

Significance
Viruses usurp the host cell machinery to replicate, disseminate, and propagate themselves. Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a viral protein kinase (vPK) also known as ORF36. Using in silico modeling and biochemistry, we report that vPK/ORF36 displays limited homology to cellular S6 kinase B1 (S6KB1). Both kinases share overlapping substrates and can phosphorylate S6. However, unlike S6KB1, vPK augments S6 phosphorylation under conditions where mammalian target of rapamycin (mTOR) is inhibited. vPK modulates cellular proliferation and protein synthesis, augments anchorage independence, and enhances angiogenesis. Depletion of vPK/ORF36 during lytic replication inhibits the production of infectious virions, thereby underscoring the importance of this kinase during the KSHV life cycle. Our collective observations suggest that vPK may function as a constitutively active mimic of S6KB1.
Keywords: cell signaling, viral protein kinase, S6K, KSHV, ORF36
Abstract
Viruses depend upon the host cell for manufacturing components of progeny virions. To mitigate the inextricable dependence on host cell protein synthesis, viruses can modulate protein synthesis through a variety of mechanisms. We report that the viral protein kinase (vPK) encoded by open reading frame 36 (ORF36) of Kaposi’s sarcoma-associated herpesvirus (KSHV) enhances protein synthesis by mimicking the function of the cellular protein S6 kinase (S6KB1). Similar to S6KB1, vPK phosphorylates the ribosomal S6 protein and up-regulates global protein synthesis. vPK also augments cellular proliferation and anchorage-independent growth. Furthermore, we report that both vPK and S6KB1 phosphorylate the enzyme 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 2 (PFKFB2) and that both kinases promote endothelial capillary tubule formation.
Protein kinases (PKs) phosphorylate cellular proteins and alter substrate localization, enzymatic activity, and protein interactions, thereby affecting signal transduction and global cellular function. The human genome encodes ∼500 PKs that modify as many as 30% of all cellular proteins (1). One vital serine/threonine kinase, the mammalian target of rapamycin (mTOR), integrates cellular demands, such as nutrient availability and environmental stress, and tightly controls protein biosynthesis via phosphorylation of a cascade of effectors (2). To facilitate protein synthesis, mTOR inhibits the repressor binding protein (4EBP), which normally sequesters eukaryotic initiation factor 4E (eIF4E), and prevents cap-dependent protein synthesis. Cap-dependent translation occurs once eIF4E binds the 5′ m7-guanosine cap of eukaryotic mRNAs. Concurrently, mTOR also phosphorylates ribosomal protein S6 kinase (S6KB1), which in turn activates protein synthesis by phosphorylating tandem serines on the ribosomal S6 protein, a component of the 40S ribosome (2).
In addition to S6, other S6KB1 targets promote cellular protein synthesis (3). For instance, S6KB1 activates eIF4B, an RNA-binding protein that facilitates the binding of mRNA to 40S preinitiation complexes (4). Moreover, S6KB1 is also known to regulate MDM2, a ubiquitin ligase responsible for degrading p53, thus preventing DNA damage-induced cell cycle arrest or apoptosis (5). By modulating its broad repertoire of targets, S6KB1 plays a key role in regulating the fundamental cellular responses to energy status, cell survival, and proliferation.
Kaposi’s sarcoma-associated herpesvirus (KSHV) is linked to Kaposi’s sarcoma (KS), primary effusion lymphoma, and multicentric Castleman’s disease. KS is a tumor driven by the proliferation of KSHV-infected endothelial cells whereas the latter are two B-cell lymphoproliferative diseases. The KSHV genome encodes several proteins that broadly alter the cellular signaling milieu and consequently impact survival and proliferation of infected cells. For example, KSHV K1 modulates B-cell receptor-mediated signaling and KSHV vFLIP inhibits apoptosis (6). KSHV also encodes two kinases: ORF21, a homolog of cellular thymidine kinase, and a viral protein kinase (vPK) encoded by ORF36. vPK is known to be incorporated into the KSHV virion and is presumed to be released into the host cell cytoplasm immediately after entry (7, 8). The phosphorylation targets of vPK include the cellular proteins c-Jun N-terminal kinase (JNK) and the mitogen-activated kinases MKK4 and MKK7 (8), and K-bZIP, a viral protein capable of repressing viral transcription (9). Other vPK targets modulate the cellular response to DNA damage (10) and are important for subverting type 1 IFN-mediated antiviral signaling (11). This wide repertoire of targets results from vPK localizing within both the cytoplasmic and nuclear compartments (12). Within the KSHV genome, vPK is located within the ORF 34–37 cluster, which contains a hypoxia-inducible factor (HIF) response element within the promoter region (13, 14). Thus, vPK expression may be induced in hypoxic environments, independent of lytic replication. Tamburro previously analyzed KS lesions derived from KS-afflicted patients in Malawi and reported that 59% of KS biopsies have detectable vPK mRNA expression (15).
In silico structural modeling of KSHV vPK revealed similarities between vPK and S6KB1. Our studies reveal that vPK mimics S6KB1, sharing several common substrate targets and modulating some similar cellular pathways.
Results
vPK Bears Structural Similarity to Human S6KB1.
The fold recognition server HHpred (16) was used to identify suitable templates for modeling structures of KSHV vPK. All top 100 hits from HHpred were kinases, with most hits belonging to the serine-threonine protein kinase family. Among these hits, one of the significant (E value < 1.0E-41) similarities was to S6KB1, also known as human p70 ribosomal S6 kinase (p70S6K), in addition to other S6 kinases as well. We used the MODELLER package and sequence alignment from HHpred to predict structures for vPK (17). Three structures of vPK were generated using each of the three structures of S6KB1 (derived using S6KB1 NCBI accession number NM_003161) available in the Protein Data Bank (PDB) database. Two of the crystal structures were of different unphosphorylated states of S6KB1 (PDB ID codes 3A60 and 3A61), and one was of the partially activated phosphorylated state of S6KB1 (PDB ID code 3A62) (18). The structural regions used as templates comprised residues 85–357 of human S6KB1, against which we modeled residues 78–369 of vPK. Over this region, vPK had four loop insertions 16, 6, 10, and 27 residues in length, and two loop deletions 8 and 5 residues in length, along with five 1- to 2-residue-long indels. We verified the sequence-structure compatibility of the model of vPK based on PDB ID code 3A62 (with the three longest inserted loops removed) using the Verify3D Structure Evaluation Server (19).
