

Significance
Responsiveness, resistance to, and speed of treatment are major problems for depression. The energetic and epigenetic agent acetyl-l-carnitine (LAC) is known to exert rapid antidepressant-like effects in LAC-deficient Flinders Sensitive Line rats. Here, we identified central metabolic-regulator genes (e.g., insulin and glucose signaling) in ventral dentate gyrus (vDG), a mood-regulatory region, as key factors predisposing to depression and LAC responsiveness. While improving central energy regulation and exerting rapid antidepressant-like effects, LAC corrects systemic metabolic markers of hyperinsulinemia. Also, acute stress during the treatment alters the responsiveness to LAC and induces some resistance to the treatment with a new gene profile, whereby, again, metabolic factors in vDG are key players. These results posit vDG energy regulation as factor to be considered for development of better therapeutics.
Keywords: metabolic factors, acetylcarnitine, dentate gyrus, insulin, RNAseq
Abstract
Although regulation of energy metabolism has been linked with multiple disorders, its role in depression and responsiveness to antidepressants is less known. We found that an epigenetic and energetic agent, acetyl-l-carnitine (LAC, oral administration), rapidly rescued the depressive- and central and systemic metabolic-like phenotype of LAC-deficient Flinders Sensitive Line rats (FSL). After acute stress during LAC treatment, a subset of FSL continued to respond to LAC (rFSL), whereas the other subset did not (nrFSL). RNA sequencing of the ventral dentate gyrus, a mood-regulatory region, identified metabolic factors as key markers predisposing to depression (insulin receptors Insr, glucose transporters Glut-4 and Glut-12, and the regulator of appetite Cartpt) and to LAC responsiveness (leptin receptors Lepr, metabotropic glutamate receptors-2 mGlu2, neuropeptide-Y NPY, and mineralocorticoid receptors MR). Furthermore, we found that stress-induced treatment resistance in nrFSL shows a new gene profile, including the metabolic regulator factors elongation of long chain fatty acids 7 (Elovl7) and cytochrome B5 reductase 2 (Cyb5r2) and the synaptic regulator NPAS4. Finally, while improving central energy regulation and exerting rapid antidepressant-like effects, LAC corrected a systemic hyperinsulinemia and hyperglicemia in rFSL and failed to do that in nrFSL. These findings establish CNS energy regulation as a factor to be considered for the development of better therapeutics. Agents such as LAC that regulate metabolic factors and reduce glutamate overflow could rapidly ameliorate depression and could also be considered for treatment of insulin resistance in depressed subjects. The approach here serves as a model for identifying markers and underlying mechanisms of predisposition to diseases and treatment responsiveness that may be useful in translation to human behavior and psychopathology.
Previous research has shown that agents that influence energy homeostasis, such as the epigenetic molecule acetyl-l-carnitine (LAC), exert rapid antidepressant-like effects by correcting imbalance of the glutamate system in the hippocampus of a genetic rat model of depression, Flinders Sensitive Line rats (FSL), and in a mouse model of stress-induced depressive-like traits (1–3). LAC, which passes through the blood–brain barrier, is a nutritional supplement and is also synthetized in the brain, liver, and kidney (4). Among its beneficial effects on the brain and the body, LAC regulates mitochondrial metabolism by facilitating transfer of fatty acids from the cytosol to the mitochondrial matrix for subsequent β-oxidation (5), needed for energy metabolism, deficits of which have been associated with a variety of diseases, including psychiatric disorders (6). However, the role of energy regulation in depression and in the responsiveness and/or resistance to antidepressants is less known.
Because the ventral dentate gyrus (vDG) has a key role in regulating resilience to stress and antidepressant actions (7), we implemented a microdissection approach to screen global gene expression, using RNA sequencing (RNAseq), in the vDG to start to identify genes predisposing to depression, genes that confer response to LAC, and genes that produce some resistance to the response to a low dose of LAC in a subset of FSL after an acute stress. Endogenously depressed FSL were subjected to an acute stress to mimic life events that could be related in humans, for example, to the stress of acute hospitalization, to study whether an acute stress could affect the responsiveness to a drug. We report that signatures of central metabolic syndrome are associated with the depressive-like phenotype of FSL and that LAC oral administration exerts rapid antidepressant-like effects while improving central energy metabolism in the vDG and systemic hyperinsulinemia and hyperglycemia. Neuroanatomically, the vDG, which corresponds to the human anterior hippocampus, appears as a “hot spot” for central energy dysregulation associated with the depressive-like phenotype of FSL as revealed by robust changes in Insr, Glut-4, and Cartpt(CART Prepropeptide). After an acute stress during LAC treatment, we identified Lepr and the previously uncovered markers of mood regulation, mGlu2 receptors (1), mineralocorticoid receptors (MR) (8), and neuropeptide-Y (NPY) (9, 10), among the genes that confer responsiveness to the antidepressant treatment, whereas elongation of long chain fatty acids 7 (Elovl7)- and cytochrome B5 reductase 2 (Cyb5r2)-mediated regulation of fatty-acid elongation and neuronal PAS domain protein 4 (NPAS4)-mediated synaptic reorganization were found to be associated with resistance to such a low dose of LAC in some FSL. Thus, treatment resistance seems to be an active process where new states emerge as a function of resistance in the brain of some animals in response to LAC antidepressant treatment after acute stress. Finally, we found that alterations in central metabolic factors were concomitant with a systemic metabolic dysregulation, whereby LAC antidepressant treatment corrected the hyperinsulinemia and hyperglycemia in responder FLS (rFSL) but not in nonresponder FSL (nrFSL). These findings show that central and systemic metabolic markers are involved in the pathophysiology of depression and are key factors to be considered for the development of better therapeutics. Our findings also suggest that FSL can be a useful model for comorbidity of depressive and central and systemic metabolic-like phenotypes (11, 12) as well as a stress-induced model of treatment resistance.
Results
Endogenously Depressed FSL Rapidly Respond to a Low Oral Dose of LAC and Some Develop Resistance to Response After Acute Stress.
Intraperitoneal injections of the epigenetic and energetic agent LAC, a molecule that epigenetically up-regulates mGlu2 expression, rapidly corrects mood abnormalities [i.e., passive behavior in the forced swim test (FST) and low sucrose preference] while correcting hippocampal deficits in a genetic rat model of risk for depression and in mice that develop depressive-like traits after chronic stress (1). We treated the endogenously depressed FSL and their controls, Flinders Resistant Line rats (FRL) (13), with a LAC oral low dose for 6 d (Supporting Information) to mimic the oral route used in humans. We assessed the treatment effects using the FST (Fig. 1A), a test of behavioral despair with pharmacological validity (14) that measures duration of swimming/struggling in a water-filled cylinder as a surrogate for psychomotor retardation which is one of the depression hallmarks in humans. As expected, vehicle-treated FSL (veh-FSL) showed increased immobility in the FST compared with vehicle-treated FRL (veh-FRL) (Fig. 1 B and C). Six days of LAC corrected the depressive-like behavior of FSL as shown by a robust decrease in the immobility of LAC-treated FSL (Fig. 1 B and C). This rapid antidepressant effect of LAC is in line with previous findings showing that LAC exerts rapid antidepressant-like effects, whereas tricyclic antidepressants (TCAs) take a longer time in the same animal model (1, 15). No difference in locomotion in an open field apparatus over 5 min was observed between FSL and FRL treated with LAC or vehicle, allowing us to exclude that changes in immobility could be due to nonspecific effects of LAC on locomotion (Fig. S1).
Fig. 1.

Rapid antidepressant-like action of LAC oral dose. (A) Experimental procedure implemented to test LAC efficacy at a low oral dose (Supporting Information) in endogenously depressed FSL rats with behavioral outcome at FST on day 6. (B) Six days of oral administration with LAC exerts clear-cut antidepressant-like effects as suggested by the rapidly decreased immobility of FSL. F1,25 = 8.54, P < 0.05 (treatment); F1,25 = 15.56, P < 0.001 (phenotype). (C) Representative FST tracing for mobility time. Bars represent mean + SEM, *significant comparisons with veh-FRL, #significant comparisons with veh-FSL. **P < 0.01, #P < 0.05.
Fig. S1.

FSL treated with vehicle showed a similar spontaneous locomotor activity compared with FSL that received LAC as suggested by the total distance traveled (A) and speed (B) over a 5-min open field (OF) test that was not altered by LAC oral dose. No difference in spontaneous locomotion is observed between strains (A and B). (C) Heat map for OF test distance traveled.
Next, to study a potential role of stress in altering the responsiveness to LAC and possibly also in inducing a resistance to the treatment, we subjected endogenously depressed FSL to a stress episode (a 15-min FST on day 6) during a 7-d-long LAC treatment (Fig. 2A). In a 5-min FST on day 7 to test LAC effects after acute stress, we found again an increased immobility of veh-FSL vs. veh-FRL (Fig. 2B). After the stress episode on day 6, LAC corrected immobility in rFSL, whereas the subset of nrFSL did not show a response to the treatment and still showed high immobility on day 7 (Fig. 2 B and C). LAC had no effect upon the immobility of FRL at day 6 (Fig. 1 B and C) and day 7 (Fig. S2), as also reported by findings showing that LAC action is specific to FSL with an inherent LAC deficiency (1). These results suggest that the acute stress on day 6 altered the responsiveness to LAC, with some FSL developing resistance to the treatment (nrFSL).
Fig. 2.

Responsiveness and some resistance to LAC in different subsets of endogenously depressed FSL rats after acute stress. (A) Experimental procedure implemented to establish a rodent model of treatment-resistance: FSL and their control FRL were subjected to 7-d-long LAC treatment and on day 6 to a prolonged 15-min FST used as acute stressor and to a 5-min FST on day 7 to test LAC effects after a stress exposure on day 6. (B) On day 7, veh-FSL continued to show increased immobility compared with veh-FRL. Likewise, LAC continued to show clear-cut antidepressant-like effects in a subset of FSL (rFSL) that had a reduced immobility at the FST, whereas a subset of FSL (nrFSL) stopped responding to the treatment. Their immobility time was not reduced by LAC on day 7, indicating that the prolonged swim stressor on day 6 induced a resistance in the brain responses to LAC. (C) Representative track traces at FST showing the clear-cut diversity of responses to LAC by rFSL and nrFSL. Bars: mean + SEM, *significant comparisons with veh-FRL, #significant comparisons with LAC-treated nrFSL. *P < 0.05, #P < 0.05.
