

Abstract
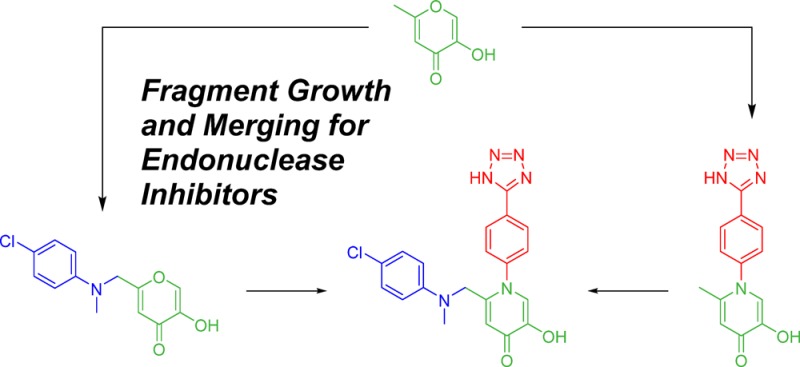
The influenza virus is responsible for millions of cases of severe illness annually. Yearly variance in the effectiveness of vaccination, coupled with emerging drug resistance, necessitates the development of new drugs to treat influenza infections. One attractive target is the RNA-dependent RNA polymerase PA subunit. Herein we report the development of inhibitors of influenza PA endonuclease derived from lead compounds identified from a metal-binding pharmacophore (MBP) library screen. Pyromeconic acid and derivatives thereof were found to be potent inhibitors of endonuclease. Guided by modeling and previously reported structural data, several sublibraries of molecules were elaborated from the MBP hits. Structure–activity relationships were established, and more potent molecules were designed and synthesized using fragment growth and fragment merging strategies. This approach ultimately resulted in the development of a lead compound with an IC50 value of 14 nM, which displayed an EC50 value of 2.1 μM against H1N1 influenza virus in MDCK cells.
Introduction
The influenza virus is responsible for annual seasonal epidemics, resulting in 3–5 million yearly cases of severe illness and an estimated 250 000–500 000 deaths worldwide.1 The past century alone has seen the advent of four influenza pandemics, each resulting in millions of deaths.2 While vaccinations are a reasonable prophylactic for healthy adults, they must be re-administered annually and are markedly less effective for individuals with compromised immunity or similar high-risk medical conditions. In addition, the efficacy of these vaccines is heavily dependent on correctly predicting the predominant infectious strains for any given year, and incorrect predictions can render vaccination less than 25% effective.3 Existing drugs, such as zanamivir (GlaxoSmithKline) and oseltamivir (Roche), which target viral neuraminidase, can be useful in treating influenza infections but must be administered within 1–2 days of infection to be effective. These therapeutics also suffer from undesirable side effects, including unusual neurologic or psychiatric events such as delirium, hallucinations, confusion, and abnormal behavior, primarily in children.4−6 M2 ion channel blockers such as rimantadine (Sun Pharma) and amantadine (Endo) were previously effective at inhibiting viral replication; however, 100% of seasonal H3N2 and 2009 pandemic H1N1 influenza strains now show resistance to these drugs.7,8 Considering this, there is an urgent need for the development of new drugs to prevent and treat influenza infection.
The influenza virus is a lipid-enveloped, negative-sense, single-strand RNA virus. The viral genome is divided into 8 distinct genomic segments, each encoding one or two of the 11 total viral proteins.9 This segmented genome allows for the exchange of segments between different viruses in infected host cells and, coupled with the low fidelity of the viral RNA polymerase, explains the high rates of drug resistance and antigenic shift seen in influenza viruses.4,10 Each RNA segment is packaged in complex with a single heterotrimeric RNA dependent RNA polymerase.11 The polymerase complex comprises three distinct subunits (PA, PB1, PB2) and is responsible for both transcription and replication of the viral genome. However, the complex is unable to synthesize the 5′-mRNA cap necessary for translation by eukaryotic host-cell translation machinery. To overcome this limitation, the polymerase hijacks a mature 5′-cap from host cell pre-mRNA. This “cap-snatching” mechanism is accomplished by the polymerase B2 subunit tightly binding the modified 5′-guanine nucleotide with subsequent cleavage 10–13 nucleotides downstream by the N-terminal endonuclease portion of the PA subunit.12,13 The sequestered, capped RNA segment is then used as a primer for viral mRNA synthesis, and the resulting hybrid RNA is translated by the host cell.14
The viral polymerase complex is an attractive target for new antiviral therapies. It is highly conserved across all influenza strains and subtypes, and inhibitors should therefore have broad efficacy against multiple serotypes. Of particular interest, the cap-snatching mechanism is essential to the virus lifecycle, is conserved in all members of the influenza virus family, and has no human analogue.4 Cap-snatching, and subsequent viral replication, has been shown to be inhibited by inactivating one of two different domains of the viral polymerase complex: either inhibiting the 5′-mRNA cap binding site on the PB2 subunit15−17 or inhibiting the N-terminal endonuclease portion of the PA subunit.4,18,19
The N-terminal domain of the PA subunit contains the endonuclease active site. Crystallographic and biochemical studies have shown the endonuclease to contain a dinuclear metal active site, employing two Mn2+ or Mg2+ cations (Figure 1).13 The structure of the endonuclease domain closely resembles type II restriction endonucleases,13 and it has been shown that coordinating the metal centers of the endonuclease active site effectively inhibits endonuclease activity. There are currently no FDA approved endonuclease inhibitors, but several classes of inhibitors have been reported in recent literature. These include diketo acids,20,21 hydroxamic acids,22 flutimide derivitives,23 green tea catechins,24,25 dihydroxypyrimidinones,26 3,2-hydroxypyridinones,27 and others. Most of these were identified by experimental or computational high-throughput screening (HTS) of large libraries and produced only modest inhibitors in spite of great synthetic effort. More recently, some very potent in vitro inhibitors have been identified.21,28,29
Figure 1.

