity for life-saving liver transplantation. Novel, non-transplant therapies are urgently needed, and understanding the role of infection and inflammation in the pathogenesis and progression of ALF is central to the development of new therapeutic strategies. Understanding the role and interplay of infection and inflammation in the pathogenesis of ALF will in addition allow better risk stratification and prognostication in ALF. In this review we detail the current thinking with regards to the role of infection and inflammation in the pathogenesis of and outcome in human ALF, their implications for the management of the ALF patient and suggest directions for future research. Comprehensive overviews of human basic science and research studies undertaken in animal models of ALF are out with the scope of this review.
IMMUNE DYSFUNCTION IN ALF
The immune dysregulation in ALF has been discussed extensively elsewhere[5-7]. Dysfunction of both the cellular and humoral innate immune system plays a role in the pathophysiological development of ALF. Defective functioning of the cellular components in particular of the innate immune system are implicated in the increased risk of infection in patients with ALF. Causes of innate immune dysfunction are proposed to include changes in gut permeability, endotoxinemia, lipoprotein and albumin dysfunction, and toll-like receptor (TLR) expression[8]. Toll like receptors are innate pattern recognition receptors, present in many cells including neutrophils and hepatocytes. Activation of neutrophil TLR can induce an inflammatory response with phagocytic activity and cytokine release, but whether this is beneficial or detrimental to the patient with ALF is not well defined[9].
The underlying mechanism of acute liver injury begins with hepatocyte necrosis. The driver of ongoing necrosis in the absence of ongoing injury is not clearly understood. Oxidative stress leads to production of reactive oxygen species, which in turn via a cascade of events results in activation of c-Jun N-terminal kinase (JNK). This is turn leads to mitochondrial dysfunction causing further hepatocyte necrosis and release of damage associated molecular patterns (DAMPs). DAMPS activate hepatic macrophages and as a further consequence the inflammasome is formed. Comprehensive reviews of the role of inflammasomes in liver disease are available elsewhere[10,11]. Briefly, inflammasomes are multiprotein complexes that sense intracellular danger signals via NOD-like receptors. The inflammasome finely controls the inflammatory response, by responding to low-threshold signals. Activation of the inflammasome by DAMPS is a result of TLR activation and activation by an inflammasome ligand, and results in caspase-1 activation and IL-1β secretion - see Figure 1. The NLRP3 inflammasome is the most well characterised member of the inflammasome family and three potential activation pathways have been proposed: (1) extracellular ATP signal resulting in potassium efflux and pannexin recruitment; (2) endocytosis of crystalized cholesterol, uric acid or amyloid with lysosomal damage following phagocytosis of these particles; and (3) activation by reactive oxygen species. Looking specifically at the role of the inflammasome in acute liver failure, inflammasome activation in APAP-ALF has been studied[11]. Necrotic hepatocytes and sinusoidal endothelial cells release DAMPS, which activate the inflammasome as described above. It remains unclear as to whether APAP directly results in inflammasome formation. The inflammasome is a determinant of hepatic inflammation in APAP-ALF, but its role in regeneration - critical in determining the outcome of the patient - is uncertain. Much of the research undertaken looking at the role of the inflammasome in APAP-ALF has been undertaken in mouse models, and therefore may not be directly applicable to human clinical practice.
Figure 1.
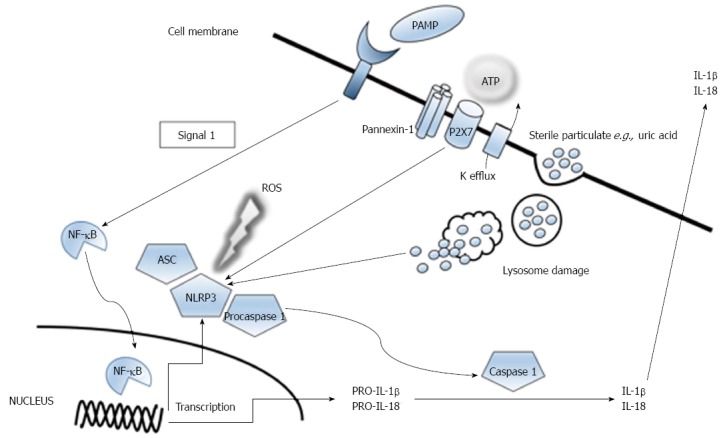
Activation of the NLRP3 inflammasome. NLRP3 inflammasome activation. Extracellular ATP is sensed by the P2X7 purinergic receptor and results in potassium efflux and recruitment of pannexin. Activation may also be induced by large particles such as uric acid, and lysosomal damage after phagocytosis of these particles induces NLRP3 inflammasome activation. Reactive oxygen species also contribute to inflammasome activation. Activation of the inflammasome results in caspase-1 and IL-1β secretion.
Subsequently, pro-inflammatory cytokines are released, leading to the recruitment of more immune cells to the site of inflammation and further advancing hepatocyte cell death. The roles of specific cell types are discussed briefly below and highlighted in more detail in Table 1[12-20]. A comprehensive overview of other pathophysiological mechanisms important in modulating inflammation and infection in ALF such as autophagic dysfunction, mitochondrial membrane potential dysregulation and the influence of calcium flux are out with the scope of this clinical review, but are extensively reviewed elsewhere[21-23].
Table 1.
Cell types and proteins involved in immune pathogenesis of acute liver failure
| Ref. | Cell type/protein study population | Finding in ALF |
| Taylor et al[12] | Neutrophils | Significant reduction in neutrophil surface expression of CD16 (causing reduced binding capacity) and neutrophil phagocytic activity compared with healthy controls. Impaired phagocytic activity predictive of death without transplantation |
| ALF | ||
| Manakkat Vijay et al[13] | Neutrophils | Toll-like receptors sense pathogens and induce inflammatory responses. Neutrophil toll-like receptor 9 expression increased on day 1 compared with healthy controls, and correlated with severity of HE and SIRS |
| APAP-ALF | ||
| Srungaram et al[14] | Osteopontin (activates neutrophils and macrophages) | Median osteopontin levels significantly elevated in the ALF group compared with comparator cohorts; median osteopontin levels were highest in patients with APAP-ALF and ischaemic hepatitis (conditions associated with a hyperacute course and better outcomes - osteopontin may have a central role to play in the resolution of ALF) |
| ALFSG | ALF | |
| Lawson et al[15] | Neutrophils | Neutrophils accumulate in liver parallel to or slightly after liver injury; number of neutrophils in liver substantial compared with baseline, with increased levels of TNF-α, KC and MIP-2 chemokines |
| APAP-ALF | ||
| Sehgal et al[16] | Monocyte-macrophages | Functionality of monocytes and macrophages impaired in pregnant patients developing ALF compared with those with ALI. |
| Hepatitis E in Pregnancy | ||
| Wigmore et al[17] | Monocytes | Peripheral blood mononuclear cells from patients with ALF show reduced potential to produce IL-6 and TNF and elicit an acute phase response in vitro |
| ALF | ||
| Leifeld et al[18] | Macrophages | CC-chemokines recruit and activate macrophages and T-cells. Elevated levels of serum and intrahepatic chemokines in ALF compared with controls, correlating with extent of infiltration by macrophages and T-cells. |
| ALF | ||
| dos Santos et al[19] | Eosinophils | High number of intrahepatic eosinophils in ALF, associated with increased expression of IL-6. |
| ALF | ||
| Wyke et al[20] | Complement system | Defective opsonisation due to complement deficiency. Complement factors reduced to below 40% of the activity of control serum |
| ALF |
ALF: Acute liver failure; APAP: Acetaminophen; SIRS: Systemic inflammatory response.