The model of vPK based on the partially activated state of S6KB1 is rendered in spheres in Fig. 1A (18). The three longest inserted loops are not shown because the loop structure cannot be predicted reliably. Residues highly conserved between S6KB1 and vPK are colored yellow, and the remainder is colored teal. Most of the conserved residues between the two kinases occur in three motifs that cross the active site pocket. The first motif—KrLGRGaFG (uppercase residues are conserved and in yellow)—consists of residues K88 to G96. The second and third motifs—DvsPDNI and LTDFG—consist of residues D201 to I207 and L223 to G227. Visible residues in these motifs are labeled in Fig. 1A. Computational modeling of vPK based on a partially activated S6KB1 identified potential functional residues of vPK.
Fig. 1.

KSHV vPK displays limited homology to cellular S6KB1. (A) In silico model of vPK based on the partially activated state of S6KB1 is rendered in spheres. Residues highly conserved between S6KB1 and vPK are colored yellow, and the rest are teal. Most of the conserved residues between the two kinases occur in three motifs that cross the active site pocket. The first motif—KrLGRGaFG (uppercase residues are conserved and in yellow)—consists of residues K88 to G96. The second and third motifs—DvsPDNI and LTDFG—consist of residues D201 to I207 and L223 to G227. (B) A PepChip array was used to identify targets of both vPK and S6KB1. Recombinant kinases were incubated with radiolabeled ATP on a glass slide arrayed with >1,000 kinase substrate peptides. (C) Twenty-four peptides are phosphorylated by both vPK and S6KB1. vPK and S6KB1 were found to uniquely phosphorylate an additional 56 and 53 peptides, respectively. (D) Scatter plot analysis of spot intensities of peptides phosphorylated either uniquely by vPK (y axis; closed circles) or S6KB1 (x axis; closed squares), dually phosphorylated by both (closed triangles), or spots phosphorylated by neither kinase (open circles).
The K123 residue of S6KB1 is involved in ATP binding and is critical for kinase activity. The corresponding residue in vPK was found to be at K108, previously reported to also be essential for ATP binding (8). We found that the K108A mutant of vPK abolished S6 phosphorylation, further highlighting the structural and functional similarity of vPK and S6KB1 (Fig. S1).
Fig. S1.
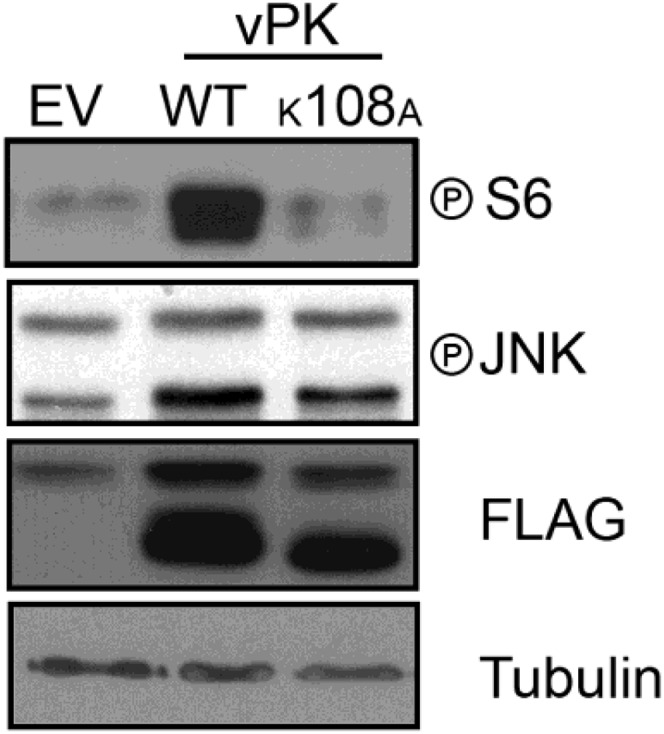
Immunoblot of 293 cells transfected with indicated plasmids. Transfection of WT vPK elevates phospho-S6 compared with the K108A mutant vPK. As previously reported, increased phosphorylation in phospho-JNK is also observed with vPK transfection. Expression of vPK constructs is confirmed by FLAG immunoblot, and tubulin is used as a loading control.
Kinase Profiling Indicates Overlapping Specificity of KSHV vPK and S6KB1.
The structural similarities between vPK and S6KB1 highlighted by computational modeling predict an overlap in substrate specificity of the two kinases. We used kinase substrate profiling (Fig. 1B) to identify peptides preferentially phosphorylated by these kinases. Among the >1,300 peptides on our peptide grid, vPK and S6KB1 respectively phosphorylated 80 and 77 peptides, with 24 common peptide targets (Fig. 1C). Among these peptides was S6, a component of the 40S ribosome complex, and the primary target of S6KB1. Peptides mapping to the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFK2/FBPase or PFKFB2) were also phosphorylated by both vPK and S6KB1. PFKFB2, normally regulated by PI3K-dependent signaling (20), controls the steady-state levels of fructose-2, 6-bisphosphate, a glycolysis intermediate. We also identified many peptide sequences that are phosphorylated by cellular S6KB1 that were not previously reported in the literature.
Scanned images of each slide were scored for phosphorylation above background, and the intensity of each spot was measured. Intensities were plotted as seen in Fig. 1D, with vPK and S6KB1 along the x and y axes, respectively. The scattered intensity data points formed four distinct groups: A large number of peptides (840) were not phosphorylated by either kinase. Every other given peptide spot was either singly phosphorylated (termed single positives) by either vPK (56 peptides) or S6KB1 (53 peptides), or phosphorylated by both (double positives, 24 peptides). Fisher’s exact test was applied to determine whether the number of double positives was enriched, given the number of single positive and negative spots. We found a significant enrichment for double positives (odds ratio 6.8; P < 1E-9), indicating that both vPK and S6KB1 share a common set of target substrates, thus supporting their underlying structural similarities.