Fig. S2.
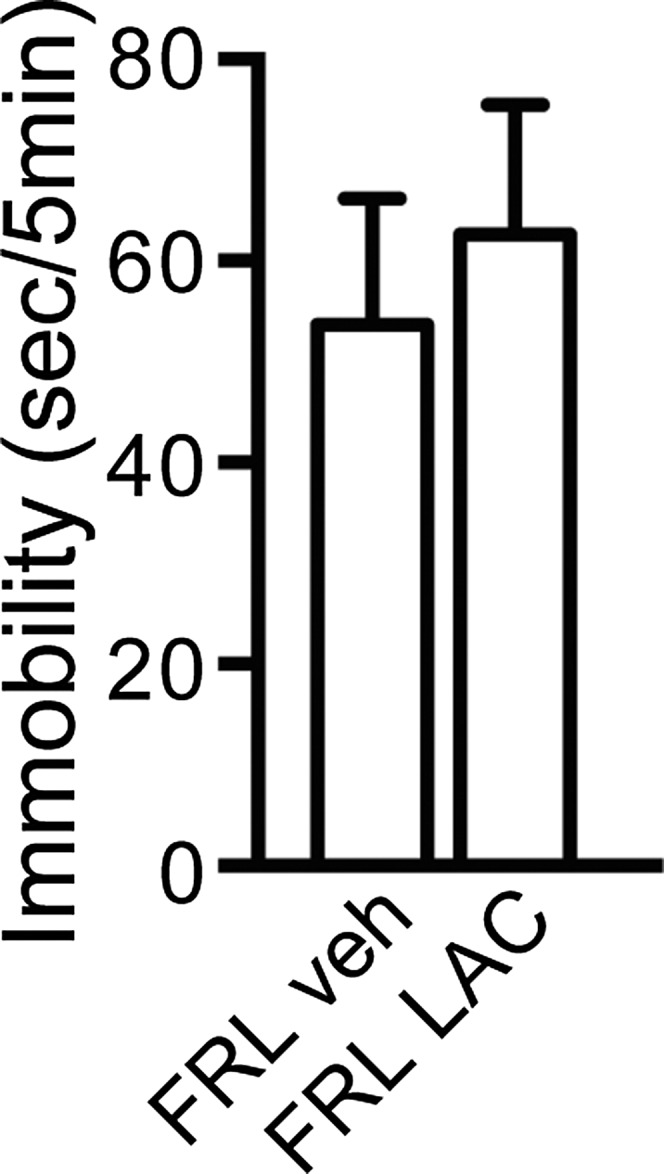
LAC had no effect on immobility time of FRL at the FST on day 7.
vDG Transcriptomes Reveal Markers Predisposing to Depression and Treatment Responsiveness and Resistance.
Recently, the vDG has emerged as a critical mediator for resilience to stress and antidepressant action, including rapid actions (7). Although gene expression changes in the hippocampus of genetic and environmentally induced animal models of depression have been well characterized within, for example, the hippocampal CA3 region, very little is known about the vDG transcriptome (Fig. 3A) (16, 17). Thus, we used an RNAseq in veh-FSL, veh-FRL, rFSL, and in nrFSL (Table S1) to begin to distinguish genes that confer response to LAC from genes that produce some resistance to response to a low dose of LAC in FSL after acute stress, as well as revealing genes responsible for the depressive phenotype of FSL. We validated our microdissection approach by examining the expression of a gene, lactase (LCT), known to be absent in the vDG (7) (Fig. S3).
Fig. 3.

vDG transcriptomes identify markers of treatment resistance, treatment response, and predisposition to depression with a key role of metabolic factors. (A) Representative coronal brain images with vDG (red) reference for microdissection. (B) Heat map and dendogram resulting from hierarchical clustering analysis displayed a clear-cut wide diversity of global RNA expression profiles between veh-FSL and veh-FRL. Such broad differences in global RNA expression profiles are nearly absent in rFSL compared with veh-FRL; in fact, rFSL seemed to show a gene expression profile almost the same as veh-FRL. Notably, the global RNA expression profile of nrFSL spanned the RNA profile of both veh-FSL and veh-FRL. (C–E) Venn diagrams from left to right narrow down the three sets of genes conferring predisposition to depression, treatment resistance, and treatment response from 749 (C, red) to 544 (E, red), from 245 (D, pink) to 205 (E, pink), and from 30 (D, gray) to 27 (E, gray). (F and G) GO analyses for biological processes of the 544 markers of genetic depression and of the 205 markers of treatment response show that the inherent passive behavior of FSL is associated with signaling pathways within the vDG implicated in energy regulation. (H and I) Functional classification analysis for biological and molecular processes for the 27 markers of the active process of treatment resistance.
Table S1.
Sequencing data summary from quality control analysis
| Group | Biological replicate | Total reads | Uniquely mapped reads | Mapping rate |
| FRL | 1 | 28,070,916 | 24,791,657 | 88.3 |
| FRL | 2 | 26,413,783 | 23,268,387 | 88.1 |
| FRL | 3 | 24,398,296 | 21,509,998 | 88.2 |
| FSL | 1 | 25,666,824 | 22,627,057 | 88.2 |
| FSL | 2 | 26,254,251 | 23,335,791 | 88.9 |
| FSL | 3 | 28,776,158 | 25,410,265 | 88.3 |
| nrFSL | 1 | 25,571,392 | 22,805,066 | 89.2 |
| nrFSL | 2 | 27,106,051 | 24,068,679 | 88.8 |
| nrFSL | 3 | 26,969,858 | 23,917,939 | 88.7 |
| rFSL | 1 | 28,792,744 | 25,439,891 | 88.4 |
| rFSL | 2 | 28,039,820 | 24,854,855 | 88.6 |
| rFSL | 3 | 27,001,088 | 23,979,092 | 88.8 |
Fig. S3.

Validation of the microdissection approach. Lct shows minimal RNAseq measured expression (cpm: count per million) in the vDG, confirming the accuracy of the microdissection approach.
The heat map and dendrogram resulting from hierarchical clustering analysis for the vDG, in three biological replicate experiments, displayed a clear-cut wide diversity of global RNA expression profiles between veh-FSL and veh-FRL (Fig. 3B). The rFSL, responding to LAC treatment, show vDG gene expression patterns in the heat maps that begin to resemble those of veh-FRL (Fig. 3B), indicating that LAC corrected many of the gene expression changes associated with the depressive-like phenotype of FSL. Notably, the global RNA expression profile of nrFSL spanned the RNA profile of both veh-FSL and veh-FRL, indicating that, although a large number of gene expression changes were rescued by LAC in nrFSL, many genes remained unaltered and other genes changed in nrFSL as a result of the acute stress. These genes rendered the nrFSL resistant to LAC treatment (Fig. 3B). These findings suggest that treatment resistance is an active process in which new clusters of genes are altered rather than a lack of effects in correcting a global RNA expression profile in unresponsive individuals.
Venn diagrams for the genes differentially expressed in veh-FSL and rFSL (Fig. 3C), either one compared with control veh-FRL, revealed that LAC treatment in rFSL rescued 749 out of the 807 genes, which were differentially expressed in veh-FSL vs. veh-FRL. In contrast, the remaining 58 genes (out of the 807) (Fig. 3C, brown) remained differentially expressed in rFSL, thus seeming not to account for the response to LAC treatment. Interestingly, the cluster of 50 genes (Fig. 3C, green), which emerged in rFSL that responded to LAC and that were not different in veh-FSL compared with veh-FRL, could be ones that compensate for the genetic predisposition of FSL. Next, Venn diagrams for the genes differentially expressed in veh-FSL and in nrFSL, either one compared with veh-FRL, showed that although lacking response nrFSL nevertheless showed gene responses to LAC similar to those in rFSL (Fig. 3D), with 562 genes rescued by LAC. However, a cluster of 245 genes (Fig. 3D, pink), which were differentially expressed in endogenously depressed veh-FSL vs. veh-FRL, was still not changed by LAC treatment in nrFSL, suggesting that such a cluster is required for the responsiveness to LAC treatment (markers of treatment response). In addition, a new set of 30 genes (Fig. 3D, gray), which were not previously differentially expressed in veh-FSL compared with veh-FRL, seemed to be altered in nrFSL, thereby suggesting an active process in the stress-mediated induction of resistance to the treatment (markers of treatment resistance) with occurrence of a new gene expression profile in nrFSL after acute stress.
Finally, Venn diagrams of the cross-comparisons (information in Fig. 3 C and D is summarized in Fig. 3E) of differentially expressed genes in veh-FSL, rFSL, and nrFSL, either one compared with control veh-FRL, further narrowed the markers of treatment resistance to 27 (Fig. 3E, gray) out of the above-mentioned 30 genes; the remaining three (Fig. 3E, dark green) genes were commonly altered in both nrFSL and rFSL rats, regardless of the response to LAC. Similarly, the markers of treatment response were restricted to 205 (Fig. 3E, pink) out of the 245 above-mentioned genes [the remaining 40 genes (Fig. 3E, inner intersection) were also altered in rFSL]. Finally, the remaining 544 (Fig. 3E, red) out of 749 above-mentioned genes were identified as markers of genetic contribution to depressive-like behavior (Fig. 3E). We note that the cluster of 205 markers of treatment response, which, as described above, was altered in both nrFSL and veh-FSL, was instead rescued by LAC treatment in rFSL rats (Fig. 3E), thus reinforcing a role of such a cluster in the responsiveness to LAC.
Genes Predisposing to Depression.
Using Gene Ontology (GO) analyses for biological processes, we found that the 544 genes here identified as markers predisposing to a depressive-like phenotype belong to several meaningful gene categories including responses to lipid and vitamin D, regulation of cell migration and protein phosphorylation, and cellular metabolic processes along with biological processes known to be affected in depression, such as regulation of gene expression, chromatin organization, inflammatory responses, NF-κB signaling, and immune system processes as well as response to stress, hormone, and growth factors (Fig. 3F and Table S2).