Neutrophils
Neutrophils - a major subset of innate immune cells- are rapidly recruited to the liver in response to liver injury. With the evidence available at present, it is unclear as to whether neutrophils directly potentiate liver injury[12]. Locally, neutrophils become activated by cytokines which may result in progressive tissue damage via release of proteolytic enzymes and reactive oxygen species. In severe sepsis, a condition which shares many clinical features with ALF, systemic neutrophil activation is associated with a functional immune paresis. Neutrophils assist in removal of necrotic cell debris in preparation for tissue repair and resolution of the inflammatory response. In ALF, neutrophils have decreased phagocytic activity - see Table 1. This decrease in phagocytosis correlates with arterial ammonia concentration, which has been shown in several studies to predict the development of cerebral edema in advanced hepatic encephalopathy[24,25]. In summary, circulating neutrophils in ALF appear to have impaired bactericidal function, and this is likely to be relevant in the increased susceptibility to infection, which may subsequently preclude life-saving liver transplantation, and hence a complete understanding of the role of cells involved in fighting infection such as neutrophils is essential.
Monocytes and macrophages
Monocytes and hepatic macrophages mediate the inflammatory response and tissue repair process occurring in ALF. Kupffer cells - resident hepatic macrophages- potentiate liver injury by sensing DAMPs and releasing pro- and anti-inflammatory mediators (with TNF-α being relevant in sensitizing hepatocytes to apoptosis); however the extent of their role remains incompletely understood[7]. Hepatic macrophages demonstrate plasticity, with their function extending from pro-inflammatory to pro-resolution. There is evidence for tolerance of circulating monocytes to bacterial endotoxins, which further impedes host immunity[26]. Liver regeneration following injury requires functional liver macrophages, the numbers of which are controlled by CSF-1 (macrophage colony stimulating factor). In patients with APAP-ALF, low serum levels of CSF-1 were associated with increased mortality[27]. Of note, administration of CSF-1 to mice increased innate immunity[27], raising the possibility that CSF-1 could be developed as a potential therapeutic target in humans. Investigating further the relationship between neutrophils, macrophages and other immune cells in human ALF would provide a clearer understanding of the pathophysiology of this condition, and potentially provide a target for urgently needed new therapies.
INFECTION AND ALF
Patients with ALF have increased susceptibility to both bacterial and fungal infection as a result of a conglomerate of factors, including reduced complement levels, impaired phagocytic function, and the need for invasive interventions. In addition, for example in those patients with cerebral edema, actions usually taken to reduce infection risk such as chest physiotherapy are contra-indicated due to the risk of exacerbating intracranial hypertension (ICH). Infection usually develops early in the course of ALF, with a median onset of 2-5 d from admission[28]. More recently, infection has been reported as a later complication in ALF, occurring at a median of 10 d[29]. Importantly, the presence of bacterial or fungal infection may preclude listing for OLT, at present the only curative option for advanced ALF.
As such, the development of infection has significant prognostic implications. There is a complex and as yet not completely understood relationship between infection, hepatic encephalopathy (HE) and outcome. Rolando demonstrated that infection is more frequent in those with higher grades of HE and higher SIRS (systemic inflammatory response) score, as a result of the mechanisms described above[30]. It is often difficult however to tease out what is cause and effect in the relationship between HE and infection. Other groups have also reported a link between the acquisition of infection and both the development and progression of HE[31]. Perhaps unsurprisingly, infection is a leading cause of death in ALF and is responsible for late death in at least a quarter of cases[28,32]. Despite this, the development of bacteremia has not been consistently shown to be independently predictive of mortality[29]. Predictors of bacteremia in ALF include admission HE grade > 2, maximum HE grade and admission SIRS score > 1[29]. The presence of a SIRS response may in fact reflect the presence of subclinical infection, or may reflect the development of an inflammatory and subsequent anti-inflammatory response, which as discussed later in this review may predispose to the acquisition of infection.
In addition, some therapies used to treat specific etiologies of ALF may also lead to the development of infectious complications, and such treatments must be used with caution when there is evidence to suggest that the development of infection is associated with a poorer outcome. One Japanese group studied the development of infectious complications in patients with autoimmune ALF treated with corticosteroids; corticosteroids were given to 19 patients, and 17 infectious complications were identified in 12 patients, at a median of 15 d[33]. Importantly, no significant differences in the clinical or biochemical features of the patients with and without infection were noted[33], reinforcing the need for a high index of clinical suspicion for infection in this group of patients. Another group reported opportunistic infection in 21.6% of ALF patients receiving steroid therapy, with cytomegalovirus and Pneumocystis jiroveci being most commonly implicated[34].
Diagnosis of infection in ALF
In patients with ALF, the clinical signs of infection such as pyrexia and elevated peripheral white cell count are absent in up to 30% of patients[35], and clinical suspicion must remain high in patients with a deteriorating course. In addition, the hemodynamic profile in ALF is similar to that observed in septic shock, adding an extra level of complexity in distinguishing between the two conditions. C-reactive protein (CRP) - a commonly used marker of infection in other clinical situations- is unhelpful in the patient with ALF. CRP is produced exclusively by hepatocytes and therefore low levels are often measured in ALF consequent to reduced hepatic parenchyma. A low CRP therefore does not reflect lack of significant inflammation or infection; Silvestre studied 7 patients with ALF and sepsis and in all septic patients, CRP levels were markedly decreased and on occasions undetectable[36], confirming the futility of CRP as a marker of infection in ALF. When meticulous microbiological surveillance is undertaken, clinical or bacteriological evidence of infection is found in up to 90% of patients with ALF[28]. Another difficulty in characterizing infections and the effect of antimicrobials in patients with ALF admitted to tertiary liver centres is that many patients are transferred from other units where they may have had microbiological cultures performed and received anti-microbial therapy. Often this information is not available and these factors may skew the results of studies of infection undertaken in tertiary liver centres.
Bacterial infection
Bacterial infections are most common, documented in 30%-80% of ALF patients[28,35,37]. Since the description of ALF as a disease entity, both the timing of the development of bacteremia and the organisms isolated has changed (Figure 2)[28,29,37]. Older data suggested that bacteremia was an early complication of ALF, occurring at a median of 3 d[28]. More recent data now support the development of bacteremia as a late complication, with Karvellas reporting a median time to bacteremia of 10 d[29]. Gram positive bacteremia was previously most common, reported in 73% of those with confirmed blood stream infection[37]. However, Gram negative bacteremia is now more common, identified in 52% of the Karvellas cohort[29]. This is of real clinical importance, as at least one study has identified a trend towards progression of HE in patients with gram negative infection compared with gram positive infection[31]. In general, critically ill patients are at increased risk of infection with antibiotic-resistant organisms such as vancomycin resistant enterococcus (VRE) and methicillin resistant Staphylococcus aureus (MRSA). Older reports of infection in ALF identified that pneumonia was the most common infection in ALF, accounting for 50% of all infections[28,35]. Urinary tract infections were the second most frequent, accounting for 22% of infections[28]. Gram positive bacteria were most commonly isolated and frequently related to pulmonary sepsis. In a more recent study of patients with ALF admitted to a Liver Intensive Care Unit, 35% developed bacteremia[29]. The most frequently isolated pathogens were Enterococcus faecium, Klebsiella spp and vancomycin-resistant Enterococcus[29], highlighting a shift in organisms most commonly implicated in bacterial infection in ALF. This may reflect changes in the choice of antimicrobial prophylaxis regimens over time, selecting out particular and resistant organisms.