S6 Is a Verified vPK Target Substrate.
Using classical in vitro kinase assays, we found that recombinant vPK and S6KB1 both efficiently phosphorylate a synthetic S6 peptide to similar levels, confirming our substrate peptide profiling data (Fig. 2A). Addition of DG2, a selective S6KB1 inhibitor, ablated phosphotransferase activity of both S6KB1 and vPK (21, 22). DG2 was designed to selectively bind the active site of S6KB1; thus, the ability of DG2 to inhibit vPK to levels similar to S6KB1 confirmed the similarity in active sites of these kinases (Fig. 2B). In contrast, neither SP-600125 (JNK inhibitor) nor MLN-8237 (aurora A inhibitor) impacted phosphorylation of S6 by either kinase (Fig. 2B).
Fig. 2.

KSHV vPK phosphorylates several S6KB1 substrates. (A) An in vitro kinase assay was performed with recombinant vPK and S6K using a synthetic S6 substrate. Data are representative of four independent experiments; error bars denote SEM. (B) Impact of S6KB1-specific and nonspecific kinase inhibitors on recombinant vPK and S6KB1 phosphorylation of S6 peptide substrate in an in vitro kinase assay. Data are representative of three independent experiments; error bars are ±SEM. (C) Ectopic expression of vPK in 293 cells increases phosphorylated levels of S6. Total S6 and tubulin are shown as loading controls. Construct expression is verified using a vPK-specific antibody. These images are representative of three independent experiments. ℗, phospho. (D) Stable HUVECs were first plated in normal media; subsequently, serum was withdrawn for 16 h, and immunoblots were performed with harvested lysates for the indicated phosphorylated and corresponding total proteins. Tubulin is shown as loading control. These images are representative of four independent experiments. (E) Metabolic labeling of de novo protein synthesis using 35S-labeled methionine and cysteine was quantified in 293 cells transiently transfected with vector, vPK, or S6KB1. Counts are normalized to total protein content. Data are representative of three independent experiments; error bars are ±SEM. (F) Metabolic labeling of de novo protein synthesis using 35S-labeled methionine and cysteine was quantified in stable HUVECs expressing vPK, S6KB1, or matched vector control. Counts are normalized to total protein content. Data are representative of three independent experiments; error bars are ±SEM, **P < 0.01, ****P < 0.0001.
To confirm the functional similarity of these proteins, eukaryotic expression vectors for vPK and matched vector control were transiently transfected into 293 cells for 36 h and either placed in normal media or serum-starved for 12 h. Lysates prepared from harvested cells were subjected to immunoblotting. Levels of phospho-S6 were significantly higher in vPK-transfected cells compared with vector controls (Fig. 2C, Left). vPK sustained phospho-S6 levels even in serum-starved cells (Fig. 2C, Right). We replicated our studies by stably expressing either vPK, S6KB1, or matched empty vector controls in immortalized human umbilical vein endothelial cells (HUVECs). Because Kaposi’s sarcoma is an angioproliferative tumor primarily driven by endothelial cells, HUVECs represent a biologically relevant cell type to test the function of vPK. Immunoblot analyses of harvested log phase cells showed elevated phospho-S6 in both vPK- and S6K-expressing HUVEC stable cells, compared with empty vector (Fig. 2D). Further confirming the peptide profiling data, we found that both vPK and S6KB1 induce phosphorylation of PFKFB2, an enzyme responsible for enhancing glycolysis. Interestingly, vPK also increased phosphorylation of the eukaryotic initiation factor 4E (eIF4E), the cap recognition protein responsible for binding to the m7-guanosine cap of eukaryotic messenger RNAs. Normally, eIF4E is sequestered via association with 4E binding protein (4EBP1); however, mTOR-dependent hyperphosphorylation of 4EBP1 releases eIF4E, which subsequently promotes translation initiation (23). vPK was also found to phosphorylate the S6KB1 target MDM2, further accentuating the high degree of substrate overlap of these two kinases (5). Despite converging substrates, both kinases also have a discrete repertoire of targets. For example, S6KB1, but not vPK, phosphorylates eIF4B, an ATP-dependent helicase essential for protein translation, and, as previously reported, vPK phosphorylates the mitogen-activated kinase JNK, whereas S6KB1 does not (8). We transfected HUVECs with nontargeting (NT) siRNA, S6KB1 siRNA, or vPK-specific siRNA. Twenty-four hours later, the cells were infected with KSHV as previously described (24). Cells were harvested 1 h postinfection (HPI), and lysates were subjected to SDS/PAGE and immunoblotting. We found that KSHV infection induces phospho-S6 and phospho-JNK in HUVECs (Fig. S2). Furthermore, prior S6KB1 knockdown does not substantially decrease phospho-S6 in the context of KSHV infection (Fig. S2), suggesting that vPK activates S6 in an S6KB1-independent manner. vPK siRNA does not reduce phospho-S6, consistent with our current understanding that vPK is a tegument protein that is packaged within the tegument layer of the virion and released into the cytoplasm after virion infection (8).
Fig. S2.

Immunoblot of HUVECs transfected for 24 h with indicated siRNAs and then infected with KSHV for 90 min. Infection was carried out as described by West and Damania (24). One hour postinfection (HPI), cells were harvested, and immunoblots were performed. KSHV infection induces S6 phosphorylation in an S6KB1-independent manner.
KSHV vPK Enhances Global Protein Synthesis.
To determine whether increased S6 phosphorylation by vPK corresponds with alteration of global protein synthesis, we measured incorporation of radioactive methionine and cysteine in 293 cells expressing vPK, S6KB1, or vector. As expected, ectopically expressed S6KB1 enhanced protein synthesis compared with vector. Interestingly, vPK also increased the basal levels of global protein synthesis compared with vector (Fig. 2E). We also measured basal protein synthesis in stable HUVECs expressing either vPK or S6KB1 and noted a statistically significant elevation in protein synthesis over vector control, when either vPK or S6KB1 was expressed (Fig. 2F).
KSHV vPK Enhances Cellular Proliferation.