Table S2.
Enrichment GO for biological process showing the most significant terms that are altered among the 544 genes that confer predisposition for depression
| GO ID | Term | No. of genes | Over(+)/under(−) | P value after Bonferroni | −log10(P value) |
| GO:0009653 | Anatomical structure morphogenesis | 137 | + | 6.06E-15 | 14.22 |
| GO:0001944 | Vasculature development | 55 | + | 8.55E-13 | 12.07 |
| GO:0051270 | Regulation of cellular component movement | 71 | + | 1.28E-12 | 11.89 |
| GO:2000145 | Regulation of cell motility | 65 | + | 8.39E-12 | 11.08 |
| GO:0040012 | Regulation of locomotion | 69 | + | 1.04E-11 | 10.98 |
| GO:0009888 | Tissue development | 109 | + | 1.20E-11 | 10.92 |
| GO:0030334 | Regulation of cell migration | 63 | + | 1.33E-11 | 10.88 |
| GO:0050896 | Response to stimulus | 278 | + | 2.27E-11 | 10.64 |
| GO:0072359 | Circulatory system development | 73 | + | 3.29E-11 | 10.48 |
| GO:0072358 | Cardiovascular system development | 73 | + | 3.29E-11 | 10.48 |
| GO:0051241 | Negative regulation of multicellular organismal process | 81 | + | 4.67E-11 | 10.33 |
| GO:0051239 | Regulation of multicellular organismal process | 144 | + | 5.38E-11 | 10.27 |
| GO:0001568 | Blood vessel development | 50 | + | 6.43E-11 | 10.19 |
| GO:0002376 | Immune system process | 88 | + | 3.42E-10 | 9.47 |
| GO:0022610 | Biological adhesion | 70 | + | 6.45E-10 | 9.19 |
| GO:0043062 | Extracellular structure organization | 30 | + | 6.84E-10 | 9.16 |
| GO:0030198 | Extracellular matrix organization | 30 | + | 6.84E-10 | 9.16 |
| GO:0048514 | Blood vessel morphogenesis | 43 | + | 8.15E-10 | 9.09 |
| GO:0033993 | Response to lipid | 80 | + | 1.33E-09 | 8.88 |
| GO:0007155 | Cell adhesion | 69 | + | 1.47E-09 | 8.83 |
| GO:0007423 | Sensory organ development | 53 | + | 1.47E-09 | 8.83 |
| GO:0044707 | Single-multicellular organism process | 238 | + | 1.72E-09 | 8.76 |
| GO:0032501 | Multicellular organismal process | 241 | + | 1.13E-08 | 7.95 |
| GO:0007165 | Signal transduction | 168 | + | 1.18E-08 | 7.93 |
| GO:0060429 | Epithelium development | 73 | + | 1.24E-08 | 7.91 |
| GO:0042127 | Regulation of cell proliferation | 93 | + | 2.41E-08 | 7.62 |
| GO:0048583 | Regulation of response to stimulus | 165 | + | 2.66E-08 | 7.58 |
| GO:0048729 | Tissue morphogenesis | 54 | + | 3.02E-08 | 7.52 |
| GO:0071310 | Cellular response to organic substance | 101 | + | 4.71E-08 | 7.33 |
| GO:0006950 | Response to stress | 149 | + | 4.72E-08 | 7.33 |
| GO:0070887 | Cellular response to chemical stimulus | 117 | + | 4.80E-08 | 7.32 |
| GO:0002009 | Morphogenesis of an epithelium | 47 | + | 5.19E-08 | 7.28 |
| GO:1901700 | Response to oxygen-containing compound | 101 | + | 6.67E-08 | 7.18 |
| GO:0001525 | Angiogenesis | 35 | + | 6.75E-08 | 7.17 |
| GO:0048584 | Positive regulation of response to stimulus | 106 | + | 9.57E-08 | 7.02 |
| GO:0009967 | Positive regulation of signal transduction | 85 | + | 1.01E-07 | 7.00 |
| GO:0030855 | Epithelial cell differentiation | 46 | + | 1.77E-07 | 6.75 |
| GO:0048646 | Anatomical structure formation involved in morphogenesis | 70 | + | 1.77E-07 | 6.75 |
| GO:0042060 | Wound healing | 37 | + | 1.80E-07 | 6.74 |
| GO:0001501 | Skeletal system development | 43 | + | 2.09E-07 | 6.68 |
| GO:0035295 | Tube development | 57 | + | 2.18E-07 | 6.66 |
| GO:0009605 | Response to external stimulus | 100 | + | 4.35E-07 | 6.36 |
| GO:0009617 | Response to bacterium | 41 | + | 4.41E-07 | 6.36 |
| GO:0006955 | Immune response | 45 | + | 5.87E-07 | 6.23 |
| GO:0002237 | Response to molecule of bacterial origin | 36 | + | 7.01E-07 | 6.15 |
| GO:0032496 | Response to lipopolysaccharide | 35 | + | 8.03E-07 | 6.10 |
| GO:0009611 | Response to wounding | 43 | + | 8.21E-07 | 6.09 |
| GO:0023056 | Positive regulation of signaling | 91 | + | 9.54E-07 | 6.02 |
| GO:0042221 | Response to chemical | 161 | + | 9.59E-07 | 6.02 |
| GO:0050678 | Regulation of epithelial cell proliferation | 34 | + | 1.04E-06 | 5.98 |
| GO:0010033 | Response to organic substance | 134 | + | 1.31E-06 | 5.88 |
| GO:0051716 | Cellular response to stimulus | 218 | + | 1.36E-06 | 5.87 |
| GO:0009719 | Response to endogenous stimulus | 98 | + | 2.31E-06 | 5.64 |
| GO:0051707 | Response to other organism | 48 | + | 2.34E-06 | 5.63 |
| GO:0009887 | Organ morphogenesis | 61 | + | 2.42E-06 | 5.62 |
| GO:0044237 | Cellular metabolic process | 155 | − | 2.56E-06 | 5.59 |
| GO:0043207 | Response to external biotic stimulus | 48 | + | 2.57E-06 | 5.59 |
| GO:0044700 | Single organism signaling | 171 | + | 3.13E-06 | 5.50 |
| GO:0007154 | Cell communication | 178 | + | 3.30E-06 | 5.48 |
| GO:0050793 | Regulation of developmental process | 120 | + | 3.36E-06 | 5.47 |
| GO:0023052 | Signaling | 171 | + | 3.37E-06 | 5.47 |
| GO:0009607 | Response to biotic stimulus | 49 | + | 3.45E-06 | 5.46 |
| GO:0044260 | Cellular macromolecule metabolic process | 109 | − | 3.52E-06 | 5.45 |
| GO:0040017 | Positive regulation of locomotion | 39 | + | 3.75E-06 | 5.43 |
| GO:1901701 | Cellular response to oxygen-containing compound | 64 | + | 4.10E-06 | 5.39 |
| GO:0035239 | Tube morphogenesis | 39 | + | 4.19E-06 | 5.38 |
| GO:0014070 | Response to organic cyclic compound | 77 | + | 5.11E-06 | 5.29 |
| GO:0009966 | Regulation of signal transduction | 126 | + | 5.13E-06 | 5.29 |
| GO:0007166 | Cell surface receptor signaling pathway | 85 | + | 5.77E-06 | 5.24 |
| GO:0044699 | Single-organism process | 399 | + | 6.10E-06 | 5.21 |
| GO:0048513 | Organ development | 144 | + | 1.07E-05 | 4.97 |
| GO:0060562 | Epithelial tube morphogenesis | 36 | + | 1.09E-05 | 4.96 |
| GO:2000147 | Positive regulation of cell motility | 37 | + | 1.25E-05 | 4.90 |
| GO:0070482 | Response to oxygen levels | 39 | + | 1.54E-05 | 4.81 |
| GO:0032502 | Developmental process | 217 | + | 1.98E-05 | 4.70 |
| GO:0044767 | Single-organism developmental process | 216 | + | 1.98E-05 | 4.70 |
| GO:0040011 | Locomotion | 64 | + | 2.00E-05 | 4.70 |
| GO:0010647 | Positive regulation of cell communication | 93 | + | 2.07E-05 | 4.68 |
| GO:0030335 | Positive regulation of cell migration | 36 | + | 2.15E-05 | 4.67 |
| GO:0051272 | Positive regulation of cellular component movement | 37 | + | 2.69E-05 | 4.57 |
| GO:0006396 | RNA processing | 1 | − | 2.84E-05 | 4.55 |
| GO:0034641 | Cellular nitrogen compound metabolic process | 63 | − | 3.12E-05 | 4.51 |
| GO:0008284 | Positive regulation of cell proliferation | 56 | + | 3.85E-05 | 4.41 |
| GO:0048856 | Anatomical structure development | 201 | + | 4.05E-05 | 4.39 |
| GO:0001101 | Response to acid chemical | 40 | + | 4.09E-05 | 4.39 |
| GO:0048598 | Embryonic morphogenesis | 45 | + | 4.11E-05 | 4.39 |
| GO:0060348 | Bone development | 24 | + | 4.44E-05 | 4.35 |
| GO:0002682 | Regulation of immune system process | 62 | + | 5.66E-05 | 4.25 |
| GO:0048468 | Cell development | 91 | + | 5.74E-05 | 4.24 |
| GO:0071495 | Cellular response to endogenous stimulus | 65 | + | 5.77E-05 | 4.24 |
| GO:0042307 | Positive regulation of protein import into nucleus | 18 | + | 7.00E-05 | 4.15 |
| GO:0061138 | Morphogenesis of a branching epithelium | 24 | + | 8.50E-05 | 4.07 |
| GO:0048518 | Positive regulation of biological process | 223 | + | 8.83E-05 | 4.05 |
| GO:0010941 | Regulation of cell death | 92 | + | 1.02E-04 | 3.99 |
| GO:0048754 | Branching morphogenesis of an epithelial tube | 22 | + | 1.02E-04 | 3.99 |
| GO:0071396 | Cellular response to lipid | 40 | + | 1.27E-04 | 3.90 |
| GO:0023051 | Regulation of signaling | 136 | + | 1.42E-04 | 3.85 |
| GO:0009628 | Response to abiotic stimulus | 75 | + | 1.42E-04 | 3.85 |