Figure 2.
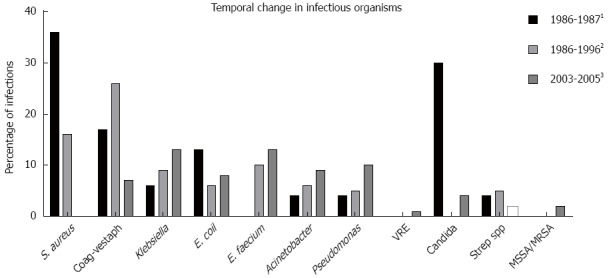
Temporal change in infections in acute liver failure. 1Adapted from Rolando et al[28]; 2Adapted from Wade et al[37]; 3Adapted from Karvellas et al[29].
Late deaths (> 7 d) in patients with ALF are often attributable to superadded bacterial infection; Rolando reported that in their cohort, all deaths occurring 7 d after admission were directly related to bacterial infection[28], highlighting the importance of remaining vigilant for the development of bacterial infection, particularly later in the course of the illness.
Fungal infection
Fungal infections are also common in the ALF patient cohort, occurring in around one third[38]. Candida species is most frequently isolated, and affected patients commonly have concurrent bacterial infection[38]. Aspergillus sp. and Pneumocystis jiroveci are also recognised to be opportunistic fungal infections in ALF[34,39]. Aggressive investigation for fungal infection should commence in the patient with deterioration in HE grade, persistent pyrexia, renal failure or a markedly elevated white cell count, in particular if the patient is already receiving and deteriorating despite broad spectrum antibiotics[38]. Without treatment, mortality with fungal infection is up to 100%[38], suggesting a possible role for prophylactic antifungal therapy. To date, no study has looked specifically and solely at the role of prophylactic antifungal therapy in ALF, and such a study may be challenging to undertake as the majority of patients will have concurrent bacterial infection which may confound results.
Viral infection
Opportunistic viral infections are common in all critically ill patients due to functional immunosuppression. In a cohort of critically ill patients due to a variety of etiologies requiring admission to the intensive care unit, cytomegalovirus (CMV) reactivation occurred in 33%, with CMV infection at any level being independently associated with death at 30 d[40]. In the ALF cohort, viral infection appears to be particularly frequent in those patients receiving steroid therapy. One study reported that of patients who were receiving steroid therapy in ALF, 25.8% developed CMV infection[34], highlighting the importance of considering the development of viral infections in appropriate patients, particularly in those who are immunosuppressed. However, the clinical relevance of opportunistic viral infection was not discussed in this study, and its impact upon outcomes remains an area for future study.
Antimicrobial prophylaxis
Despite the plausible rationale behind giving prophylactic antimicrobials to reduce risk of infection and the associated impact on HE and outcome, there is no published evidence to confirm that this approach results in a clear mortality benefit. The Acute Liver Failure Study Group (ALFSG) retrospectively assessed the impact of antimicrobial prophylaxis (physician dependent and including both anti-bacterial and anti-fungal agents) on rates of blood steam infection (BSI) and 21-d survival in ALF[32]. Of a cohort of 1551 patients with ALF (the most common etiology being APAP), 34% had at least one episode of culture-positive infection and 14.6% had at least one episode of BSI. 39% of all patients received antimicrobial prophylaxis; 19% of this cohort received antifungal therapy. Those patients receiving prophylaxis had higher HE grade and had a higher requirement for organ support, generally reflecting a sicker group of patients requiring more invasive therapy, and therefore potentially at higher risk of infection. However, there was no significant difference in the probability of developing BSI in patients receiving prophylaxis compared with those without prophylaxis (P = 0.12). In the APAP subgroup, patients receiving prophylaxis were more likely to proceed to transplantation, but there was no significant difference in overall 21-d survival. Looking at the whole cohort on multivariate analysis, antimicrobial prophylaxis did not confer a benefit on 21-d survival. It must also be borne in mind that as discussed above, empirical use of antibiotics may lead to the development of multidrug resistant organisms.
Changes in gut permeability may contribute to the development of infection in ALF, and therefore it has been proposed that selective gut decontamination with poorly absorbable oral antibiotics may be an effective method to reduce the risk of bacterial translocation and infection in ALF. Salmeron reported that in a small group of patients, the administration of poorly absorbable oral antibiotics significantly reduced the likelihood of developing infection; this reduction was predominantly related to a difference in the rate of infection from enterobacteria[41]. The clinical relevance of infection with this specific organism is not clear, and the routine use of selective intestinal decontamination cannot be advocated at present.
In summary, data regarding antimicrobial prophylaxis in ALF are limited. The ALFSG recommend that prophylactic antimicrobials and antifungals cannot be advocated in all patients - particularly those with mild HE - as these have not been consistently shown to improve overall survival rates[42]. The ALFSG advise that periodic surveillance cultures are undertaken and therapy initiated promptly according to culture results at the earliest indication of active infection or clinical deterioration[42]. Many units therefore commence antimicrobials at the development of higher grade HE requiring intubation and ventilation, the development of SIRS or otherwise unexplained clinical deterioration.
Infection and outcome
Rolando has reported upon the impact of BSI in ALF, with an attributable mortality of up to 60%-76%[28,30]. Late deaths (i.e., greater than 7 d after admission) may all be attributable to the acquisition of infection[28]. Karvellas recently demonstrated a significant increase in 21-d mortality in patients developing BSI, and this risk was higher in those patients with non-APAP etiology[32]. This is likely related to the often sub-acute and prolonged course of this illness and development of the compensatory anti-inflammatory response, both of which result in predisposition to infection. However, in an earlier study, Karvellas failed to demonstrate a significant association between the development of bacteremia and poorer outcome[29]. The data regarding the association between infection and outcome in ALF are therefore are somewhat conflicting, however clinically and pathophysiologically it is logical that the acquisition of infection results in a deteriorating course and poorer outcome.
SIRS
SIRS is the result of a clinical response to an insult of infectious or non-infectious origin, and occurs as a result of systemic pro-inflammatory (e.g., TNF-α, IL-1, IL-6) and anti-inflammatory (e.g., IL-10) cytokine release. The source of this systemic cytokine release may be from hepatocyte cell death and the necrotic liver itself, secondary to endotoxemia or impaired hepatic cytokine metabolism[43-45]. SIRS is defined as two or more of: temperature < 36 °C or > 38 °C, heart rate > 90 beats/min, leucocyte count < 4 × 109/L or > 12 × 109/L and tachypnea > 20 breaths/min or PaCO2 < 4.3 kPa. In a general population of acutely ill hospitalized patients, patients with SIRS response had a 6.9 times higher 28-d mortality than those without SIRS, and the severity of SIRS correlated with the severity of organ dysfunction and mortality rate[46]. In ALF specifically, SIRS is believed to be an important factor in the development of renal impairment, HE, multi-organ failure and death.
Difficulties in utilizing SIRS in studies of ALF
In studies of SIRS in patents with ALF, the respiratory component is often not included as many patients are ventilated and accepted respiratory parameters set. However, this approach is not uniform and not always commented upon in study methodology; this must be borne in mind when analyzing the results of any study assessing the utility of the SIRS score in ALF. In addition, many studies assess SIRS at a single time point only, whereas the true value of the score may come with serial measurements and assessment of trends. Furthermore, the appropriate cut off SIRS score for predicting prognosis in ALF has not been defined. Therefore there are limitations currently in using the SIRS score to predict prognosis in ALF, and further work is required before this score can be accepted as a prognostic marker in isolation, or be included in other validated prognostic models.