To determine whether vPK-mediated enhancement of S6 phosphorylation impacts the physiology of the host cell, we assayed cellular metabolic rates. Stable HUVECs were transfected with either scrambled or vPK-specific siRNA for 24 h and expression after knockdown was confirmed by immunoblot (Fig. S3). Equivalent numbers of cells were seeded onto 96-well plates, and cellular proliferation was quantified using Cell Titer Aqueous One MTS reagent. By comparing the absorbance values of each stable cell line over a course of 72 h, we found that vPK-expressing HUVECs have a higher absorbance compared with vector cells. However, when transfected with vPK-specific siRNA, cellular proliferation rates of HUVEC-vPK were reduced to those of vector (Fig. 3A), which indicates that a vPK-mediated increase in S6 phosphorylation may translate into accelerated cell proliferation rates.
Fig. S3.

Immunoblot analysis of lysates prepared from stable HUVECs transfected with either nontargeting control (NTC) or vPK-specific siRNA (siVPK). Blot is probed with vPK-specific antibody, and shows significant knockdown in HUVEC-vPK transfected with vPK-specific siRNA (siVPK). Ku70 is shown as a loading control.
Fig. 3.

KSHV vPK enhances cellular proliferation. (A) Stable HUVECs transfected with indicated siRNAs were plated in a 96-well plate, and their basal metabolic rate was measured 72 h afterward using the Cell Titer Aqueous One Assay. Data are representative of three independent experiments; error bars are ±SEM, ****P < 0.001. NTC, nontargeting control. (B) Immunoblot analyses of indicated proteins with lysates prepared from 293 cells transiently transfected with empty vector, vPK, or S6KB1 and then treated with either LY294002 or rapamycin (10 nM), each for 1 h. Transgene expression was confirmed using vPK- and FLAG-specific antibodies. Tubulin is shown as a loading control. Images represent one of three independent experiments. (C) Stable HUVECs were treated with increasing doses of rapamycin (10, 50 nM) for 1 h, and harvested lysates were subject to immunoblotting with indicated phospho-specific and total antibodies. Although phospho-S6 remains elevated after rapamycin treatment in HUVEC-vPK, it is diminished in vector- and S6KB1-expressing HUVECs. Images are representative of three independent experiments. (D) Stable 293 cells were transfected with indicated siRNAs for 48 h, and immunoblots were performed after 12-h serum withdrawal. mT, pooled Raptor- and mTOR-specific siRNA; Scr, scrambled. (E) Immunoblot analysis of both latent (No Dox) and reactivated (Dox) iSLK.219 cells transfected with either vPK-directed or a nonspecific (NS) siRNA. Representative of four independent experiments. (F) Infectivity of 293 cells infected with KSHV derived from the experiment described in E; **** P < 0.0001.
S6 phosphorylation is tightly regulated by the upstream kinase, S6KB1, which itself is regulated by the PI3K/AKT/mTOR signaling pathway. mTOR activity can be subdued either by inhibition of upstream regulatory kinases, such as PI3K using the reversible inhibitor LY294002, or by treatment with rapamycin, a macrolide that directly binds to and inhibits the mTOR complex. Diminished S6 phosphorylation (particularly at S235, S236) is a prognostic indicator of rapamycin’s efficacy (25) and a signature of S6KB1 inhibition. We have previously shown that the PI3K/AKT/mTOR pathway is critical for the survival of KSHV-infected cells (25, 26). We treated transfected 293 cells with 10 nM LY294002 or rapamycin, or the respective vehicle, for 1 h, after which cells were harvested and immunoblotting was performed with the lysates. Interestingly, we found that after LY294002 and rapamycin treatment, S6 phosphorylation was sustained in vPK-expressing cells and was at a higher level than the vector- and S6K-transfected cells, where phospho-S6 was diminished (Fig. 3B). We also treated stable HUVECs with increasing doses of rapamycin (10 and 50 nM) for 1 h and performed immunoblots. Similar to transfected 293 cells, S6 phosphorylation after rapamycin treatment was still higher in vPK-expressing stable HUVECs (Fig. 3C), compared with vector- and S6KB1-expressing HUVECs. These observations indicate that, despite mTORC1 inhibition, vPK can sustain phosphorylation of S6.
Next, we transfected stable vPK-expressing 293 cells with either scrambled or a pool of both mTOR- and Raptor-specific siRNAs (denoted “mT”) and analyzed phospho-S6 levels (Fig. 3D). A reduction in phospho-S6 levels was observed in mTOR-regulated S6KB1 (Thr389) concurrent with mTOR/Raptor knockdown. vPK-expressing 293 cells had higher levels of phospho-S6 even in the context of mTOR/Raptor knockdown, confirming that vPK can sustain phosphorylation of S6 even when mTORC1 expression is depleted.
To dissect the role of vPK in the viral life cycle, we used the latent KSHV-infected iSLK cell line, which can be induced into lytic replication by doxycycline-dependent expression of the replication and transcription activator (RTA) (27). This cell line fluoresces green when latently infected with KSHV; productive viral replication leads to RFP expression under control of a lytic promoter. iSLK.219 cells were transfected with either nontargeting (NT) or vPK-directed siRNA, with simultaneous doxycycline-mediated reactivation. Cells transfected with NT siRNA had maximal vPK expression within 48 h after reactivation, with a concurrent elevation in S6 phosphorylation (Fig. 3E). We found that after vPK-specific siRNA knocked down vPK expression, S6 phosphorylation was greatly reduced. To gauge the degree of infectivity of newly synthesized virions, supernatants from latent and reactivated iSLK.219 cells containing either vPK-specific or NS siRNA were collected, clarified, and applied for 48 h to naive 293 cells; infection was monitored by fluorescence microscopy. Compared with virus derived from NT siRNA-transfected cells, KSHV derived from vPK-knockdown cells was significantly attenuated, indicated by the small number of GFP+ 293 cells (Fig. 3F).
vPK Expression Augments Anchorage Independence.