Structural model of the influenza RNA-dependent RNA polymerase PA subunit (PDB code 4M5Q). The endonuclease active site employs two divalent metal cations to facilitate the hydrolytic cleavage of the phosphodiester backbone of nucleic acids. Key active site residues and binding pockets are highlighted. Pocket 2 is obscured by residue Tyr24. Divalent metal cations are shown as orange spheres with the bridging hydroxide anion shown as a red sphere.
In an effort to efficiently identify novel, potent inhibitors of influenza PA endonuclease, we carried out a fragment-based drug discovery (FBDD) campaign, using a designer metal-binding pharmacophore (MBP) library. This library is an expansion of our prior report30 and consists of ∼300 fragment molecules designed to interact with metal ions found in metalloenzyme active sites. From this screening, pyromeconic acid was identified as a ligand-efficient scaffold for inhibitor development. Guided by modeling and SAR identified in the initial library screen, a modest sublibrary of molecules was elaborated from the MBP hits. Our metalloenzyme-focused FBDD approach ultimately leads to molecules with potent in vitro inhibitory activity (IC50 < 50 nM) against viral PA endonuclease. The most potent of these, 1-(4-(1H-tetrazol-5-yl)phenyl)-2-(((4-chlorophenyl)(methyl)amino)methyl)-5-hydroxypyridin-4(1H)-one (71), was found to inhibit endonuclease activity with an IC50 of 14 nM in enzymatic assays. Compound 71 further showed good antiviral activity against H1N1 influenza A virus in Marvin–Darby canine kidney (MDCK) cells with an EC50 value of 2.1 μM and a CC50 value of 280 μM. These findings demonstrate that a metal-centric FBDD approach can result in a rapid and efficient identification of leadlike molecules, especially when compared to FBDD using unbiased libraries or high-throughput screening.
Results
MBP Library Screening
Fragments in the MBP library were initially screened for endonuclease inhibition against the PA subunit of the influenza polymerase complex. The screen was performed using a FRET-labeled DNA oligonucleotide substrate based on previously reported protocols with some minor modifications (Supporting Information).13,28 Compounds that exhibited >80% inhibition at a concentration of 200 μM were re-evaluated at a concentration of 50 μM. Dose–response curves were compiled and IC50 values determined for fragments exhibiting >50% inhibition at 50 μM. This screen identified pyromeconic acid (1) as a moderate inhibitor of endonuclease activity (IC50 = 22.5 μM; LE = 0.79) and unveiled important structure–activity trends associated with simple modifications to the hydroxypyrone ring (Table 1). A proposed mode of metal coordination for 1 is shown in Figure 2. This model is based on superposition of the donor atoms in 1 with crystallographic water molecules present in X-ray structures of uninhibited influenza PA endonuclease and superposition of the pyrone ring with other similar cyclic metal-binding inhibitors that have been crystallographically validated against the endonuclease.28
Table 1. Select MBP Fragments That Displayed Potent Inhibition against Influenza PA Endonuclease31,a.