| GO:0007275 | Multicellular organismal development | 191 | + | 1.47E-04 | 3.83 |
| GO:0001763 | Morphogenesis of a branching structure | 24 | + | 2.14E-04 | 3.67 |
| GO:0048705 | Skeletal system morphogenesis | 24 | + | 2.14E-04 | 3.67 |
| GO:0030278 | Regulation of ossification | 24 | + | 2.14E-04 | 3.67 |
| GO:0016477 | Cell migration | 50 | + | 2.27E-04 | 3.64 |
| GO:1901342 | Regulation of vasculature development | 24 | + | 2.48E-04 | 3.61 |
| GO:0006952 | Defense response | 45 | + | 2.54E-04 | 3.60 |
| GO:0072001 | Renal system development | 31 | + | 2.58E-04 | 3.59 |
| GO:0030155 | Regulation of cell adhesion | 44 | + | 2.85E-04 | 3.55 |
| GO:1903034 | Regulation of response to wounding | 32 | + | 2.91E-04 | 3.54 |
| GO:0048731 | System development | 177 | + | 3.22E-04 | 3.49 |
| GO:0051674 | Localization of cell | 52 | + | 3.23E-04 | 3.49 |
| GO:0048870 | Cell motility | 52 | + | 3.23E-04 | 3.49 |
| GO:2000026 | Regulation of multicellular organismal development | 93 | + | 3.29E-04 | 3.48 |
| GO:0009725 | Response to hormone | 68 | + | 3.41E-04 | 3.47 |
| GO:0060485 | Mesenchyme development | 20 | + | 3.89E-04 | 3.41 |
| GO:0090596 | Sensory organ morphogenesis | 26 | + | 3.92E-04 | 3.41 |
| GO:0001655 | Urogenital system development | 33 | + | 3.93E-04 | 3.41 |
| GO:0098602 | Single organism cell adhesion | 36 | + | 4.18E-04 | 3.38 |
| GO:0048519 | Negative regulation of biological process | 189 | + | 4.41E-04 | 3.36 |
| GO:1900182 | Positive regulation of protein localization to nucleus | 18 | + | 4.78E-04 | 3.32 |
| GO:0051094 | Positive regulation of developmental process | 73 | + | 5.26E-04 | 3.28 |
| GO:1903035 | Negative regulation of response to wounding | 18 | + | 5.81E-04 | 3.24 |
| GO:0008285 | Negative regulation of cell proliferation | 46 | + | 6.04E-04 | 3.22 |
| GO:0001654 | Eye development | 32 | + | 6.06E-04 | 3.22 |
| GO:0051240 | Positive regulation of multicellular organismal process | 81 | + | 6.11E-04 | 3.21 |
| GO:0032879 | Regulation of localization | 121 | + | 6.27E-04 | 3.20 |
| GO:0048545 | Response to steroid hormone | 47 | + | 6.57E-04 | 3.18 |
| GO:0060541 | Respiratory system development | 28 | + | 6.60E-04 | 3.18 |
| GO:0036293 | Response to decreased oxygen levels | 34 | + | 6.84E-04 | 3.16 |
| GO:0007507 | Heart development | 41 | + | 6.99E-04 | 3.16 |
| GO:0030154 | Cell differentiation | 145 | + | 7.30E-04 | 3.14 |
| GO:0001503 | Ossification | 28 | + | 8.31E-04 | 3.08 |
| GO:0070848 | Response to growth factor | 39 | + | 8.63E-04 | 3.06 |
| GO:0043067 | Regulation of programmed cell death | 83 | + | 1.11E-03 | 2.95 |
| GO:0031589 | Cell-substrate adhesion | 20 | + | 1.26E-03 | 2.90 |
| GO:0050679 | Positive regulation of epithelial cell proliferation | 20 | + | 1.26E-03 | 2.90 |
| GO:0001666 | Response to hypoxia | 33 | + | 1.28E-03 | 2.89 |
| GO:0043410 | Positive regulation of MAPK cascade | 37 | + | 1.35E-03 | 2.87 |
| GO:0060560 | Developmental growth involved in morphogenesis | 18 | + | 1.47E-03 | 2.83 |
| GO:0060349 | Bone morphogenesis | 15 | + | 1.75E-03 | 2.76 |
| GO:0046824 | Positive regulation of nucleocytoplasmic transport | 18 | + | 1.76E-03 | 2.75 |
| GO:0045785 | Positive regulation of cell adhesion | 29 | + | 1.81E-03 | 2.74 |
| GO:0002684 | Positive regulation of immune system process | 39 | + | 1.94E-03 | 2.71 |
| GO:0009790 | Embryo development | 60 | + | 2.26E-03 | 2.65 |
| GO:0010646 | Regulation of cell communication | 137 | + | 2.34E-03 | 2.63 |
| GO:0071363 | Cellular response to growth factor stimulus | 36 | + | 2.67E-03 | 2.57 |
| GO:0046483 | Heterocycle metabolic process | 56 | − | 2.69E-03 | 2.57 |
| GO:0040013 | Negative regulation of locomotion | 26 | + | 2.74E-03 | 2.56 |
| GO:0048869 | Cellular developmental process | 149 | + | 2.83E-03 | 2.55 |
| GO:0022603 | Regulation of anatomical structure morphogenesis | 58 | + | 2.86E-03 | 2.54 |
| GO:0045765 | Regulation of angiogenesis | 21 | + | 2.88E-03 | 2.54 |
| GO:0071559 | Response to transforming growth factor beta | 19 | + | 2.89E-03 | 2.54 |
| GO:0061298 | Retina vasculature development in camera-type eye | 9 | + | 3.01E-03 | 2.52 |
| GO:0030324 | Lung development | 25 | + | 3.33E-03 | 2.48 |
| GO:0006139 | Nucleobase-containing compound metabolic process | 53 | − | 3.49E-03 | 2.46 |
| GO:0048568 | Embryonic organ development | 33 | + | 3.82E-03 | 2.42 |
| GO:0051271 | Negative regulation of cellular component movement | 24 | + | 4.01E-03 | 2.40 |
| GO:0030323 | Respiratory tube development | 25 | + | 4.19E-03 | 2.38 |
| GO:2000146 | Negative regulation of cell motility | 23 | + | 4.23E-03 | 2.37 |
| GO:0048839 | Inner ear development | 22 | + | 4.36E-03 | 2.36 |
| GO:0032967 | Positive regulation of collagen biosynthetic process | 8 | + | 4.87E-03 | 2.31 |
| GO:0010714 | Positive regulation of collagen metabolic process | 8 | + | 4.87E-03 | 2.31 |
| GO:0048522 | Positive regulation of cellular process | 194 | + | 5.30E-03 | 2.28 |
| GO:0043583 | Ear development | 23 | + | 5.41E-03 | 2.27 |
| GO:0001822 | Kidney development | 27 | + | 5.85E-03 | 2.23 |
| GO:0006725 | Cellular aromatic compound metabolic process | 58 | − | 6.23E-03 | 2.21 |
| GO:0061035 | Regulation of cartilage development | 12 | + | 6.57E-03 | 2.18 |
| GO:0043408 | Regulation of MAPK cascade | 46 | + | 6.62E-03 | 2.18 |
| GO:0006807 | Nitrogen compound metabolic process | 79 | − | 7.38E-03 | 2.13 |
| GO:0043065 | Positive regulation of apoptotic process | 44 | + | 7.40E-03 | 2.13 |
| GO:0001775 | Cell activation | 34 | + | 7.46E-03 | 2.13 |
| GO:0002460 | Adaptive immune response based on somatic recombination of immune receptors built from Ig superfamily domains | 14 | + | 7.81E-03 | 2.11 |
| GO:0051704 | Multiorganism process | 78 | + | 8.33E-03 | 2.08 |
| GO:0043068 | Positive regulation of programmed cell death | 44 | + | 8.43E-03 | 2.07 |
| GO:0042306 | Regulation of protein import into nucleus | 21 | + | 8.49E-03 | 2.07 |
| GO:0090304 | Nucleic acid metabolic process | 42 | − | 8.57E-03 | 2.07 |
| GO:0048523 | Negative regulation of cellular process | 172 | + | 8.97E-03 | 2.05 |
| GO:0030336 | Negative regulation of cell migration | 22 | + | 9.27E-03 | 2.03 |
| GO:0042981 | Regulation of apoptotic process | 79 | + | 9.30E-03 | 2.03 |
| GO:0010942 | Positive regulation of cell death | 46 | + | 9.65E-03 | 2.02 |
| GO:0006954 | Inflammatory response | 24 | + | 1.12E-02 | 1.95 |
| GO:0048771 | Tissue remodeling | 16 | + | 1.13E-02 | 1.95 |
| GO:0032102 | Negative regulation of response to external stimulus | 23 | + | 1.23E-02 | 1.91 |
| GO:0044093 | Positive regulation of molecular function | 83 | + | 1.24E-02 | 1.91 |
| GO:0016337 | Single organismal cell–cell adhesion | 31 | + | 1.28E-02 | 1.89 |
| GO:0030326 | Embryonic limb morphogenesis | 16 | + | 1.34E-02 | 1.87 |
| GO:0035113 | Embryonic appendage morphogenesis | 16 | + | 1.34E-02 | 1.87 |
| GO:0045087 | Innate immune response | 23 | + | 1.38E-02 | 1.86 |
| GO:0051276 | Chromosome organization | 5 | − | 1.57E-02 | 1.80 |
| GO:0051093 | Negative regulation of developmental process | 54 | + | 1.59E-02 | 1.80 |
| GO:0071407 | Cellular response to organic cyclic compound | 37 | + | 1.63E-02 | 1.79 |
| GO:1901698 | Response to nitrogen compound | 62 | + | 1.68E-02 | 1.77 |
| GO:0010467 | Gene expression | 42 | − | 1.72E-02 | 1.76 |
| GO:0043010 | Camera-type eye development | 27 | + | 1.73E-02 | 1.76 |
| GO:1902533 | Positive regulation of intracellular signal transduction | 53 | + | 1.79E-02 | 1.75 |
| GO:1901360 | Organic cyclic compound metabolic process | 64 | − | 1.83E-02 | 1.74 |