To investigate whether increases in S6 phosphorylation and protein synthesis functionally impact the growth of vPK-expressing cells, we performed a soft agar assay using stable Rat1 fibroblasts. Five hundred stable cells were suspended in agarose-containing media. Three weeks later, colonies were stained with crystal violet and counted in a blinded manner. Rat1 fibroblasts expressing empty vector or GFP (negative control) formed very few colonies whereas S6KB1 expression modestly increased the number of colonies. In contrast, vPK-expressing cells had a significantly higher number of colonies. Simian virus 40 (SV40) T antigen was used as a positive control (Fig. 4A).
Fig. 4.

KSHV vPK augments tubule formation. (A) Rat1 fibroblasts stably expressing the indicated transgenes were plated in soft agar for 3 wk. Colonies were stained with crystal violet and quantified to measure anchorage independence. Data are representative of four independent experiments; error bars are ±SEM; ****P < 0.0001. (B) When placed in growth factor-reduced Matrigel, vPK-stable HUVECs form tubular networks within 4 h. Also shown are EV- and S6KB1-expressing cells. (C) Significantly increased branching was observed at 4 h (Left) and 8 h (Right) in vPK-expressing HUVECs, compared with EV- and S6KB1-HUVEC. Tubule formation data are representative of three independent experiments. (D) Proposed model for vPK’s mechanism of action.
vPK Induces Endothelial Tubule Formation in HUVECs.
Endothelial tubule formation assays measure the ability of endothelial cells to induce tubule formation in vitro (28). Stable HUVECs expressing either vPK or empty vector were used. Growth factor-reduced Matrigel was used to mitigate any cytokines and growth factors contained in standard Matrigel. Stable HUVEC-expressing empty vector formed small tubules 4 h after placement on Matrigel, with maximal tubular networks formed within 12 h. Tubule formation was accelerated in vPK-expressing HUVECs, with intricate tubular networks forming as early as 4 h after plating (Fig. 4B; quantified in Fig. 4C, Left). By 8 h, extensive tubular matrices could be seen in vPK- and S6KB1-expressing HUVECs (Fig. 4C, Right). At all time points analyzed, tubule branching was significantly higher in vPK-expressing cells, compared with EV- and S6KB1-expressing HUVECs. S6KB1-expressing HUVECs showed increased tubule formation at and after 8 h compared with the vector-control HUVECs (Fig. 4C). These observations suggest that protein synthesis induced by vPK may drive angiogenic remodeling in endothelial cells. Alternatively, it is also possible that vPK and S6KB1 modulate expression and/or activation of proteins that impact endothelial cell remodeling and angiogenesis.
Discussion
Herpesviruses establish lifelong latency in the host, with sporadic bouts of viral reactivation and replication. These viruses commandeer host cell machinery to synthesize viral proteins needed for successful replication. For example, UL13 and UL97, orthologous kinases encoded by herpes simplex type I (HSV1) and human cytomegalovirus (HCMV), respectively, are functionally similar to the cellular kinase CDC2/CDK1 and phosphorylate several cellular functional targets of cyclin-dependent kinases, including elongation factor 1δ (EF1δ) (29) and pRB (30). Herpesviral kinases are also known to phosphorylate several different members of the cellular DNA damage response pathway. Notably, the regulatory protein TIP60 is targeted by herpesvirus kinases and is known to be essential for efficient herpesvirus replication (10).
Like other herpesviruses, KSHV depends on the host for replication, propagation, and dissemination. Using in silico structural modeling approaches, we discovered that vPK displays limited structural similarity to the cellular S6KB1 (also known as p70S6K or RPS6KB1) protein. Given that vPK homologs are present in all human herpesviruses and that it is predicted to be a core protein in the common ancestor to the Herpesviridae (31), acquisition of the vPK ancestral protein possibly occurred more than 400 million y ago. S6KB1 expression is known to enhance protein synthesis and is itself tightly controlled via phosphorylation by mTORC1. We hypothesize that vPK’s ability to mimic S6KB1 is advantageous for the virus because it provides a mechanism for activating protein synthesis despite host antiviral signaling, which normally down-regulates protein synthesis as a defensive strategy.
We identified overlapping and unique cellular targets of both vPK and S6KB1 using a peptide array. We identified S6 as a significant target of vPK-mediated phosphorylation. As a component of the eukaryotic 40S ribosome, S6 is phosphorylated at tandem serines in an mTOR-dependent manner by its cognate kinase S6KB1. Phosphorylated S6 is a well-established signature of active protein translation of capped mRNAs. Using an in vitro kinase assay, we found that vPK phosphorylates S6 peptide at the same residues phosphorylated by S6KB1 (S235, S236), which denotes activation of protein translation. Moreover, DG2, a S6KB1-specific kinase inhibitor (21, 22), abolished phosphotransferase activity of both S6KB1 and vPK, underscoring the similarity of the active sites of these two kinases. Through ectopic vPK expression in both epithelial (293) and endothelial (HUVEC) cells, we confirmed that vPK phosphorylates S6, the furthest downstream effector of PI3K/AKT/mTOR signaling. Overall, our data suggest that there are a set of common targets of vPK and S6KB1. Additionally, vPK can phosphorylate proteins that S6K cannot, thereby expanding the repertoire of cellular proteins that vPK can target.
We report that levels of phospho-S6 are reduced when vPK expression was depleted during lytic reactivation. Moreover, virions generated from vPK-depleted cells were less infectious, compared with nonspecific siRNA-transfected cells. These observations suggest that activation of cellular pathways (e.g., protein synthesis) by vPK is important for virion production during the replicative life cycle.