pIC50 is defined as pIC50 = −log(IC50) and is included to allow a linear comparison between IC50 values. Ligand efficiency (LE) provides a measure of binding energy per non-hydrogen atom in the fragment molecule.
Figure 2.
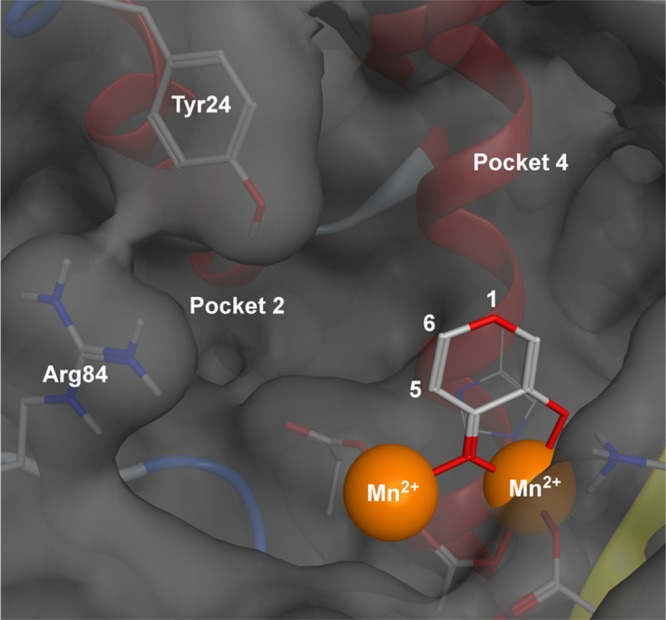
Proposed binding of pyromeconic acid (1) to the PA subunit active site. Ring positions 5 and 6 were deemed amenable to further derivatization for targeting hydrophobic pocket 2 and key residues Tyr24 and Arg84. Derivatization at the ring position 1 was proposed to probe interactions with pocket 4.
It was found that the addition of a methyl group to the 6-position of the pyromeconic acid ring (2, allomaltol) had little effect on the potency of the fragment, while the addition of a methyl group to the 2-position (3, maltol) effectively abolished activity. This is consistent with other endonuclease inhibitors that employ a similar MBP motif, which demonstrates that addition of substituents α to the hydroxyl donor atom reduces potency.26,27 Addition of hydrophilic substituents in the 6-position (4, 5) was well tolerated. The addition of a carboxylic acid moiety to the 5-position, as in compound 6, afforded an increase in potency, as did a methylamide derivative. Other derivatives of 2 (6-position substituent) gave rise to modest improvements in potency (8, 9).
Conversion of the pyrone ring to an N-methylpyridinone resulted in a marked increase in potency, as seen in 9 and 10. Pyridinones, as compared to pyrones, have more aromatic character and greater electron density on the donating oxygen atoms.32 This change in the electronic structure of the ligand typically results in greater ligand basicity, which can lead to better interactions with hard Lewis acidic metal centers, such as the Mg2+ or Mn2+ found in the endonuclease active site.32 On the basis of this preliminary SAR, synthetically feasible elaborations were designed using pyromeconic acid derivatives as an MBP core. Substitutions at the 1-, 5-, and 6-positions were deemed most accessible and were evaluated independently.
5-Position Derivatives
On the basis of the SAR outlined in Table 1, initial efforts focused on exploring amide derivatives of compound 6/7. Guided by modeling and prior crystallographic evidence,18,28 it was proposed that aryl substituents at the 5-position could favorably interact with residue Tyr24 or residue Arg84 or make favorable hydrophobic interactions with pocket 2 (Figure 1). A sublibrary of 11 arylamides derived from 6 were synthesized and evaluated for inhibitory activity (Table 2). While the methylamide 7 was equipotent to the parent fragment 6, all arylamide derivatives were found to be less active. This trend persisted regardless of additional functionalization of the aryl ring or the introduction of methylene spacers between the amide and aryl ring. The only exception to this trend was compound 20, a tryptamine amide, but this derivative was not viewed as an attractive lead based on the substantial increase in molecular weight for little gain in activity. The resulting SAR suggested that 5-position amide functionalization was not a productive route to more active inhibitors.
Table 2. Inhibition Values of Various 5- and 6-Position Derivatives.