| GO:0006928 | Movement of cell or subcellular component | 60 | + | 1.88E-02 | 1.73 |
| GO:0001934 | Positive regulation of protein phosphorylation | 53 | + | 1.89E-02 | 1.72 |
| GO:0043604 | Amide biosynthetic process | 1 | − | 1.91E-02 | 1.72 |
| GO:0033280 | Response to vitamin D | 10 | + | 1.92E-02 | 1.72 |
| GO:0006325 | Chromatin organization | 2 | − | 2.01E-02 | 1.70 |
| GO:0002250 | Adaptive immune response | 17 | + | 2.06E-02 | 1.69 |
| GO:0010935 | Regulation of macrophage cytokine production | 7 | + | 2.09E-02 | 1.68 |
| GO:0010811 | Positive regulation of cell-substrate adhesion | 15 | + | 2.21E-02 | 1.66 |
| GO:0032989 | Cellular component morphogenesis | 57 | + | 2.38E-02 | 1.62 |
| GO:0042345 | Regulation of NF-κB import into nucleus | 10 | + | 2.58E-02 | 1.59 |
| GO:0071560 | Cellular response to transforming growth factor beta stimulus | 17 | + | 2.77E-02 | 1.56 |
| GO:0048562 | Embryonic organ morphogenesis | 23 | + | 2.95E-02 | 1.53 |
| GO:0045595 | Regulation of cell differentiation | 83 | + | 3.00E-02 | 1.52 |
| GO:0044253 | Positive regulation of multicellular organismal metabolic process | 8 | + | 3.05E-02 | 1.52 |
| GO:0046822 | Regulation of nucleocytoplasmic transport | 23 | + | 3.28E-02 | 1.48 |
| GO:0044267 | Cellular protein metabolic process | 69 | − | 3.32E-02 | 1.48 |
| GO:0000904 | Cell morphogenesis involved in differentiation | 38 | + | 3.47E-02 | 1.46 |
| GO:0042346 | Positive regulation of NF-κB import into nucleus | 7 | + | 3.50E-02 | 1.46 |
| GO:0010243 | Response to organonitrogen compound | 58 | + | 3.57E-02 | 1.45 |
| GO:0090100 | Positive regulation of transmembrane receptor protein serine/threonine kinase signaling pathway | 14 | + | 3.63E-02 | 1.44 |
| GO:0061041 | Regulation of wound healing | 15 | + | 3.65E-02 | 1.44 |
| GO:0001817 | Regulation of cytokine production | 31 | + | 3.70E-02 | 1.43 |
| GO:0098609 | Cell–cell adhesion | 37 | + | 3.75E-02 | 1.43 |
| GO:0042327 | Positive regulation of phosphorylation | 54 | + | 4.01E-02 | 1.40 |
| GO:0032101 | Regulation of response to external stimulus | 48 | + | 4.19E-02 | 1.38 |
| GO:0010712 | Regulation of collagen metabolic process | 9 | + | 4.24E-02 | 1.37 |
| GO:0060173 | Limb development | 18 | + | 4.26E-02 | 1.37 |
| GO:0048736 | Appendage development | 18 | + | 4.26E-02 | 1.37 |
| GO:0002064 | Epithelial cell development | 21 | + | 4.52E-02 | 1.34 |
| GO:0050680 | Negative regulation of epithelial cell proliferation | 16 | + | 4.70E-02 | 1.33 |
| GO:0097305 | Response to alcohol | 38 | + | 4.81E-02 | 1.32 |
| GO:0048585 | Negative regulation of response to stimulus | 70 | + | 4.93E-02 | 1.31 |
Over, overrepresented; under, underrepresented.
Given the importance of energy regulation for any biological process (18), we started by screening genes known to be involved in such regulatory processes in our RNAseq databank for the vDG and performed subsequent quantitative PCR (qPCR) validation on larger biological replicate cohorts. Among the 544 markers predisposing to depression, we identified meaningful up-regulation of many biological markers involved in energy metabolism. Such up-regulated genes that are associated with the depressive-like phenotype of FSL include Lepr, Insr, Cartpt (a prohormone involved in the regulation of appetite through NPY signaling), and Glut-4 with 2-, 10-, 2-, and 4-fold change up-regulation in qPCR validations, respectively (Fig. 4 A–D). The magnitude of such changes suggests that endogenously depressed FSL show central alterations in energy homeostasis with a development of a resistance to the action centrally of insulin through a mechanism that is not linked to Glut-4, which is up-regulated. The unexpected direction of these robust changes provides a starting point for the study of the mechanisms of comorbid depression and metabolic syndromes, for example, whether the increase in Glut-4 may be a compensatory mechanism in response to changes in other glucose transporters that may lead to a deficit in central glucose levels, such as Glut-12, which show a fivefold up-regulation in veh-FSL compared with veh-FRL and the direction of this change is reversed in rFSL with a reduced 1.8-fold change compared with veh-FRL (Fig. S4).
Fig. 4.

Metabolic factors in vDG as important targets of responsiveness to LAC antidepressant treatment, treatment resistance, and depressive-like phenotype. (A–C) qPCR validation confirmed changes in the genes unveiled by RNAseq as markers of the depressive-like phenotype of FSL rats showing up-regulations of Insr, Glut-4, and Cartpt transcripts in the vDG of veh-FSL that were rapidly corrected by LAC in both rFSL and nrFSL. (D–G) qPCR validation confirmed increase in Lepr and MR along with decrease in mGlu2 and NPY in the vDG of veh-FSL as unveiled by RNAseq. Such changes were corrected in rFSL and not in nrFSL after stress. (H and I) The regulators of fatty-acid elongation, Elovl7 and Cyb5r2, emerged as important targets in development of resistance to LAC, low oral dose, in some FSL after stress as confirmed by qPCR validation. (J) Network analysis identifies NPY, among other genes such as BDNF and TrkB (Ntrk2), as critical mediators of the interaction between Lepr and Grm2 (mGlu2: previously identified targets for LAC rapid antidepressant effects). Bars: mean + SEM, *significant comparisons with veh-FRL, #significant comparisons with LAC-treated nrFSL. *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05, ##P < 0.01.
Fig. S4.

Among the large number of glucose transporters of class I, II, and III, only Glut-4 and Glut-12, which have been previously implicated in insulin resistance, seem to be associated with predisposition to depression, LAC rapid antidepressant action, and resistance to LAC treatment after stress in a subset of FSLs, as indicated by RNAseq measured expression (cpm: count per million) in the vDG. n.s., not statistically significant.
Fig. S5.

No change in the metabolic marker of treatment responsiveness Lepr (A) or in the metabolic marker of treatment resistance Elovl7 (B) was observed in the dorsal dentate gyrus, in line with a key role of the vDG as a target of antidepressants with functions distinctly different from that of the dorsal part, which is more involved in spatial orientation.
Genes of Treatment Responsiveness.
Biological processes rendering animals responsive to the oral administration of LAC treatment were elucidated using GO analysis for the above-identified 205 markers of treatment response. We found meaningful gene categories that belong to peptide cross-linking, regulation of cell migration and gene expression, gland development and response to estrogen and steroid hormones, and, again, processes of metabolic regulation (Fig. 3G and Table S3). We note that about 55 out of the 205 markers of treatment response were mediators of metabolic processes, indicating a critical role of energy homeostasis in the response to the antidepressant candidate, LAC. Interestingly, among the metabolic regulators above identified as genes that account for the depressive-like phenotype of FSL, only Lepr emerged in the cluster of 205 genes that confer responsiveness to LAC. Indeed, subsequent qPCR validation on larger biological replicate cohorts for the vDG confirmed that only nrFSL continued to show the same up-regulation of Lepr as veh-FSL (Fig. 4D). Such differential regulation of Lepr in the vDG of rFSL and nrFSL indicates that adjustment in the hyperactivation of the leptin signaling is a feature of responsiveness to LAC treatment.
Table S3.