Furthermore, vPK-mediated S6 phosphorylation translates into enhanced global protein synthesis in cells of both epithelial and endothelial origin. A recent report used quantitative phosphoproteomics to identify strictly nuclear targets of BGLF4, the Epstein–Barr virus homolog of KSHV vPK. BGLF4 was found to affect the cellular phosphoprotein milieu, including the phosphorylation of mitotic kinases (32).
vPK was also found to phosphorylate an essential regulator of glycolysis, PFKFB2, which we found is also a direct S6KB1 target. Glycolysis is a frequent bioenergetic adaptation of cancer cells, both in aerobic conditions and in hypoxic environments unfavorable for oxidative phosphorylation. Notably, vPK is induced by hypoxia (13, 14), and, furthermore, KSHV is known to up-regulate glycolysis in both lytic and latent conditions (33, 34). Thus, vPK may modulate the induction of the glycolytic phenotype. We found that vPK-expressing HUVECs established a larger number of tubule networks compared with control cells at all time points. A recent report underscores the importance of glycolysis for driving capillary endothelial tubule formation in vivo (35). Since PFKFB2 was found to be a target for phosphorylation by vPK and S6KB1, it is likely that the mechanism for tubule formation by these two kinases is similar and may be independent of their impact on protein synthesis. Nonetheless, vPK-induced enhancement of tubule formation suggests that vPK may play a role in the angiogenesis apparent in Kaposi’s sarcoma.
We propose a model wherein vPK circumvents regulatory control by upstream kinases and activates the ribosomal protein S6, the furthest downstream effector of PI3K/AKT/mTOR signaling (Fig. 4D). By directly phosphorylating S6, vPK abolishes the need for S6KB1-dependent phosphorylation, thus preempting inhibitory signaling that may result from growth factor withdrawal, nutrient scarcity, or active antiviral signaling. In conclusion, based on our in silico findings and biochemical data, we propose that vPK mimics S6KB1 function and, as such, induces infected cells to up-regulate protein synthesis, proliferate, and vascularize, despite upstream inhibitory signals. These findings bear significant implications for KSHV-associated malignancies including Kaposi’s sarcoma.
Materials and Methods
Human embryonic kidney-293 (293) cells were cultured in DMEM supplemented with 10% (vol/vol) heat-inactivated FBS, 100 IU/mL penicillin G and 100 μg/mL streptomycin (P/S), and 2 mM l-glutamate. Primary human umbilical vein endothelial cells were cultured in endothelial cell growth medium supplemented with 10% heat-inactivated FBS, as previously described (28, 36). Rat1 fibroblasts were cultured in DMEM with 10% heat-inactivated calf serum, P/S, and 2 mM l-glutamate. The iSLK.219 clone 10 cells were cultured as previously described (27). Additional methods are described in SI Materials and Methods.
SI Materials and Methods
Cell Culture, Plasmids, and Inhibitors.
Human embryonic kidney-293 (HEK293) cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, 100 IU/mL penicillin G, 100 μg/mL streptomycin, and 2 mM l-glutamate. Primary human umbilical vein endothelial cells (HUVECs) were cultured in endothelial cell growth medium supplemented with 10% heat-inactivated FBS, as previously described (28). Rat1 fibroblasts (gift of Adrienne Cox, University of North Carolina at Chapel Hill) were cultured in DMEM with 10% heat-inactivated calf serum, 100 IU/mL penicillin G, 100 μg/mL streptomycin, and 2 mM l-glutamate. The iSLK.219 clone 10 cells, which harbor latent rKSHV.219 and express RTA in a doxycycline-dependent manner, were kindly provided by D. Ganem, Novartis Institutes for BioMedical Research, Emeryville, CA, and cultured as previously described (27). KSHV generated from siRNA-transfected, reactivated iSLK.219 cells was used to infect HEK293 cells using previously described methods (37). All cells were cultured at 37 °C in 5% CO2. Eukaryotic expression vector for SV40 T antigen (22297) was purchased from Addgene. S6KB1 construct (RC217324) was purchased from Origene; the C-terminal FLAG and Myc tags were deleted, and an N-terminal FLAG tag was inserted using standard PCR. Rapamycin was purchased from LC Biosciences; DG2 and LY294002 were purchased from Calbiochem. Moloney leukemia retrovirus (MLV) vector TK502-eGFP, encoding a neomycin resistance gene, was provided by Tal Kafri, University of North Carolina at Chapel Hill. eGFP was excised using HindIII restriction endonuclease (New England Biolabs) and replaced with 3x-FLAG-vPK and -S6KB1 sequences generated by PCR. Clones were sequence-verified.
RNA Interference.
Small interfering RNAs (siRNAs) against vPK were designed using BLOCK-iT RNAi Designer (Life Technologies). mTOR and Raptor siRNA sequences have been previously reported (38, 39). A nonspecific siRNA was also used as previously described (37). siRNAs were transfected into either stable HUVECs, HEK293, or iSLK.219 cells using Lipofectamine RNAiMAX reagent per the manufacturer’s guidelines (Life Technologies); iSLK.219 cells were reactivated for 48 h (0.2 µg/mL doxycycline), concurrent with siRNA transfection. HUVECs were transfected for 24 h, after which they were split and seeded for downstream viability assays for a further 48 h (described in Cell Proliferation and Viability Assays; total transfection time was 72 h), or infected with KSHV using spinoculation, as described in KSHV Infectivity Assay. The siRNA sequences used are as follows: vPK siRNA 1, 5′-AUGACACUCAGGUUGACCUGACUCC-3′; vPK siRNA 2, 5′-UGUCCUUGAUCUCCGAAGAGGAGUG-3′; mTOR siRNA, 5′-GAGCCUUGUUGAUCCUUAA-3′; Raptor siRNA, 5′-CGAGAUUGGACGACCAAAU-3′; S6KB1 siRNA 1, 5′-CCUUGAGUAUCUCAGUGGAGGAGAA-3′; S6KB1 siRNA 2, 5′-UUCUCCUCCACUGAGAUACUCAAGG-3′.
KSHV Infectivity Assay.
Supernatants from reactivated iSLK.219 cells transfected with either NS or vPK siRNA were collected and clarified by centrifugation for 5 min at 2,300 × g. Polybrene (8 µg/mL) was added to clarified supernatants, which were overlaid on naive HEK293 cells and spinoculated at 30 °C for 90 min at 3,000 × g as previously described (24). Cells were imaged 48 h later using a Nikon Eclipse Ti inverted microscope, and the number of GFP+ cells was quantified in a minimum of 10 fields per condition; statistical analyses were performed as described in Statistical Analyses.
Immunoblot.