6-Position Derivatives
Initial SAR (8, 9) indicated that hydrophilic substituents at the 6-position were well tolerated. To continue probing interactions with pocket 2 (Figure 2), a series of 6-aminomethyl derivatives were synthesized. Starting with commercially available kojic acid, derivatives of the 6-position could be readily prepared with amine, amide, and sulfonamide linkages. Amine, amide, and sulfonamide derivatives were between 3- and 18-fold more active than kojic acid (8), with compound 31 being the most potent of the set. Arylamine and ether, as well as benzylamine derivatives, displayed similar activity, with tertiary amine derivatives being slightly more potent than secondary amine derivatives (compound 26 vs 27; 32 vs 34). Functionalization of the aryl ring substituent in some cases provided added potency; however, the introduction of a pyridine substituent drastically decreased activity. Ultimately, the SAR at the 6-position was relatively flat among aryl derivatives but did indicate some gains could be made, with compound 35 identified as the most potent derivative of this set with an IC50 value of 0.94 ± 0.08 μM.
Pyridinone Derivatives
Conversion of the pyrone ring to a pyridinone ring not only increases ligand basicity (and hence metal binding) but also affords a chemical handle for facile exploration of the large pocket located above the metal center active site of endonuclease (Figure 2). To probe this pocket, a sublibrary of N-substituted pyridinones was synthesized and screened against endonuclease (Table 3). It was found that both N-aryl- and N-cycloalkylpyridinones (compounds 44 and 47) were significantly more active than the parent fragment compound 10. Simple extension of the aryl substituent (compounds 46 and 50) did not improve activity. Interestingly, compounds 45 and 48 showed a significant loss in activity, consistent with the previously observed negative effect of substituents at the 2-position (compound 3, Table 1).
Table 3. Inhibition Values of Various N-Functionalized Hydroxypyridinones.

In addition to elongation of the substituent, a series of more polar, saturated, heterocyclic derivatives of 47 were prepared. As detailed in Table 3, all of these derivatives showed reduced activity, with modifications to the cyclohexyl moiety not being well tolerated. The simple phenyl ring in compound 44 was deemed more suitable for further synthetic elaboration.
A series of derivatives were synthesized exploring substituents at either the 3′ or 4′ positions of the N-inserted phenyl ring (Table 4). In general, 3′ modifications yielded a flat SAR, with only small changes in activity. The SAR around the 4′ derivatives was more varied and revealed that the addition of an aryl ether (compound 60) or carboxylic acid (compound 62) both afforded increases in activity. While several other polar 4′ derivatives resulted in a reduced potency (64, 65, 66), the addition of a tetrazole group to the 4′ position (63) resulted in a substantial increase in potency. This finding is consistent with previous reports that phenyltetrazoles make very favorable interactions with pocket 4.26−28 Compound 63 was identified as the most potent derivative of this series with an IC50 value of 36 ± 7 nM against PA endonuclease.
Table 4. Inhibition Values and SAR of 3′ and 4′ Derivatives of 5-Hydroxy-2-methyl-1-phenylpyridin-4(1H)-one.

Fragment Merging
In an effort to maximize potency from derivatives of pyromeconic acid (1), a fragment-merging strategy was pursued. Computational modeling suggested that derivatives at the 5- and 6-position were likely interacting with pocket 2 in the endonuclease active site and that N-substituted derivatives at the 1-position were interacting with pocket 4. It was decided to merge the most potent 5- or 6-position derivatives with the most potent 1-position N-aryl derivative to interact with both pockets simultaneously. Because amide derivatization of compound 6 showed effectively no improvement to potency, incorporation of a 5-position carboxylic acid to N-substituted compounds 44, 60, and 63 was pursued. Ethyl esters 67, 68, and 69 surprisingly showed markedly decreased potency after the addition of the carboxylate group at the 5-position. Base catalyzed hydrolysis of these compounds to produce the free carboxylic acid resulted in complete degradation of these molecules; however, 70 was synthesized via an alternative route. Disappointingly, 70 showed no improvement over the parent compound 44. Due to the poor inhibition shown by 70, further efforts toward 5-position carboxylic acids were abandoned.
Extensive SAR of the 6-position revealed that arylamine and sulfonamide derivatives were the most potent elaborations at this position, likely due to favorable hydrophobic interactions with pocket 2 and possible π-stacking interactions with Tyr24. As detailed in Table 5, the addition of a 4-chloro-N-methylaniline substituent to the already potent 63 resulted in a one-half log increase in potency from an IC50 value of 36 ± 7 nM to 14 ± 3 nM for compound 71. While fragment merging did result in an overall more potent molecule, the fact that the observed potency increase was not more additive suggests either a conformational change in binding mode as compared to the original fragments or a possible overlap in binding pockets between the two substituents.
Table 5. Inhibition Values of Fragment Merged Inhibitors.