Enrichment GO for biological process showing the most significant terms that are altered among the 205 genes that confer responsiveness to LAC oral treatment
| GO ID | Term | No. of genes | Over(+)/under(−) | P value after Bonferroni | −log10(P value) |
| GO:0009888 | Tissue development | 56 | + | 4.38E-10 | 9.36 |
| GO:0044260 | Cellular macromolecule metabolic process | 25 | − | 7.82E-08 | 7.11 |
| GO:0042060 | Wound healing | 22 | + | 5.93E-07 | 6.23 |
| GO:0048513 | Organ development | 72 | + | 1.09E-06 | 5.96 |
| GO:0009611 | Response to wounding | 25 | + | 1.51E-06 | 5.82 |
| GO:0044237 | Cellular metabolic process | 46 | − | 1.30E-05 | 4.89 |
| GO:0048545 | Response to steroid hormone | 28 | + | 3.35E-05 | 4.47 |
| GO:0048732 | Gland development | 23 | + | 2.01E-04 | 3.70 |
| GO:0009887 | Organ morphogenesis | 30 | + | 3.41E-04 | 3.47 |
| GO:0034641 | Cellular nitrogen compound metabolic process | 16 | − | 5.81E-04 | 3.24 |
| GO:0001568 | Blood vessel development | 21 | + | 6.99E-04 | 3.16 |
| GO:0033993 | Response to lipid | 34 | + | 8.79E-04 | 3.06 |
| GO:0060429 | Epithelium development | 32 | + | 9.31E-04 | 3.03 |
| GO:0044267 | Cellular protein metabolic process | 15 | − | 1.18E-03 | 2.93 |
| GO:0043627 | Response to estrogen | 18 | + | 1.33E-03 | 2.88 |
| GO:0001944 | Vasculature development | 21 | + | 2.09E-03 | 2.68 |
| GO:0001655 | Urogenital system development | 18 | + | 2.94E-03 | 2.53 |
| GO:0006807 | Nitrogen compound metabolic process | 20 | − | 3.05E-03 | 2.52 |
| GO:0042476 | Odontogenesis | 10 | + | 3.17E-03 | 2.50 |
| GO:0014070 | Response to organic cyclic compound | 35 | + | 4.21E-03 | 2.38 |
| GO:0010467 | Gene expression | 8 | − | 4.44E-03 | 2.35 |
| GO:0022612 | Gland morphogenesis | 11 | + | 4.54E-03 | 2.34 |
| GO:0043170 | Macromolecule metabolic process | 41 | − | 5.83E-03 | 2.23 |
| GO:0044238 | Primary metabolic process | 54 | − | 7.20E-03 | 2.14 |
| GO:0072359 | Circulatory system development | 28 | + | 7.49E-03 | 2.13 |
| GO:0072358 | Cardiovascular system development | 28 | + | 7.49E-03 | 2.13 |
| GO:1901342 | Regulation of vasculature development | 13 | + | 7.86E-03 | 2.10 |
| GO:0048608 | Reproductive structure development | 19 | + | 9.57E-03 | 2.02 |
| GO:0046483 | Heterocycle metabolic process | 14 | − | 1.02E-02 | 1.99 |
| GO:0006139 | Nucleobase-containing compound metabolic process | 13 | − | 1.18E-02 | 1.93 |
| GO:0061458 | Reproductive system development | 19 | + | 1.19E-02 | 1.92 |
| GO:0048731 | System development | 77 | + | 1.40E-02 | 1.85 |
| GO:0048771 | Tissue remodeling | 10 | + | 1.52E-02 | 1.82 |
| GO:0045765 | Regulation of angiogenesis | 12 | + | 1.63E-02 | 1.79 |
| GO:0009725 | Response to hormone | 32 | + | 1.77E-02 | 1.75 |
| GO:0030335 | Positive regulation of cell migration | 17 | + | 1.89E-02 | 1.72 |
| GO:0061448 | Connective tissue development | 13 | + | 2.13E-02 | 1.67 |
| GO:0030334 | Regulation of cell migration | 23 | + | 2.23E-02 | 1.65 |
| GO:0048729 | Tissue morphogenesis | 22 | + | 2.24E-02 | 1.65 |
| GO:0071704 | Organic substance metabolic process | 59 | − | 2.45E-02 | 1.61 |
| GO:0044707 | Single-multicellular organism process | 92 | + | 2.56E-02 | 1.59 |
| GO:2000147 | Positive regulation of cell motility | 17 | + | 2.65E-02 | 1.58 |
| GO:0097305 | Response to alcohol | 21 | + | 2.74E-02 | 1.56 |
| GO:0007275 | Multicellular organismal development | 81 | + | 2.80E-02 | 1.55 |
| GO:0018149 | Peptide cross-linking | 5 | + | 2.82E-02 | 1.55 |
| GO:0051270 | Regulation of cellular component movement | 25 | + | 3.01E-02 | 1.52 |
| GO:0051272 | Positive regulation of cellular component movement | 17 | + | 3.89E-02 | 1.41 |
| GO:2000145 | Regulation of cell motility | 23 | + | 4.56E-02 | 1.34 |
| GO:0040017 | Positive regulation of locomotion | 17 | + | 4.80E-02 | 1.32 |
Over, overrepresented; under, underrepresented.
Genes Involved in Treatment Resistance: An Active Process with a New Gene Profile.
To explore which biological processes (Fig. 3H) and molecular functions (Fig. 3I) confer resistance in some FSL rats to low-dose LAC after exposure to a stress episode, we performed a functional classification analysis for the 27 genes involved in the resistance to treatment response. Meaningful gene categories include processes of biogenesis, cellular development, biological adhesion, and immune and metabolic regulation, and meaningful molecular functions include transcription factor, receptor and transporter activities, and enzyme regulator and binding activities (Fig. 3 H and I). Because of the key role of LAC in mediating transfer of fatty acids from cytosol to mitochondria for subsequent β-oxidation (5) that is needed for energy homeostasis, it is noteworthy that nrFSL were characterized by alterations in fatty acid signaling because subsequent qPCR analysis confirmed threefold up-regulation in the vDG of Elovl7, a fatty-acid elongase, and Cyb5r2, a cytochrome-b NAD(P)H oxidoreductases implicated in fatty acid elongation, whereas such genes were not altered in both veh-FSL and rFSL compared with veh-FRL in vDG (Fig. 4 H and I). Thus, development of resistance to antidepressant action is an active process with occurrence of a new gene expression profile, whereby altered regulation of fatty acid signaling seems to link resistance to response to LAC action in nrFSL. We also found a strong down-regulation of NPAS4 among the 27 treatment resistance genes; NPAS4 is an energy-demanding process that mediates reorganization of inhibitory synapses (19). Such NPAS4 impairments are consistent with neuronal hyperactivity in the vDG of nrFSL that is in line with a hyperactivity of the glutamate system in the vDG as a result of mGlu2 decrease.
Our analysis of rFSL vs. nrFSL showed other changes that remain to be fully explored. One, in particular, is that whereas in rFSL transcription of the MR (Fig. 4E), mGlu2 and InsR genes seem to be at the levels of veh-FRL, nrFSL showed a strong up-regulation in MR and reductions in InsR in the vDG compared with FRL. These findings are in line with previous studies reporting a link between increased mineralocorticoid activity and insulin resistance via a decreased transcription of the InsR, an effect that is blocked by treatment with MR antagonists (20).
vDG As a Hot Spot of Depressive-Like Behavior and Rapid Antidepressant-Like Action.
Because previous studies showed that hippocampal mGlu2 regulation is involved in depressive-like phenotypes and rapid antidepressant-like action, we screened such a target in our RNAseq databank and validated with subsequent qPCR. Indeed, FSL had reduced mGlu2 expression as previously reported (1) and this decrease was still observed in nrFSL, whereas mGlu2 levels were corrected in rFSL (Fig. 4F) along with a decrease in the immobility at the FST (Fig. 1 B–D). A dynamic mGlu2 regulation has been shown recently in the vDG of chronically stressed mice, where windows of epigenetic plasticity that control behavior are seen in the immediate aftermath of stress with a glutamatergic hyperactivity of the vDG based on the continued down-regulation of the regulators of glutamate release, mGlu2 receptors, which normally inhibit neuronal glutamate release (21, 22). Of note, nrFSL, which had lower mGlu2 in the vDG and high immobility at the FST compared with rFSL, also showed increased MR levels, in line with previous findings showing an MR-mediated down-regulation of mGlu2 that leads to glutamate overflow (8, 23) and contributes to depression in mice prone to develop anxiety and depressive-like traits after stress. Next, we performed an interaction network (IN) analysis to uncover the genes that connect mGlu2, MR, and Lepr because they seemed to be differentially regulated in response to LAC treatment in rFSL vs. nrFSL. The IN analysis identified NPY in a signaling associated with Lepr and mGlu2-mediated glutamate regulation along with neutrophic factors, such as tyrosine receptor kinase B (TrkB) and BDNF, that may play a role in the depressive-like phenotype of FSL and LAC action (Fig. 4J). Indeed, NPY gene expression was decreased in the vDG of FSL, suggesting that the NPY decrease may be a consequence of the hyperactivation of the glutamate and leptin systems due to the MR-mediated decrease in mGlu2 and concomitant increase in Lepr, respectively. In rFSL, but not in nrFSL, the decrease in NPY was rapidly corrected by LAC (Fig. 4G) along with increased mGlu2 levels, indicating that regulation of NPY and regulation of mGlu2 and Lepr in the vDG are markers of responsiveness to treatment. Previous studies also showed an interaction between leptin and NPY in that leptin activates NPY signaling to regulate energy metabolism. In mice, NPY injections into the brains stimulate feeding, whereas specific inhibition of NPY neurons induces a loss of appetite (24); these are factors that can be relevant to anhedonia, a hallmark of depression.
LAC Corrects Systemic Hyperinsulinemia and Hyperglycemia Associated with Endogenously Depressed FSL: Implications for Insulin Resistance.
Because of the central metabolic features we described in the vDG and because of the reciprocal communication between the brain and the body (25), we studied the effects of LAC treatment on plasma glucose, insulin, and triglyceride. Endogenously depressed FSL showed high plasma triglycerides levels vs. veh-FRL; this was corrected by a short LAC treatment in both rFSL and nrFSL (Fig. 5A), suggesting that the system is attempting to respond to the treatment. Also, veh-FSL showed increased plasma glucose and insulin levels vs. veh-FRL. The increased glucose and insulin levels seemed to be corrected to the levels of veh-FRL only in rFSL, whereas nrFSL still had high glucose and insulin levels (Fig. 5 B and C). These results suggest that nrFSL showed systemic insulin resistance that reflects central metabolic changes in the vDG, and vice versa.
Fig. 5.

LAC rapidly corrects a systemic hyperinsulemia and hyperglycemia associated with endogenously depressed FSL while improving depressive-like traits and central deficits in the vDG: implications for insulin-resistance. (A) FSL show high plasma triglyceride levels compared with FRL, and this increase is rapidly corrected by LAC in both rFSL and nrFSL. (B and C) FSL show high plasma glucose and insulin levels compared with FRL, and this increases are rapidly corrected by LAC in rFSL only, whereas nrFSL show still high glucose and insulin levels that reflect occurrence of an insulin-resistant state after stress exposure that is associated with a still-high immobility time at the FST time in such subset of animals not responding to the treatment. Bars: mean + SEM, *significant comparisons with veh-FRL, #significant comparisons with LAC-treated nrFSL. **P < 0.01, #P < 0.05.