Immunoblotting was performed as previously described (26). Briefly, cells were harvested by washing with ice-cold PBS and lysed in a buffer containing 150 mM NaCl, 50 mM Tris⋅HCl (pH 8), 0.1% Nonidet P-40, 50 mM NaF, 30 mM β-glycerophosphate, 1 mM Na3VO4 and 1× Complete Protease Inhibitor Mixture (Roche). Proteins were electrophoresed on 10% or 12% SDS/PAGE gels and transferred onto a Hybond-ECL nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% fat-free milk for 1 h and then incubated overnight at 4 °C with the indicated antibodies directed against either phosphorylated or total protein. The following primary antibodies against phosphorylated proteins were used: eIF4B (S422), eIF4E (S209), JNK (T183, Y185), S6 ribosomal protein (S235, S236), MDM2 (S166), with corresponding total antibodies (Cell Signaling Technology). Tubulin (Cell Signaling Technology) was used as a loading control, and anti-FLAG (Sigma) was used for detecting ectopically expressed proteins. Anti-vPK antibody was generated as described previously (9). Bands were visualized by chemiluminescence. Photoshop CS6 was used for minimal image processing.
Recombinant Protein Production and Purification.
Large-scale cultures (10 T150 flasks) of Spodoptera fugiperda SF9 cells, maintained at 27 °C in EX-CELL 420 medium (Invitrogen), were infected with baculoviruses encoding FLAG-vPK for 48 h. Expression of vPK was confirmed by immunoblot. Cells were harvested with FLAG lysis buffer [50 mM Tris⋅HCl (pH 7.5), 500 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with Complete protease inhibitor mixture. Cell lysate was cleared by centrifugation, and FLAG-vPK was immunoprecipitated using anti-FLAG antibody-conjugated agarose beads (Sigma), per the manufacturer’s instructions. Protein was eluted with 150 μg/mL 3× FLAG peptide (Sigma) in TBS buffer. Purification was confirmed by immunoblot. Silver staining (Pierce) of SDS/PAGE gels was carried out to determine protein concentration by comparison with a BSA standard.
Phosphopeptide Screen.
The PepChip Kinomics v2 array containing 1,024 peptides (16 controls and 1,008 experimental) was used to reveal target substrates of vPK and S6KB1. Phosphopeptide arrays were performed as previously described (40). Briefly, 100 ng of each purified kinase was placed on peptide-embedded glass slides and incubated with [\x{03b3}-33P]ATP in a humidified chamber at 37 °C for 90 min. Slides were consecutively washed in three different buffers: Tris-buffered saline containing Tween 20, 2 M NaCl, and, finally, demineralized water. Slides were exposed to a phosphoimager screen, which was scanned, and spot intensity was quantified in a MATLAB environment. Results were analyzed using R v3.1.1. Overlapping targets of vPK and S6KB1 are as follows: ribosomal protein S6; 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2; ADAM 12; APC; ATP2B1; beta-2-adrenergic receptor; c-Fos; c-Myb; cAMP response element-binding protein 1; CHK2; coilin; connexin 32; cyclin-dependent kinase inhibitor 1B; cytohesin 2; cytohesin 1; eIF2 alpha; ezrin; lamin A/C; LcK; LKB1; neurogranin; protein tyrosine phosphatase, receptor type, alpha; RAP1 GTPase activating protein 1; ribosomal S6 kinase 1; SHP2; small nuclear ribonucleoprotein 70 kD; sperm associated antigen 1; splicing factor 1; and vitronectin.
In Vitro Kinase Assay.
As previously described, purified kinases were first adjusted to 100 ng/μL in kinase dilution buffer [50 mM Tris⋅HCl (pH 7.5), 0.1 mM EGTA, 1 mg/mL BSA, 0.1% (vol/vol) 2-mercaptoethanol] (9). Kinase (100 ng) was incubated with 2 μg of S6 peptide (carboxyl terminus-STSKSESSQK-amino terminus) in kinase reaction buffer [20 mM Hepes (pH 7.5), 5 mM MnCl2, 10 mM 2-mercaptoethanol] supplemented with 10 μCi [\x{03b3}-32P]ATP (Perkin-Elmer) at 37 °C for 30 min, after which the entire reaction was spotted onto P81 phosphocellulose paper and plunged into 75 mM H3PO4 to terminate the reaction. Phosphocellulose papers were washed and dried, and radioactivity was measured using liquid scintillation counting (Perkin-Elmer).
Quantification of Protein Synthesis.
Log phase cells were placed in methionine- and cysteine-free DMEM (MP Bio) with 2% (vol/vol) dialyzed FBS (Sigma) for 1 h. Then 10 μCi/μL EXpress EasyTag labeling mix (35S-labeled methionine and cysteine; Perkin-Elmer) was diluted in dialyzed FBS and added to cells, bringing total FBS concentration to 10% (vol/vol) for 30 min. Cells were subsequently washed and placed in standard growth medium for a further 4 h, washed again, and harvested in radioimmunoprecipitation assay (RIPA) buffer, and lysates were clarified by centrifugation. Incorporated radioactivity in cleared lysates was quantified using liquid scintillation counting (Perkin-Elmer). Counts were normalized to total protein content.
Cell Proliferation and Viability Assays.
To compare the basal rates of proliferation in stable cells expressing transgenes, 2.5 × 103 cells of each were plated in quadruplicate in 96-well dishes. Cellular metabolic activity, measured per the manufacturer’s directions using a CellTiter 96Aqueous One Solution Cell Proliferation assay (Promega), was used to quantify proliferation. Absorbance was measured using a FLUOstar OPTIMA plate reader.
Colony Formation Assay.
Rat1 fibroblasts were transduced using MLV encoding the indicated transgenes. After confirming transduction by immunoblot, 500 cells were suspended in complete growth medium containing 0.375% agarose, layered over growth medium containing 0.5% agarose, and allowed to grow for 3 wk. Colonies were stained with 0.006% crystal violet and counted in a blinded fashion using a histology microscope.
Statistical Analyses.
All statistical analyses were performed using Prism software (GraphPad Inc.). Except for Fig. 2E, all data were analyzed using ordinary one-way ANOVA with α of 0.05; Dunnett’s test was performed to correct for multiple comparisons. For Fig. 2E, an unpaired two-tailed Student’s t test was performed to compare means of each sample to the vector mean. Where indicated, *P < 0.05, **P < 0.01, ****P < 0.0001.