Development of SIRS and CARS
Local tissue injury is the initiating factor in the development of SIRS, and this can arise from any number of insults in the setting of ALF. Initially, this injury triggers a marked release of a number of pro-inflammatory mediators (e.g., DAMPS, TNF-α, IL-6 and IL-8)[5] through the activation of macrophages, polymorphonuclear cells, endothelial cells and the complement system. These pro-inflammatory mediators are now recognized to be associated with both tissue regeneration and tissue damage. Circulating monocytes and lymphocytes are attracted to the area of injury, and act to increase the local response to injury. Platelet and coagulation cascade activation occurs with an increase in vascular permeability and spill over of pro-inflammatory mediators, which may in turn lead to the SIRS. Systemic pro-inflammatory cytokine release following local tissue injury subsequently results in an increase in the level of circulating anti-inflammatory mediators. The point at which and the stimulus for the switch to the production of anti-inflammatory mediators is not well defined, but this counter response aims to prevent overwhelming systemic inflammation. This compensatory anti-inflammatory response syndrome (CARS) is associated with raised levels of anti-inflammatory mediators (IL-4, IL-10, TGF-β), and an impairment in cellular immune function[5]. Monocyte deactivation is key in the development of CARS[47]. This CARS phase results in an increased risk of and decreased clearance of infection, and multiorgan failure[5].
The development of SIRS is associated with poorer outcomes. SIRS is associated with progression of HE[31,48], development of bacteremia[29] and in some studies a poorer outcome independent of the presence of infection[48,49]. Karvellas reported that on multivariate analysis, SIRS was not predictive of mortality either in ALF patients (all etiologies) developing bacteremia or in non-transplanted ALF patients[29]. In contrast, Craig demonstrated that SIRS occurred significantly earlier and was of a greater magnitude in patients with APAP- ALF who died compared with patients who survived, with the number of SIRS components fulfilled post overdose being significantly associated with increased mortality at 48, 72 and 96 h time points[49]. These data suggest that perhaps the clinical effect of developing SIRS is etiology specific. SIRS develops more frequently in APAP-ALF compared with non-APAP ALF[50], and this may reflect the hyperacute course of APAP-ALF. The role of SIRS in non-APAP ALF is less well defined and is an area worthy of future research. Analysing all etiologies of ALF, Leithead identified that the presence of SIRS was associated with a reduced chance of spontaneous survival[50]. Miyake et al[48] assessed the relationship between SIRS and outcome in 99 patients with non-APAP ALF. In this cohort, increasing SIRS score was significantly associated with the development of adult respiratory distress syndrome (ARDS), disseminated intravascular coagulation (DIC), acute renal failure and multi-organ failure. Patients with a SIRS score of 0 or 1 had a significantly higher chance of survival than patients with a SIRS score of 2. This group also demonstrated - albeit in small numbers - that using SIRS as a predictor of prognosis had better specificity and positive predictive values when compared with the Kings College Hospital Poor Prognostic Criteria (KCC)[48]. The same group analyzed the association between SIRS and outcome in fulminant hepatitis B and reported that a SIRS response was independently associated with 1 wk and overall mortality[51]. The role of SIRS in the development of HE and renal dysfunction will be discussed further below.
NEUROINFLAMMATION AND HEPATIC ENCEPHALOPATHY
Progression of HE is a major determinant of outcome in ALF, and increasingly the presence of infection and/or a SIRS response is recognised to predispose to increasing HE grade and a poorer prognosis[30,31]. Following progression, patients with deeper HE are at greater risk of developing infection, as a result of poorer liver function and the invasive interventions required to manage their condition.
Key mechanisms in the development of central nervous system (CNS) complications of ALF, including cerebral edema, are incompletely understood, and much of the basic research has been undertaken in animal models of ALF. Raised arterial and brain ammonia levels- as a consequence of decreased removal by the liver- have been shown to correlate with the severity of CNS complications in ALF[52]. Ammonia sensitizes macrophages to activating stimuli, increasing the secretion of pro-inflammatory cytokines, thereby perpetuating the progression of HE and cerebral edema, yet the use of ammonia lowering therapies have not been consistently proven to prevent or treat such complications[53,54].
The concept of the “inflamed brain” in ALF has been proposed[55], and TNF-α, IL-Ib and IL-6 are thought to be key mediators in this process. Neuro-inflammation may occur as a result of a direct effect of systemically derived pro-inflammatory cytokines and/or effects of local cerebral accumulation of lactate which subsequently causes a direct neuroinflammatory response[56]. Neuro-inflammation is characterized by microglial activation and local release of pro-inflammatory cytokines. Increased cerebral lactate levels correlate with microglial activation and cerebral pro-inflammatory cytokine production, confirming its importance in the pathophysiology of HE[57]. TNF-α levels have been shown to be elevated in relation to the severity of HE in ALF[58]. Furthermore, TNF-α gene polymorphisms influence outcome in human ALF, and in the setting of APAP-ALF, decreased TNF-α production appears to protect against the development of severe HE[59].
There has been some debate as to whether the blood-brain barrier (BBB) remains intact in ALF[56,60]. Both permeability changes and complete breakdown of the BBB have been suggested. In human ALF, studies have failed to demonstrate structural BBB breakdown in contrast to the findings in murine studies[61,62]. However, TNF-α is known to disrupt the BBB and in septic encephalopathy structural BBB breakdown in reported[63], therefore it seems likely that some degree or form of BBB breakdown is intrinsically involved in the pathogenesis of HE in ALF. Transfer of systemic pro-inflammatory cytokines across a disrupted blood-brain barrier is one proposed mechanism for the development of CNS complications. Further work is required to establish the role of the blood-brain barrier - and its particular its relationship with TNF-α in the pathogenesis of HE in human ALF, to allow the development of targeted therapies.
In ALF, a significant correlation is seen between both the presence and severity of systemic inflammation, and the development of CNS complications, in particular intracranial hypertension (ICH)[51,52]. This appears to be particularly relevant in cases of APAP-ALF and in non-infected patients, a higher number of SIRS components present at admission (reflecting either sterile inflammation or occult infection) was associated with a progression to deeper stage HE[31]. In patients requiring intervention for raised intracranial pressure (ICP), SIRS score and levels of IL-1β, TNF-α and IL-6 were significantly higher than in patients not requiring therapy for raised ICP[52]. TNF-α levels correlated with cerebral blood flow (rapid increases in which are proposed to underlie the development of raised ICP in ALF)[52]. Furthermore, in the group of patients who did not require specific therapy for raised ICP, increases in the number of SIRS components fulfilled, CRP and TNF-α were associated with the development of surges of increased ICP[52]. These findings confirm that both systemic and local inflammation is important in the pathogenesis of the raised ICP of ALF. The acquisition of infection (with or without the development of SIRS) during grade I-II HE has been shown to be predictive of worsening HE in a group of patients with APAP-ALF, but interestingly not in patients with non-APAP ALF[31]. Despite this, Vaquero reported that detection of infection during the early stage of HE was not associated with a poorer outcome[31]. These findings raise the possibility that prophylactic antibiotics might benefit patients with APAP-ALF and early HE.