Docking
In lieu of a cocrystal structure of 71 with endonuclease (efforts are currently underway), docking studies were performed to gain insight about the probable binding mode of 71. Previously reported X-ray crystal structures of inhibitors with similar MBPs bound to the endonuclease active site were used as a starting point for these docking experiments.28 Using Molecular Operating Environment (MOE) software, constrained docking and minimization studies were performed starting from a reported endonuclease crystal structure with a bound hydroxypyridinone-based inhibitor (PDB code 4M4Q, Figure 3). The bound inhibitor was removed from the structure and replaced with compound 71, utilizing the position of the metal-coordinating donor atoms of the cocrystallized inhibitor as an initial guide for the binding mode of 71. Constrained minimizations were then performed on this model in which the metal-binding oxygen atoms of 71 were fixed in optimal geometries based on several parameters, primarily previous crystal structures of inhibitors with similar ligand donor atom identity and geometry.26,27 These constrained simulations ensure that a reasonable metal binding geometry is maintained, as MM-docking simulations do not appropriately account for metal–ligand interactions.33,34 Flexible receptor modeling (induced fit) was employed in which active site residue side chains proximal to the inhibitor molecule were allowed to move, while residue backbones were more constrained. In the simulation the coordinating phenolic oxygen atom on inhibitor 71 was deprotonated, and the charge on each metal center was assigned as 2+ (i.e., Mn2+). Two water molecules coordinated to one of the Mn2+ centers in the template structure (PDB code 4M4Q, Mn302) were retained in the simulation, and their positions were fixed relative to the metal center.
Figure 3.

Docking analysis of 71 in influenza PA endonuclease (PDB code 4M4Q). (a) Docked structure of 71 bound to endonuclease. Hydrophobic interactions were found between the 6-position phenylaminomethyl moiety and hydrophobic pocket 2, as well as halogen bonding with Arg82. The N-phenyltetrazole moiety was found to hydrogen-bond simultaneously to Arg124 and Lys34. (b) Ligand interaction diagram detailing ligand/protein/solvent interactions rendered in two dimensions. Interactions between the ligand and protein active site are displayed as colored dotted lines: coordination bonds in purple, hydrogen bonds and π–π interactions in green, and halogen bonds in blue. Blue halos indicate a measure of ligand solvent-exposure, with larger halos indicating greater exposure.
Analysis showed that the MBP chelates one of the active-site metal ions, with the carbonyl oxygen atom replacing the bridging water molecule and with the hydroxyl oxygen atom binding one metal center and forming a hydrogen bond with Lys134. The phenylaminomethyl moiety at the 6-position was found to be too short to allow for π-stacking with Tyr24 but allowed for hydrophobic interactions with pocket 2. The 4-chloro substituent is also positioned to allow for halogen bonding with the backbone carbonyl of Arg82. The N-phenyltetrazole moiety at the 1-position fits well into pocket 4, with the aryl ring making hydrophobic interactions with the hydrophobic wall of the pocket and the tetrazole making favorable contacts with the basic residues found at the end of the pocket (Figure 3). It was found that the 4′ tetrazole was able to hydrogen-bond with two nitrogen atoms from the guanidine moiety of Arg124 and simultaneously hydrogen-bond with the terminal nitrogen of Lys34. The larger size and greater number of available hydrogen bond acceptors in the tetrazole ring, when compared to a carboxylic acid group, may account for the observed difference in activity between the compounds containing these isosteres (62 vs 63).
Cytotoxicity and Antiviral Activity
Cytotoxicity and antiviral activity studies were performed in MDCK cells (Supporting Information). MDCK cells were incubated for a period of 48 h in the presence of varying concentrations of compound 71 or the commercially available diketo acid compound 89 (L-742,001; Supporting Information), as a control. Compound 89 is one of the most potent inhibitors of influenza PA endonuclease reported, in both cellular and protein based assays.19,35 Cell viability was then determined using CellTiter-Glo luminescent assay (Promega). Antiviral activity was determined by exposing MDCK cells to a lethal challenge of influenza virus, in the presence of varying concentrations of inhibitor. Influenza A virus obtained from the American Type Culture Collection (H1N1, ATCC VR-1737) was used to infect healthy MDCK cells, and inhibitor molecules were coadministered in varying concentrations with the viral challenge. The cells were then incubated for 48 h and analyzed for viability via luminescence assay. Compound 71 was found to have an EC50 value of 2.1 μM and a CC50 value of 280 μM (Figure 4). Both of these values were comparable to the positive control (89), with 71 being 4- to 5-fold more potent than the control in the viral challenge assay.
Figure 4.