Discussion
Although impairments in energy processes have been associated with multiple disorders, including neurological and psychiatric diseases, less is known about the role that regulation of energy metabolism plays in depression and in the responsiveness and/or resistance to antidepressants. We report that signatures of central and systemic metabolic syndrome are associated with the depressive-like phenotype of endogenously depressed FSL and that LAC oral administration exerts rapid antidepressant-like effects while improving energy regulation in the vDG and reducing hyperinsulinemia and hyperglycemia systemically. Such understanding may be critical for the development of novel antidepressants with a higher rate of response and rapid onset of therapeutic effects, considering that patients suffering with depression show a variety of symptoms that are features of impaired energy homeostasis, such as abnormalities in appetite, and physical inactivity, among other symptoms (26).
Rapid Impact of LAC on Central and Systemic Energy Regulation and Mood Abnormalities.
Here, we show that energy regulation via metabolic pathways is a target of LAC antidepressant treatment in the vDG, a limbic brain region important for depression and resilience to stress (22). While improving central deficits in the vDG and depressive-like behavior, LAC rapidly corrects systemic metabolic alterations in insulin and glucose levels associated with FSL. Previous studies have shown that systemic injections of LAC rapidly correct behavioral abnormalities (i.e., low sucrose preference and passive behavior at FST) associated with FSL via an epigenetic regulation of acetylation of histone H3K27 bound to the promoter of Grm2, which encodes for the regulator of glutamate release mGlu2 in hippocampus (1). Neuroanatomically, the present study and other evidence (7) reveal that the vDG is a hot spot for an increased glutamate overflow due to mGlu2 decrease. Such glutamate overflow is concomitant with central metabolic alterations [e.g., up-regulation in Insr, Lepr, Glut-4, Glut-12, and Cartpt, a prohormone involved in the regulation of appetite through NPY signaling (24)]. The occurrence of these central metabolic alterations along with systemic metabolic changes in endogenously depressed FSL indicates that the FSL can be a useful model for future research to study the mechanisms of comorbidity of depression and central and systemic metabolic impairments that is of great interest because of the high incidence of metabolic syndrome in depressed subjects (11, 12). Here, the dysregulation in central insulin levels in FSL does not seem to involve the canonical mechanism of insulin resistance linked to Glut-4, which, instead, is up-regulated in the vDG of FSL, thereby suggesting that there may be an upstream mechanism in the insulin resistance associated with the depressive-like phenotype of FSL that could involve other glucose transporters, such as Glut-12. A similar link between insulin resistance and increased Glut-4 (27) was shown previously in hyperinsulemic-hyperglycemic rats, which show increased Glut-4 levels in the cerebellum (28). Future work should study whether Glut-4 changes may reflect a compensatory response to deficits in central glucose levels and to the hyperactivation of the glutamate and leptin systems.
Role of Stress in Development of Treatment Resistance in Some Individuals: Implications for Adjunctive Therapies.
Although stress is well recognized as a key factor in precipitating a variety of disorders (29), including depression, the role of stress in altering the responsiveness to antidepressants is less known. Recently, we showed that stress opens windows of epigenetic plasticity in at-risk individuals (22) and these temporary slots of plasticity can be manipulated by LAC pharmacological intervention to increase resilience, whereby structural plasticity of the medial amygdala stellate neurons plays an important role (30). Here, our findings in endogenously depressed FSL subjected to an acute stress during LAC treatment show that an individual responsiveness to stress needs to be factored in toward the development of better therapeutics. Indeed, the nrFSL showed 27 genes that confer resistance to treatment after stress exposure, interfering with LAC-induced gene changes evident in nrFSL that resemble those that LAC produces in rFSL. Among those 27 genes are two metabolic regulators of fatty acid elongation, namely Elovl7 and Cyb5r2, suggesting that altered metabolic processes, including fatty acid metabolism, play a yet-to-be-determined role in resistance to LAC after stress exposure. These changes are concomitant with impairments in high-energy demanding processes of inhibitory synaptic reorganization as indicated by NPAS4 down-regulation in the vDG of nrFSL. Such regulation of NPAS4, and mGlu2, points to regulation of neuronal hyperactivity of the vDG as a target for overcoming treatment resistance. Moreover, nrFSL with still elevated systemic levels of glucose and insulin showed up-regulation of MR, and decreased mGlu2, in the vDG along with a degradation of the InsR, whereas rFSL did not. Previous findings have shown that MR activation mediates, in hippocampus, stress-induced down-regulation of mGlu2 that further exacerbates overflow of glutamate (8, 23), which is known to contribute to depression (16). Also, previous studies have reported a link between increased MR activity and insulin resistance via a decreased transcription of the InsR and consequent inability to lower glucose levels, an effect that is blocked by treatment with MR antagonists (20). Hence, based on our findings and previous evidence, agents such as LAC, by reducing the MR-mediated mGlu2-driven glutamate overflow, could be useful in the treatment of insulin resistance with comorbid depression. This idea is supported by previous findings showing that an NMDA-mediated hyperactivity of the glutamate system causes phosphatases to inactivate insulin receptor with consequent development of insulin resistance (31). Human studies have also shown that LAC therapy ameliorates insulin resistance (32). Future studies are warranted to investigate LAC efficacy in humans with depression and LAC could also be considered for treatment of insulin resistance in depressed subjects. FSL are also known to be deficient in leptin and adiponectin, and adiponectin is related to some symptoms of depression in humans (33). Previous studies have associated a hyperactivity of the HPA axis with haploinsufficiency in adiponectin (34), which stimulates fatty-acid oxidation (35), whose metabolism seems to be impaired in only nrFSL based on alterations in Cyb5r2- and Elovl7-mediated regulation of fatty-acid elongation. Therefore, our results suggest that adiponectin (33) may be relevant in determining occurrence of resistance to antidepressant treatment, and this is an interesting topic for future investigation.
In conclusion, we elucidated a previously unrecognized role of metabolic-regulator factors in depressive-like behavior and responsiveness to a putative rapid-acting antidepressant agent, LAC. These results further support a key role for the vDG. Furthering our understanding of how stress affects the responsiveness to therapeutics will ultimately contribute to improved treatments where an individual responsiveness to stress needs to be factored in.
Methods
All procedures were carried out in accordance with the National Institutes of Health, The Rockefeller University Institutional Animal Care and Use Committee guidelines, and the European (86/609/EEC) guidelines of animal care. All in vivo experiments were approved by the Karolinska Institutet Ethical Committee, according to Swedish and European guidelines. All efforts were made to minimize animal distress and to reduce the number of animals used in this study. See SI Methods for details of stress procedure, behavioral tests, gene expression, ELISA, RNAseq, and Bioinformatics analysis.
SI Methods
Animals.
Male FSL and FRL from rat colonies maintained at the Karolinska Institutet were housed in standard Macrolon type 3 cages in a temperature- and humidity-controlled room (22 ± 1 °C) with standard 12-h light/dark cycle and ad libitum access to food and water. To control intake of water/drug, while avoiding isolation, adult male rats (average of 430 g) were housed in groups of four of the same genotype. Cages were randomly assigned to a treatment group, for a total of n = 8 animals per group. From five to eight biological replicates were used for the behavioral and molecular experiments.
FSL rats were divided into rFSL and nrFSL to LAC treatment based on their immobility time at the forced swim, similarly to published reports (8). LAC-treated FSL rats, which fell into the SD from the mean of the control veh-FRL group, were considered rFSL because they showed a behavior comparable to that of veh-FRL rats. FSL rats that fell outside the SD of the mean of the control veh-FRL group were designated as nrFSL. Briefly, a cutoff is given from the sum of the mean and SD of veh-FRL.
Experimental Design, Pharmacological Treatment, and Behavioral Tests.
There was a habituation period of 5 d, where each animal was handled for ∼2 min/d. Tap water (vehicle) or LAC (Sigma), dissolved in tap water, at a concentration of 0.3% (wt/vol), was available ad libitum in standard rat sipping bottles. The treatment was available for a total of 7 d, with fresh water and LAC prepared after 3 d. LAC intake (30 mL) did not differ from water intake (31 mL) per day. This corresponded to a daily oral LAC consumption of ∼210 mg/kg body weight.
At day 6, we measured performance in an open field arena (60 × 60 × 60 cm) for 5 min, immediately followed by a 15-min forced swim (1) used as a behavioral test over the first 5 min of swim and as a stressor over the prolonged 15 min of swim. Animals were placed in 30-cm-diameter, 50-cm-tall tanks filled with water (at 24 ± 1 °C) at 35-cm depth. Rats were gently dried before being placed back in their home cages. At day 7, rats performed a 5-min FST. After drying, rats were again placed in their respective home cages before being killed by decapitation following brief exposure to CO2 in a saturated chamber. The order of testing was randomized, to alternate between groups at each trial.
Performance in the open field (1) was tracked and analyzed using an automated video tracking system (Noldus Ethovision XT11), which measured average velocity and cumulative distance moved. Performance in the FST was recorded with a video camera and was subsequently scored by Ethovision, using 10% as immobility threshold setting.
Gene Expression Analysis.
vDG tissues were dissected and weighed before homogenization and about 130 mg of tissue was lysed. Total RNA was extracted using Qiazol reagent and RNeasy mini Kit (Qiagen), according to the manufacturer’s instructions. RNA quality was checked using the 2100 Bioanalyzer (Agilent Technologies) and quantified spectrophotometrically. The acceptance criteria for RNA quality were a 260/280 ratio ≥1.80 and ≤2.20. Two micrograms of total RNA was then used for cDNA synthesis according to the manufacturer’s instructions of the high-capacity cDNA reverse transcription kit (Life Technologies). The reverse-transcribed reaction was carried out according the follow condition steps: 25 °C for 10 min, 37 °C for 120 min, and 85 °C for 5 min. qPCR was performed according to the protocols of the manufacturer (Life Technologies), using TaqMan Universal PCR Master Mix and gene-specific primers, synthetized by Applied Biosystem Company (Life Technologies). We validated our microdissection approach for isolating the vDG by examining the expression of a gene, lactate (LCT), which is known to be absent in the vDG (Fig. S3). GAPDH was used as a reference control for qPCR analysis. The following PCR conditions were used: an initial incubation of 50 °C for 2 min and a denaturation step of 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. All reactions were performed in triplicate. The threshold cycle (CT), which correlates inversely with the levels of target mRNA, was measured as the number of cycles at which the reporter fluorescence emission exceeds the preset threshold level. The amplified transcripts were quantified using the comparative CT method with the formula for relative fold change =2ΔΔCT.