Recognition of the role of neuroinflammation and infection in the progression of HE allows the development of pathophysiologically based management strategies. As infection and SIRS appear to be intrinsically linked to the development and progression of HE, stringent microbiological surveillance and prompt treatment of infection are a cornerstone in management of HE in ALF. With regards to novel therapies, as TNF-α has been demonstrated to be involved in the development of HE, the use of albumin dialysis to facilitate removal of TNF-α has been proposed[64]. In animal models of ALF, etanercept (an anti TNF-α molecule) prevents microglial activation and IL-6 accumulation, delaying the onset of HE and cerebral edema[65]. It may be that etanercept provides a new therapeutic target for the patient with ALF, but primarily the role of TNF-α and related genetic polymorphisms in human ALF needs to be confirmed.
Other novel therapies targeting neuroinflammation in ALF include N-acetylcysteine (NAC) and minocycline. NAC crosses the BBB, and is reported to have anti-inflammatory properties[66]. NAC presumably has both peripheral and central actions, and has been shown to slow progression of HE and prevent cerebral edema[67,68]. Minocycline is a tetracycline antibiotic that appears to prevent microglial activation, which is known to be a major factor in the development of HE[69]. The role of indomethacin - a cyclo-oxygenase inhibitor - has also been studied; it appears to prevent the effects of cytokines on cerebrovascular endothelial cells, which form the BBB[70]. The latter two therapies have been studied predominantly in animal models of ALF, and as yet their therapeutic benefit has not been translated to human practice.
INFECTION, INFLAMMATION AND RENAL DYSFUNCTION
Renal failure is a common complication of ALF (more common in APAP-ALF than non-APAP ALF, at least in part due to direct renal toxicity caused by APAP)[50], and is associated with increased mortality[71]. Despite the clear prognostic importance of the development of renal dysfunction, the pathogenesis is not well understood. Some schools of thought believe that the renal dysfunction of ALF shares similar pathophysiological mechanisms to the hepatorenal syndrome of chronic liver disease[72]. As SIRS has been shown to contribute to the development and progression of HE and multiorgan failure, it has been proposed that the SIRS response may also be implicated in the specific development of renal dysfunction in ALF. The systemic inflammatory response stimulates apoptotic death of renal tubular cells[73], and both TNF-α and IL-6 contribute to hemodynamic disturbance in ALF, which may lead to pre-renal acute kidney injury (AKI)[74].
Studying a cohort of 308 patients with ALF, Leithead reported that 70% of patients demonstrated a SIRS response; overall, SIRS was more prevalent in the APAP-ALF cohort and was not affected by the presence of infection[50]. Those patients with APAP-ALF were more likely to develop renal dysfunction (as defined by the RIFLE criteria) than those with non-APAP ALF and this likely relates in part to a direct nephrotoxic effect of acetaminophen. Overall, patients who developed renal dysfunction demonstrated a greater systemic inflammatory response, with an increasing number of SIRS components fulfilled being associated with an increased probability of renal dysfunction. 78% of AKI patients developed SIRS, compared with 53% of the non-AKI patients and on multivariate analysis SIRS remained an independent risk factor for the development of AKI[50]. Patients with renal dysfunction were also were also more likely to demonstrate infection and superadded infection was associated with development of AKI; in both those with and without infection, SIRS was more common in the AKI patients. The risk of developing AKI and the impact of SIRS is different in the APAP-ALF and non APAP-ALF cohorts. In APAP-ALF, patients developing AKI were not more likely to have a SIRS response, and they were not more likely to have infection. In contrast, in non-APAP ALF there was a strong association between SIRS and renal dysfunction[50].
The relationship between pro-inflammatory mediators, SIRS and renal dysfunction in ALF suggest that the inflammatory cascade is central to the development of renal dysfunction. Therapies which target this inflammatory response may therefore be vital in preventing the development and halting the progression of AKI in ALF, affording a subsequent mortality benefit.
BIOMARKERS OF INFECTION AND INFLAMMATION IN ALF
There remains an urgent need for dynamic, easy to apply ALF- specific biomarkers that can be used for predicting prognosis in ALF, as currently applied prognostic scoring systems fall short on negative predictive value and specificity. Recently, a multitude of biomarkers reflecting the important role of inflammation and infection in the pathogenesis and prognosis of ALF have been proposed and studied- see Table 2[75-80]. The majority of biomarkers described in the table remain research tools at present. However, the neutrophil-lymphocyte ratio (NLR) has been proposed as an important marker of systemic inflammation. This marker is rapidly available and cost effective as it can be calculated from routine laboratory tests. The NLR has been studied in other inflammatory conditions, and found to be of prognostic value[81,82]. In ALF, NLR has been studied in single time point and staggered APAP overdose[83]. Craig reported that median NLR was higher at 72 and 96 h post overdose in single time point overdoses in patients who subsequently either died or were transplanted compared with those who spontaneously survived. A NLR > 16.7 during first 96 h following overdose correlated with the development of HE. Interestingly however, in the staggered overdose cohort, NLR was not predictive of adverse outcomes either at admission or subsequently[83]. Future human research studies should make identification and validation of a pathophysiologically based biomarker a priority, to assist in decision making regarding need for urgent liver transplantation.
Table 2.
Proposed biomarkers in acute liver failure with an immune basis
| Ref. | Biomarker study population | Relevance | Finding in ALF |
| Antoniades et al[75] | Monocyte HLA-DR expression | Monocytes are key in the immune dysregulation of ALF | Percentage of monocyte HLA-DR expression significantly lower in ALF patients compared with healthy controls, correlating with poor prognosis |
| APAP-ALI | |||
| Koch et al[76] | Soluble urokinase plasminogen activator receptor (suPAR) | suPAR related to immune activation in systemic inflammation | suPAR levels significantly increased in ALF patients, correlating with parameters reflecting hepatocyte injury |
| ALF (all aetiologies) | |||
| Craig et al[77] | Pentraxin-3 | Pentraxins are soluble pattern recognition receptors forming part of the humoral innate immune system | Admission levels of pentraxin-3 significantly higher in patients with APAP-ALI than those with non-APAP ALI. Pentraxin-3 levels significantly higher in APAP-ALI patients who died/required transplantation vs spontaneous survivors. |
| APAP-ALI | |||
| Rule et al[78] | Procalcitonin | Biomarker of bacterial infection studied in other clinical conditions | No difference in procalcitonin levels in pre-defined severity groups, non-SIRS and SIRS groups with no documented infection and no correlation with presence of infection |
| ALFSG | ALF (all aetiologies) | ||
| Antoniades et al[79] | Secretory Leukocyte Protease Inhibitor | Stimulates epithelial cell proliferation and modulates macrophage function | Higher SLPI levels were associated with a greater liver injury, infection and adverse outcome |
| APAP-ALI | |||
| Craig et al[80] | Neopterin and soluble CD163 (sCD163) | Markers of macrophage activation | Levels of both markers significantly higher in APAP-ALI compared with CLD and healthy controls. No association between biomarker level and presence of infection. |
| APAP-ALI |
ALF: Acute liver failure; APAP: Acetaminophen.
REMAINING PROBLEMS AND FUTURE RESEARCH DIRECTIONS
A better understanding of the pathophysiology of ALF- including the individual components involved in immune dysregulation and the role of infection- is vital to the development of novel treatments. Increasing understanding of the underlying pathophysiological mechanisms of acute liver injury and resolution in mouse models gives rise to the hope of developing new treatment options in humans, in whom currently liver transplantation is the only curative treatment, yet limited by organ availability and patient suitability. Monocyte and macrophage numbers and function may be the key and should be a priority focus in human research. Targeting cytokines proven to play a role in human ALF is also pivotal. However, some concern has also been raised about trialling such therapies due to a potential risk of infection, in patients who are already at higher risk of acquiring bacterial or fungal infection. Much work needs to be undertaken in the field of immunology and infection before ALF becomes a treatable disease without the requirement for organ transplantation.