Tubule Formation Assay.
As previously described, 5 × 104 HUVECs suspended in complete growth media were layered upon growth factor-reduced Matrigel and incubated at 37 °C with 5% CO2 (28). Cells were subsequently visualized every 2 h with a Nikon Eclipse Ti microscope equipped with a 4× objective.
Acknowledgments
We thank Joel Parker from the UNC Bioinformatics core for phosphopeptide screen analyses and Adrienne Cox, Don Ganem, and Tal Kafri for cell lines and reagents. We also thank Dirk Dittmer for advice. B.D. is supported by NIH Grants CA096500, CA163217, AI109965, AI107810, DE018281, and CA019014. H.-j.K. and Y.I. are supported by Grant CA14779 from NIH and RSG-13-383 from the American Cancer Society. B.D. is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in Infectious Disease.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1600587113/-/DCSupplemental.
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 3.Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43(1):47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Shahbazian D, et al. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25(12):2781–2791. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai KP, et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010;29(17):2994–3006. doi: 10.1038/emboj.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giffin L, Damania B. KSHV: Pathways to tumorigenesis and persistent infection. Adv Virus Res. 2014;88:111–159. doi: 10.1016/B978-0-12-800098-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park J, Lee D, Seo T, Chung J, Choe J. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) open reading frame 36 protein is a serine protein kinase. J Gen Virol. 2000;81(Pt 4):1067–1071. doi: 10.1099/0022-1317-81-4-1067. [DOI] [PubMed] [Google Scholar]
- 8.Hamza MS, et al. ORF36 protein kinase of Kaposi’s sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem. 2004;279(37):38325–38330. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- 9.Izumiya Y, et al. Kaposi’s sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J Virol. 2007;81(3):1072–1082. doi: 10.1128/JVI.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li R, et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011;10(4):390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang S, et al. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe. 2009;5(2):166–178. doi: 10.1016/j.chom.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuny CV, Chinchilla K, Culbertson MR, Kalejta RF. Cyclin-dependent kinase-like function is shared by the beta- and gamma- subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010;6(9):e1001092. doi: 10.1371/journal.ppat.1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. Genetic organization and hypoxic activation of the Kaposi’s sarcoma-associated herpesvirus ORF34-37 gene cluster. J Virol. 2006;80(14):7037–7051. doi: 10.1128/JVI.00553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis DA, Singer KE, Reynolds IP, Haque M, Yarchoan R. Hypoxia enhances the phosphorylation and cytotoxicity of ganciclovir and zidovudine in Kaposi’s sarcoma-associated herpesvirus infected cells. Cancer Res. 2007;67(14):7003–7010. doi: 10.1158/0008-5472.CAN-07-0939. [DOI] [PubMed] [Google Scholar]
- 15.Tamburro KM. 2013. Profiling for KSHV and other herpesviruses in vivo in clinical specimens. PhD dissertation (University of North Carolina at Chapel Hill, Chapel Hill, NC)
- 16.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server issue):W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;50:2.9.1–2.9.31. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 18.Sunami T, et al. Structural basis of human p70 ribosomal S6 kinase-1 regulation by activation loop phosphorylation. J Biol Chem. 2010;285(7):4587–4594. doi: 10.1074/jbc.M109.040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 20.Moritz A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3(136):ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okuzumi T, et al. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5(7):484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang BT, et al. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc Natl Acad Sci USA. 2011;108(37):15201–15206. doi: 10.1073/pnas.1103746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 24.West J, Damania B. Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J Virol. 2008;82(11):5440–5449. doi: 10.1128/JVI.02590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sin SH, et al. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood. 2007;109(5):2165–2173. doi: 10.1182/blood-2006-06-028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatt AP, et al. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood. 2010;115(22):4455–4463. doi: 10.1182/blood-2009-10-251082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myoung J, Ganem D. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: Maintenance of tight latency with efficient reactivation upon induction. J Virol Methods. 2011;174(1-2):12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Damania B. Kaposi’s sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008;68(12):4640–4648. doi: 10.1158/0008-5472.CAN-07-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawaguchi Y, Kato K. Protein kinases conserved in herpesviruses potentially share a function mimicking the cellular protein kinase cdc2. Rev Med Virol. 2003;13(5):331–340. doi: 10.1002/rmv.402. [DOI] [PubMed] [Google Scholar]
- 30.Hume AJ, et al. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 2008;320(5877):797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- 31.McGeoch DJ, Rixon FJ, Davison AJ. Topics in herpesvirus genomics and evolution. Virus Res. 2006;117(1):90–104. doi: 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Li R, et al. Phosphoproteomic profiling reveals Epstein-Barr virus protein kinase integration of DNA damage response and mitotic signaling. PLoS Pathog. 2015;11(12):e1005346. doi: 10.1371/journal.ppat.1005346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delgado T, et al. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc Natl Acad Sci USA. 2010;107(23):10696–10701. doi: 10.1073/pnas.1004882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhatt AP, et al. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc Natl Acad Sci USA. 2012;109(29):11818–11823. doi: 10.1073/pnas.1205995109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Bock K, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 36.Wang L, et al. The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res. 2004;64(8):2774–2781. doi: 10.1158/0008-5472.can-03-3653. [DOI] [PubMed] [Google Scholar]
- 37.Dillon PJ, et al. Tousled-like kinases modulate reactivation of gammaherpesviruses from latency. Cell Host Microbe. 2013;13(2):204–214. doi: 10.1016/j.chom.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, et al. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res Treat. 2012;134(3):1057–1066. doi: 10.1007/s10549-012-2036-2. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, et al. Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncol Rep. 2014;32(1):382–388. doi: 10.3892/or.2014.3182. [DOI] [PubMed] [Google Scholar]
- 40.Diks SH, et al. Kinome profiling for studying lipopolysaccharide signal transduction in human peripheral blood mononuclear cells. J Biol Chem. 2004;279(47):49206–49213. doi: 10.1074/jbc.M405028200. [DOI] [PubMed] [Google Scholar]