RNAseq and Bioinformatics Analysis.
RNA integrity was checked using the Agilent 2100 Bioanalyzer. Four micrograms of total RNA were used for total RNA library constructs (TruSeq Stranded Total RNA with Ribo-Zero Human/Mouse/Rat according to the manufacturer instructions) and sequenced on the Illumina HiSeq2500 machine at the Genomic Core facility at The Rockefeller University. In total, three biological replicates were carried out for each condition. Each sample was provided with a unique adapter and all samples were put in the same pool and run in multiple lanes to control for lane effects and sequenced with a sequencing depth of about 30 M (100 bp, single) (Table S1). The Fastq files were evaluated for quality control using FastQC and trimmed with Trimmomatic (36). Alignment was performed using the Tophat2 (37) against a reference gene database (RGSC 5.0/rn5). Raw counts were calculated using htseq-count (38). Statistical analyses for differential expression (DE) were performed with EdgeR (39); genes with more than 1 count per million in at least three samples were retained in the EdgeR analysis to control for low-expressed genes. A gene is considered differentially expressed when reaching the following criteria: false discovery rate <15% and fold change >1.3. Table S2 reports the enrichment GO for biological process showing the most significant terms that are altered among the 544 genes that confer predisposition for depression, and Table S3 reports the enrichment GO for biological process showing the most significant terms that are altered among the 205 genes that confer responsiveness to LAC oral treatment. Heat maps were generated with an R script created in-house. Statistical overrepresentation for GOs was generated with Panther (40).
Systemic Triglyceride, Insulin, and Glucose Measurements.
Plasma levels of triglyceride, insulin, and glucose were measured in samples taken from trunk blood collected at time of killing, according to the manufacturer’s instructions (triglyceride: 10010303, Cayman; insulin: EZRMI-13K, Millipore; glucose: 10009582, Cayman) with the following dilution factors: triglyceride: 1/2, insulin: 1/5, and glucose: 1/10.
Statistics.
Statistical analyses were performed using two-tailed unpaired Student’s t tests or two-way analysis of variances (ANOVA) followed by Tukey’s test for the post hoc analysis.
Acknowledgments
This work was supported by American Foundation for Suicide Prevention, Hope for Depression Research Foundation, Grant UL1 TR000043 from the National Center for Advancing Translational Sciences, the NIH Clinical and Translational Science Award program, The Wenner-Gren Foundations, and Swedish Medical Research Council Grant 10414.
Footnotes
The authors declare no conflict of interest.
Data deposition: Raw and processed RNA-sequencing data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database (accession no. GSE83336).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1603111113/-/DCSupplemental.
References
- 1.Nasca C, et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA. 2013;110(12):4804–4809. doi: 10.1073/pnas.1216100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russo SJ, Charney DS. Next generation antidepressants. Proc Natl Acad Sci USA. 2013;110(12):4441–4442. doi: 10.1073/pnas.1301593110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flight MH. Antidepressant epigenetic action. Nat Rev Neurosci. 2013;14(4):226. doi: 10.1038/nrn3466. [DOI] [PubMed] [Google Scholar]
- 4.Pettegrew JW, Levine J, McClure RJ. Acetyl-L-carnitine physical-chemical, metabolic, and therapeutic properties: Relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol Psychiatry. 2000;5(6):616–632. doi: 10.1038/sj.mp.4000805. [DOI] [PubMed] [Google Scholar]
- 5.Fritz IB, McEWEN B. Effects of carnitine on fatty-acid oxidation by muscle. Science. 1959;129(3345):334–335. doi: 10.1126/science.129.3345.334. [DOI] [PubMed] [Google Scholar]
- 6.Kishi T, Horiguchi J. [Psychiatric disorders and neural mechanisms underlying energy intake and expenditure: A review] Yakubutsu Seishin Kodo. 2003;23(5):197–203. Japanese. [PubMed] [Google Scholar]
- 7. Zelli D, McEwen BS, Nasca C (2015) xCT epigenetically promotes homeostatic regulation of the glutamate system in the responses to stress: Implications for next-generation treatments. Available at www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=c7563ab4-bcde-44ef-9924-2530e02f4e24&cKey=7b8af88d-bc80-41ec-903e-7b0cb48bedd3&mKey=%7bD0FF4555-8574-4FBB-B9D4-04EEC8BA0C84%7d. Accessed June 15, 2016.
- 8.Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS. Mind the gap: Glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry. 2015;20(6):755–763. doi: 10.1038/mp.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann ID, et al. Animal models of depression and anxiety: What do they tell us about human condition? Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(6):1357–1375. doi: 10.1016/j.pnpbp.2010.11.028. [DOI] [PubMed] [Google Scholar]
- 10.Wu G, et al. Central functions of neuropeptide Y in mood and anxiety disorders. Expert Opin Ther Targets. 2011;15(11):1317–1331. doi: 10.1517/14728222.2011.628314. [DOI] [PubMed] [Google Scholar]
- 11.McIntyre RS, et al. Brain volume abnormalities and neurocognitive deficits in diabetes mellitus: Points of pathophysiological commonality with mood disorders? Adv Ther. 2010;27(2):63–80. doi: 10.1007/s12325-010-0011-z. [DOI] [PubMed] [Google Scholar]
- 12.Wroolie TE, Kenna HA, Singh MK, Rasgon NL. Association between insulin resistance and cognition in patients with depressive disorders: Exploratory analyses into age-specific effects. J Psychiatr Res. 2015;60:65–72. doi: 10.1016/j.jpsychires.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Overstreet DH, Friedman E, Mathé AA, Yadid G. The Flinders Sensitive Line rat: A selectively bred putative animal model of depression. Neurosci Biobehav Rev. 2005;29(4-5):739–759. doi: 10.1016/j.neubiorev.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13(10):1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang SM, et al. A review of current evidence for acetyl-l-carnitine in the treatment of depression. J Psychiatr Res. 2014;53:30–37. doi: 10.1016/j.jpsychires.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 16.McEwen BS, et al. Mechanisms of stress in the brain. Nat Neurosci. 2015;18(10):1353–1363. doi: 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McEwen BS, Gray JD, Nasca C. 60 YEARS OF NEUROENDOCRINOLOGY: Redefining neuroendocrinology: Stress, sex and cognitive and emotional regulation. J Endocrinol. 2015;226(2):T67–T83. doi: 10.1530/JOE-15-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nat Rev Endocrinol. 2014;10(5):303–310. doi: 10.1038/nrendo.2014.22. [DOI] [PubMed] [Google Scholar]
- 19.Bloodgood BL, Sharma N, Browne HA, Trepman AZ, Greenberg ME. The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature. 2013;503(7474):121–125. doi: 10.1038/nature12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg R, Adler GK. Role of mineralocorticoid receptor in insulin resistance. Curr Opin Endocrinol Diabetes Obes. 2012;19(3):168–175. doi: 10.1097/MED.0b013e3283533955. [DOI] [PubMed] [Google Scholar]
- 21.McEwen BS, Nasca C, Gray JD. Stress effects on neuronal structure: Hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology. 2016;41(1):3–23. doi: 10.1038/npp.2015.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nasca C, et al. Stress dynamically regulates behavior and glutamatergic gene expression in hippocampus by opening a window of epigenetic plasticity. Proc Natl Acad Sci USA. 2015;112(48):14960–14965. doi: 10.1073/pnas.1516016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karst H, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA. 2005;102(52):19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalra SP, Kalra PS. NPY and cohorts in regulating appetite, obesity and metabolic syndrome: Beneficial effects of gene therapy. Neuropeptides. 2004;38(4):201–211. doi: 10.1016/j.npep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 25.van Praag H, Fleshner M, Schwartz MW, Mattson MP. Exercise, energy intake, glucose homeostasis, and the brain. J Neurosci. 2014;34(46):15139–15149. doi: 10.1523/JNEUROSCI.2814-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaidanovich-Beilin O, Cha DS, McIntyre RS. Crosstalk between metabolic and neuropsychiatric disorders. F1000 Biol Rep. 2012;4:14. doi: 10.3410/B4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106(2):171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alquier T, Leloup C, Lorsignol A, Pénicaud L. Translocable glucose transporters in the brain: Where are we in 2006? Diabetes. 2006;55(Suppl 2):S131–S138. [Google Scholar]
- 29.McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 30.Lau TBB, Zelli D, McEwen BS, Nasca C. Stress-induced structural plasticity of medial amygdala stellate neurons and rapid prevention by a candidate antidepressant. Mol Psych. May 31, 2016. [DOI] [PMC free article] [PubMed]
- 31.De Felice FG, Lourenco MV, Ferreira ST. 2014. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10(Suppl 1):S26–S32.
- 32.Ruggenenti P, et al. Ameliorating hypertension and insulin resistance in subjects at increased cardiovascular risk: effects of acetyl-L-carnitine therapy. Hypertension. 2009;54(3):567–574. doi: 10.1161/HYPERTENSIONAHA.109.132522. [DOI] [PubMed] [Google Scholar]
- 33.Wilhelm CJ, Choi D, Huckans M, Manthe L, Loftis JM. Adipocytokine signaling is altered in Flinders sensitive line rats, and adiponectin correlates in humans with some symptoms of depression. Pharmacol Biochem Behav. 2013;103(3):643–651. doi: 10.1016/j.pbb.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, et al. Adiponectin is critical in determining susceptibility to depressive behaviors and has antidepressant-like activity. Proc Natl Acad Sci USA. 2012;109(30):12248–12253. doi: 10.1073/pnas.1202835109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padmalayam I, Suto M. Role of adiponectin in the metabolic syndrome: Current perspectives on its modulation as a treatment strategy. Curr Pharm Des. 2013;19(32):5755–5763. doi: 10.2174/13816128113199990360. [DOI] [PubMed] [Google Scholar]
- 36.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;15:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;4 doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;2:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;1:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucl Acids Res. 2016;44:D336–D342. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]