Footnotes
Conflict-of-interest statement: All authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Peer-review started: March 28, 2016
First decision: May 12, 2016
Article in press: June 15, 2016
P- Reviewer: Possamai LA, Sergi CM, Staufer K S- Editor: Ma YJ L- Editor: A E- Editor: Ma S
References
- 1.Hoofnagle JH, Carithers RL, Shapiro C, Ascher N. Fulminant hepatic failure: summary of a workshop. Hepatology. 1995;21:240–252. [PubMed] [Google Scholar]
- 2.Lee WM, Squires RH, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401–1415. doi: 10.1002/hep.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernal W, Hyyrylainen A, Gera A, Audimoolam VK, McPhail MJ, Auzinger G, Rela M, Heaton N, O’Grady JG, Wendon J, et al. Lessons from look-back in acute liver failure? A single centre experience of 3300 patients. J Hepatol. 2013;59:74–80. doi: 10.1016/j.jhep.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Liou IW, Larson AM. Role of liver transplantation in acute liver failure. Semin Liver Dis. 2008;28:201–209. doi: 10.1055/s-2008-1073119. [DOI] [PubMed] [Google Scholar]
- 5.Antoniades CG, Berry PA, Wendon JA, Vergani D. The importance of immune dysfunction in determining outcome in acute liver failure. J Hepatol. 2008;49:845–861. doi: 10.1016/j.jhep.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg Nutr. 2014;3:331–343. doi: 10.3978/j.issn.2304-3881.2014.11.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Possamai LA, Antoniades CG, Anstee QM, Quaglia A, Vergani D, Thursz M, Wendon J. Role of monocytes and macrophages in experimental and human acute liver failure. World J Gastroenterol. 2010;16:1811–1819. doi: 10.3748/wjg.v16.i15.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leber B, Spindelboeck W, Stadlbauer V. Infectious complications of acute and chronic liver disease. Semin Respir Crit Care Med. 2012;33:80–95. doi: 10.1055/s-0032-1301737. [DOI] [PubMed] [Google Scholar]
- 9.Prince LR, Whyte MK, Sabroe I, Parker LC. The role of TLRs in neutrophil activation. Curr Opin Pharmacol. 2011;11:397–403. doi: 10.1016/j.coph.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 11.Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 12.Taylor NJ, Nishtala A, Manakkat Vijay GK, Abeles RD, Auzinger G, Bernal W, Ma Y, Wendon JA, Shawcross DL. Circulating neutrophil dysfunction in acute liver failure. Hepatology. 2013;57:1142–1152. doi: 10.1002/hep.26102. [DOI] [PubMed] [Google Scholar]
- 13.Manakkat Vijay GK, Ryan JM, Abeles RD, Ramage S, Patel V, Bernsmeier C, Riva A, McPhail MJ, Tranah TH, Markwick LJ, et al. Neutrophil Toll-Like Receptor 9 Expression and the Systemic Inflammatory Response in Acetaminophen-Induced Acute Liver Failure. Crit Care Med. 2016;44:43–53. doi: 10.1097/CCM.0000000000001309. [DOI] [PubMed] [Google Scholar]
- 14.Srungaram P, Rule JA, Yuan HJ, Reimold A, Dahl B, Sanders C, Lee WM. Plasma osteopontin in acute liver failure. Cytokine. 2015;73:270–276. doi: 10.1016/j.cyto.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawson JA, Farhood A, Hopper RD, Bajt ML, Jaeschke H. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol Sci. 2000;54:509–516. doi: 10.1093/toxsci/54.2.509. [DOI] [PubMed] [Google Scholar]
- 16.Sehgal R, Patra S, David P, Vyas A, Khanam A, Hissar S, Gupta E, Kumar G, Kottilil S, Maiwall R, et al. Impaired monocyte-macrophage functions and defective Toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology. 2015;62:1683–1696. doi: 10.1002/hep.28143. [DOI] [PubMed] [Google Scholar]
- 17.Wigmore SJ, Walsh TS, Lee A, Ross JA. Pro-inflammatory cytokine release and mediation of the acute phase protein response in fulminant hepatic failure. Intensive Care Med. 1998;24:224–229. doi: 10.1007/s001340050554. [DOI] [PubMed] [Google Scholar]
- 18.Leifeld L, Dumoulin FL, Purr I, Janberg K, Trautwein C, Wolff M, Manns MP, Sauerbruch T, Spengler U. Early up-regulation of chemokine expression in fulminant hepatic failure. J Pathol. 2003;199:335–344. doi: 10.1002/path.1298. [DOI] [PubMed] [Google Scholar]
- 19.dos Santos DC, da Silva Gomes Martinho JM, Pacheco-Moreira LF, Carvalho Viana de Araújo C, Caroli-Bottino A, Pannain VL, Soares Trinta K, Gandini M, da Costa Neves PC, de Souza Matos DC, et al. Eosinophils involved in fulminant hepatic failure are associated with high interleukin-6 expression and absence of interleukin-5 in liver and peripheral blood. Liver Int. 2009;29:544–551. doi: 10.1111/j.1478-3231.2008.01872.x. [DOI] [PubMed] [Google Scholar]
- 20.Wyke RJ, Rajkovic IA, Eddleston AL, Williams R. Defective opsonisation and complement deficiency in serum from patients with fulminant hepatic failure. Gut. 1980;21:643–649. doi: 10.1136/gut.21.8.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Y, Jin L, Wang J, Yan Z, Chen T, Zhao Y. Mechanisms of fibrosis in acute liver failure. Liver Int. 2015;35:1877–1885. doi: 10.1111/liv.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 23.Salas VM, Corcoran GB. Calcium-dependent DNA damage and adenosine 3’,5’-cyclic monophosphate-independent glycogen phosphorylase activation in an in vitro model of acetaminophen-induced liver injury. Hepatology. 1997;25:1432–1438. doi: 10.1002/hep.510250621. [DOI] [PubMed] [Google Scholar]
- 24.Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648–653. doi: 10.1002/hep.510290309. [DOI] [PubMed] [Google Scholar]
- 25.Strauss GI, Knudsen GM, Kondrup J, Møller K, Larsen FS. Cerebral metabolism of ammonia and amino acids in patients with fulminant hepatic failure. Gastroenterology. 2001;121:1109–1119. doi: 10.1053/gast.2001.29310. [DOI] [PubMed] [Google Scholar]
- 26.West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- 27.Stutchfield BM, Antoine DJ, Mackinnon AC, Gow DJ, Bain CC, Hawley CA, Hughes MJ, Francis B, Wojtacha D, Man TY, et al. CSF1 Restores Innate Immunity After Liver Injury in Mice and Serum Levels Indicate Outcomes of Patients With Acute Liver Failure. Gastroenterology. 2015;149:1896–1909.e14. doi: 10.1053/j.gastro.2015.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Rolando N, Harvey F, Brahm J, Philpott-Howard J, Alexander G, Gimson A, Casewell M, Fagan E, Williams R. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11:49–53. doi: 10.1002/hep.1840110110. [DOI] [PubMed] [Google Scholar]
- 29.Karvellas CJ, Pink F, McPhail M, Cross T, Auzinger G, Bernal W, Sizer E, Kutsogiannis DJ, Eltringham I, Wendon JA. Predictors of bacteraemia and mortality in patients with acute liver failure. Intensive Care Med. 2009;35:1390–1396. doi: 10.1007/s00134-009-1472-x. [DOI] [PubMed] [Google Scholar]
- 30.Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 31.Vaquero J, Polson J, Chung C, Helenowski I, Schiodt FV, Reisch J, Lee WM, Blei AT. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125:755–764. doi: 10.1016/s0016-5085(03)01051-5. [DOI] [PubMed] [Google Scholar]
- 32.Karvellas CJ, Cavazos J, Battenhouse H, Durkalski V, Balko J, Sanders C, Lee WM. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol. 2014;12:1942–9.e1. doi: 10.1016/j.cgh.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujiwara K, Yasui S, Yonemitsu Y, Arai M, Kanda T, Fukuda Y, Nakano M, Oda S, Yokosuka O. Analysis of infectious complications and timing for emergency liver transplantation in autoimmune acute liver failure. J Hepatobiliary Pancreat Sci. 2016;23:212–219. doi: 10.1002/jhbp.326. [DOI] [PubMed] [Google Scholar]
- 34.Arai M, Kanda T, Yasui S, Fujiwara K, Imazeki F, Watanabe A, Sato T, Oda S, Yokosuka O. Opportunistic infection in patients with acute liver failure. Hepatol Int. 2014;8:233–239. doi: 10.1007/s12072-013-9514-4. [DOI] [PubMed] [Google Scholar]
- 35.Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis. 1996;16:389–402. doi: 10.1055/s-2007-1007252. [DOI] [PubMed] [Google Scholar]
- 36.Silvestre JP, Coelho LM, Póvoa PM. Impact of fulminant hepatic failure in C-reactive protein? J Crit Care. 2010;25:657.e7–657.12. doi: 10.1016/j.jcrc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Wade J, Rolando N, Philpott-Howard J, Wendon J. Timing and aetiology of bacterial infections in a liver intensive care unit. J Hosp Infect. 2003;53:144–146. doi: 10.1053/jhin.2002.1363. [DOI] [PubMed] [Google Scholar]
- 38.Rolando N, Harvey F, Brahm J, Philpott-Howard J, Alexander G, Casewell M, Fagan E, Williams R. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol. 1991;12:1–9. doi: 10.1016/0168-8278(91)90900-v. [DOI] [PubMed] [Google Scholar]
- 39.Arai M, Imazeki F, Yonemitsu Y, Kanda T, Fujiwara K, Fukai K, Watanabe A, Sato T, Oda S, Yokosuka O. Opportunistic infection in the patients with acute liver failure: a report of three cases with one fatality. Clin J Gastroenterol. 2009;2:420–424. doi: 10.1007/s12328-009-0108-6. [DOI] [PubMed] [Google Scholar]
- 40.Limaye AP, Kirby KA, Rubenfeld GD, Leisenring WM, Bulger EM, Neff MJ, Gibran NS, Huang ML, Santo Hayes TK, Corey L, et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA. 2008;300:413–422. doi: 10.1001/jama.300.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salmerón JM, Titó L, Rimola A, Mas A, Navasa MA, Llach J, Ginès A, Ginès P, Arroyo V, Rodés J. Selective intestinal decontamination in the prevention of bacterial infection in patients with acute liver failure. J Hepatol. 1992;14:280–285. doi: 10.1016/0168-8278(92)90171-k. [DOI] [PubMed] [Google Scholar]
- 42.Lee WM, Larson AM, Stravitz RT. AASLD Position Paper: The management of acute liver failure: Update 2011. Available from: http: //www.aasld.org/publications/practice-guidelines-0.
- 43.Bone RC. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med. 1996;24:163–172. doi: 10.1097/00003246-199601000-00026. [DOI] [PubMed] [Google Scholar]
- 44.Wilkinson SP, Arroyo V, Gazzard BG, Moodie H, Williams R. Relation of renal impairment and haemorrhagic diathesis to endotoxaemia in fulminant hepatic failure. Lancet. 1974;1:521–524. doi: 10.1016/s0140-6736(74)92711-1. [DOI] [PubMed] [Google Scholar]
- 45.Boermeester MA, Houdijk AP, Meyer S, Cuesta MA, Appelmelk BJ, Wesdorp RI, Hack CE, Van Leeuwen PA. Liver failure induces a systemic inflammatory response. Prevention by recombinant N-terminal bactericidal/permeability-increasing protein. Am J Pathol. 1995;147:1428–1440. [PMC free article] [PubMed] [Google Scholar]
- 46.Comstedt P, Storgaard M, Lassen AT. The Systemic Inflammatory Response Syndrome (SIRS) in acutely hospitalised medical patients: a cohort study. Scand J Trauma Resusc Emerg Med. 2009;17:67. doi: 10.1186/1757-7241-17-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 48.Miyake Y, Yasunaka T, Ikeda F, Takaki A, Nouso K, Yamamoto K. SIRS score reflects clinical features of non-acetaminophen-related acute liver failure with hepatic coma. Intern Med. 2012;51:823–828. doi: 10.2169/internalmedicine.51.6686. [DOI] [PubMed] [Google Scholar]
- 49.Craig DG, Reid TW, Martin KG, Davidson JS, Hayes PC, Simpson KJ. The systemic inflammatory response syndrome and sequential organ failure assessment scores are effective triage markers following paracetamol (acetaminophen) overdose. Aliment Pharmacol Ther. 2011;34:219–228. doi: 10.1111/j.1365-2036.2011.04687.x. [DOI] [PubMed] [Google Scholar]
- 50.Leithead JA, Ferguson JW, Bates CM, Davidson JS, Lee A, Bathgate AJ, Hayes PC, Simpson KJ. The systemic inflammatory response syndrome is predictive of renal dysfunction in patients with non-paracetamol-induced acute liver failure. Gut. 2009;58:443–449. doi: 10.1136/gut.2008.154120. [DOI] [PubMed] [Google Scholar]
- 51.Miyake Y, Iwasaki Y, Terada R, Takaguchi K, Sakaguchi K, Shiratori Y. Systemic inflammatory response syndrome strongly affects the prognosis of patients with fulminant hepatitis B. J Gastroenterol. 2007;42:485–492. doi: 10.1007/s00535-007-2029-9. [DOI] [PubMed] [Google Scholar]
- 52.Jalan R, Olde Damink SW, Hayes PC, Deutz NE, Lee A. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol. 2004;41:613–620. doi: 10.1016/j.jhep.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 53.Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology. 2009;136:2159–2168. doi: 10.1053/j.gastro.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 54.Stravitz RT, Larsen FS. Therapeutic hypothermia for acute liver failure. Crit Care Med. 2009;37:S258–S264. doi: 10.1097/CCM.0b013e3181aa5fb8. [DOI] [PubMed] [Google Scholar]
- 55.Butterworth RF. The concept of “the inflamed brain” in acute liver failure: mechanisms and new therapeutic opportunities. Metab Brain Dis. 2015 doi: 10.1007/s11011-015-9747-0. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 56.Bémeur C, Butterworth RF. Liver-brain proinflammatory signalling in acute liver failure: role in the pathogenesis of hepatic encephalopathy and brain edema. Metab Brain Dis. 2013;28:145–150. doi: 10.1007/s11011-012-9361-3. [DOI] [PubMed] [Google Scholar]
- 57.Jiang W, Desjardins P, Butterworth RF. Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: protective effect of hypothermia. J Cereb Blood Flow Metab. 2009;29:944–952. doi: 10.1038/jcbfm.2009.18. [DOI] [PubMed] [Google Scholar]
- 58.Odeh M. Pathogenesis of hepatic encephalopathy: the tumour necrosis factor-alpha theory. Eur J Clin Invest. 2007;37:291–304. doi: 10.1111/j.1365-2362.2007.01778.x. [DOI] [PubMed] [Google Scholar]
- 59.Bernal W, Donaldson P, Underhill J, Wendon J, Williams R. Tumor necrosis factor genomic polymorphism and outcome of acetaminophen (paracetamol)-induced acute liver failure. J Hepatol. 1998;29:53–59. doi: 10.1016/s0168-8278(98)80178-5. [DOI] [PubMed] [Google Scholar]
- 60.Scott TR, Kronsten VT, Hughes RD, Shawcross DL. Pathophysiology of cerebral oedema in acute liver failure. World J Gastroenterol. 2013;19:9240–9255. doi: 10.3748/wjg.v19.i48.9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kato M, Hughes RD, Keays RT, Williams R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology. 1992;15:1060–1066. doi: 10.1002/hep.1840150615. [DOI] [PubMed] [Google Scholar]
- 62.Livingstone AS, Potvin M, Goresky CA, Finlayson MH, Hinchey EJ. Changes in the blood-brain barrier in hepatic coma after hepatectomy in the rat. Gastroenterology. 1977;73:697–704. [PubMed] [Google Scholar]
- 63.Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat. 2002;200:639–646. doi: 10.1046/j.1469-7580.2002.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ilonen I, Koivusalo AM, Höckerstedt K, Isoniemi H. Albumin dialysis has no clear effect on cytokine levels in patients with life-threatening liver insufficiency. Transplant Proc. 2006;38:3540–3543. doi: 10.1016/j.transproceed.2006.10.058. [DOI] [PubMed] [Google Scholar]
- 65.Chastre A, Bélanger M, Beauchesne E, Nguyen BN, Desjardins P, Butterworth RF. Inflammatory cascades driven by tumor necrosis factor-alpha play a major role in the progression of acute liver failure and its neurological complications. PLoS One. 2012;7:e49670. doi: 10.1371/journal.pone.0049670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walsh TS, Hopton P, Philips BJ, Mackenzie SJ, Lee A. The effect of N-acetylcysteine on oxygen transport and uptake in patients with fulminant hepatic failure. Hepatology. 1998;27:1332–1340. doi: 10.1002/hep.510270520. [DOI] [PubMed] [Google Scholar]
- 67.Stravitz RT, Sanyal AJ, Reisch J, Bajaj JS, Mirshahi F, Cheng J, Lee WM. Effects of N-acetylcysteine on cytokines in non-acetaminophen acute liver failure: potential mechanism of improvement in transplant-free survival. Liver Int. 2013;33:1324–1331. doi: 10.1111/liv.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bémeur C, Vaquero J, Desjardins P, Butterworth RF. N-acetylcysteine attenuates cerebral complications of non-acetaminophen-induced acute liver failure in mice: antioxidant and anti-inflammatory mechanisms. Metab Brain Dis. 2010;25:241–249. doi: 10.1007/s11011-010-9201-2. [DOI] [PubMed] [Google Scholar]
- 69.Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, Kuiper J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. 1996;64:37–43. doi: 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- 71.O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97:439–445. doi: 10.1016/0016-5085(89)90081-4. [DOI] [PubMed] [Google Scholar]
- 72.O’Grady J. Clinical disorders of renal function in acute liver failure. In: Gines P, Arroyo V, Rodes J, Schrier RW, editors. Ascites and renal dysfunction in liver disease. Oxford: Blackwell Publishing; 2005. pp. 383–393. [Google Scholar]
- 73.Wan L, Bagshaw SM, Langenberg C, Saotome T, May C, Bellomo R. Pathophysiology of septic acute kidney injury: what do we really know? Crit Care Med. 2008;36:S198–S203. doi: 10.1097/CCM.0b013e318168ccd5. [DOI] [PubMed] [Google Scholar]
- 74.Sheron N, Goka J, Wendon J, Keane H, Keays R, Alexander G, Williams R. Plasma cytokine levels in fulminant hepatic failure: associations with multiple organ failure [abstract] J Hepatol. 1991;13(Suppl):S71. [Google Scholar]
- 75.Antoniades CG, Berry PA, Davies ET, Hussain M, Bernal W, Vergani D, Wendon J. Reduced monocyte HLA-DR expression: a novel biomarker of disease severity and outcome in acetaminophen-induced acute liver failure. Hepatology. 2006;44:34–43. doi: 10.1002/hep.21240. [DOI] [PubMed] [Google Scholar]
- 76.Koch A, Zimmermann HW, Gassler N, Jochum C, Weiskirchen R, Bruensing J, Buendgens L, Dückers H, Bruns T, Gerken G, et al. Clinical relevance and cellular source of elevated soluble urokinase plasminogen activator receptor (suPAR) in acute liver failure. Liver Int. 2014;34:1330–1339. doi: 10.1111/liv.12512. [DOI] [PubMed] [Google Scholar]
- 77.Craig DG, Lee P, Pryde EA, Walker SW, Beckett GJ, Hayes PC, Simpson KJ. Elevated levels of the long pentraxin 3 in paracetamol-induced human acute liver injury. Eur J Gastroenterol Hepatol. 2013;25:359–367. doi: 10.1097/MEG.0b013e32835ac77a. [DOI] [PubMed] [Google Scholar]
- 78.Rule JA, Hynan LS, Attar N, Sanders C, Korzun WJ, Lee WM. Procalcitonin Identifies Cell Injury, Not Bacterial Infection, in Acute Liver Failure. PLoS One. 2015;10:e0138566. doi: 10.1371/journal.pone.0138566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Antoniades CG, Khamri W, Abeles RD, Taams LS, Triantafyllou E, Possamai LA, Bernsmeier C, Mitry RR, O’Brien A, Gilroy D, et al. Secretory leukocyte protease inhibitor: a pivotal mediator of anti-inflammatory responses in acetaminophen-induced acute liver failure. Hepatology. 2014;59:1564–1576. doi: 10.1002/hep.26933. [DOI] [PubMed] [Google Scholar]
- 80.Craig DG, Lee P, Pryde EA, Hayes PC, Simpson KJ. Serum neopterin and soluble CD163 as markers of macrophage activation in paracetamol (acetaminophen)-induced human acute liver injury. Aliment Pharmacol Ther. 2013;38:1395–1404. doi: 10.1111/apt.12530. [DOI] [PubMed] [Google Scholar]
- 81.Zou ZY, Liu HL, Ning N, Li SY, DU XH, Li R. Clinical significance of pre-operative neutrophil lymphocyte ratio and platelet lymphocyte ratio as prognostic factors for patients with colorectal cancer. Oncol Lett. 2016;11:2241–2248. doi: 10.3892/ol.2016.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun XD, Shi XJ, Chen YG, Wang CL, Ma Q, Lv GY. Elevated Preoperative Neutrophil-Lymphocyte Ratio Is Associated with Poor Prognosis in Hepatocellular Carcinoma Patients Treated with Liver Transplantation: A Meta-Analysis. Gastroenterol Res Pract. 2016;2016:4743808. doi: 10.1155/2016/4743808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Craig DG, Kitto L, Zafar S, Reid TW, Martin KG, Davidson JS, Hayes PC, Simpson KJ. An elevated neutrophil-lymphocyte ratio is associated with adverse outcomes following single time-point paracetamol (acetaminophen) overdose: a time-course analysis. Eur J Gastroenterol Hepatol. 2014;26:1022–1029. doi: 10.1097/MEG.0000000000000157. [DOI] [PubMed] [Google Scholar]