Potency and cytotoxicity analysis of 71 and a reported diketo acid inhibitor 89 in MDCK cell lines. Cytotoxicity was determined by incubating cells in the presence of inhibitors for 48 h, followed by evaluating cell viability. Potency was determined by coadministration of inhibitor and a lethal challenge of virus particles, followed by a 48 h incubation and subsequent analysis of cell viability.
Discussion and Conclusions
Of the reported binding pockets in the influenza PA endonuclease active site, many inhibitors that have been structurally characterized in the active site interact with pocket 4. This common interaction space is due to the open nature of pocket 4 (Figure 1, Figure 3), coupled with its proximity to the catalytic metal ions that many inhibitors also interact with.18,25,28 A number of reported inhibitors interact with pocket 3 for similar reasons.26,36 Fewer reported inhibitors have been shown to interact with pocket 1 or pocket 2, with the only examples being dioxobutanoic acid derivatives.18 Examples of inhibitors with good interactions with pockets 3 and 4 include compounds containing a phenyltetrazole moiety for pocket 4 (Figure 5).26,28 Arnold and LaVoie initially identified a 5-chloro-3,2-hydroxypyridinone as a fragment hit against endonuclease using a crystallography-based fragment screening of 775 compounds.27 Fragment elaboration resulted in the development of Parhi-6 (Figure 5), which contains the aforementioned phenyltetrazole moiety and displays a reported IC50 value of 368 ± 41 nM. In an effort to develop even more potent inhibitors of endonuclease, additional efforts lead to additional MBP heterocycle analogues in combination with a phenyltetrazole substituent (Figure 5, Figure 6).
Figure 5.

Chemical structure and inhibitory activity of several reported influenza endonuclease inhibitors that contain a phenyltetrazole moiety. Reported IC50 values for each compound are provided. The MBP portion of each molecule is highlighted in blue. Compounds were initially reported by Parhi et al.27 and Sagong et al.26
Figure 6.

Structural comparison of several phenyltetrazole-containing influenza endonuclease inhibitors. Left: Crystal structure of Sagong-11 (PDB code 4W9S). Middle: Crystal structure of a derivative of Parhi-6 (PDB code 4M4Q). Right: Docking analysis of 71 (PDB code 4M4Q). Crystal structures of phenyltetrazole-containing inhibitors show the binding orientation of the phenyltetrazole moiety to be essentially conserved. Docking analysis of 71 predicts a very similar binding mode for this moiety to that observed in crystallographically validated inhibitors.
The MPB scaffolds and elaborated inhibitors shown in Figure 5 were the result of a “scaffold hopping” campaign, which is common in inhibitor development.37 Successful scaffold hopping requires the identification of more potent, yet functionalizable, core molecules. As such, the FBDD approach reported here was able to further advance this strategy, using new MBP fragments, resulting in compound 63 that exhibits improved activity over all of these other derivatives shown in Figure 5. Therefore, the MBP-based FBDD approach facilitates the identification of potent core molecules and can allow for more effective and rapid scaffold hopping, which also serves to further validate the pharmacophore model and SAR observed in these prior studies.26,28 The MBP library used here helps complement the previous screening efforts by exploring a broader chemical space of metal-binding functionality, rapidly identifying novel MBPs, and providing significant improvements in activity. Furthermore, by examination of other fragment sublibraries, a “fragment growth” strategy was applied here, as demonstrated by compound 71, which led to even greater enhancements in activity by exploiting less-targeted active site pockets (pocket 2).
By employment of a FBDD strategy and use of a metalloenzyme-targeted MBP library, pyromeconic acid (1) and similar fragments were identified as active inhibitors against the influenza PA endonuclease. Prior work in the field, along with modeling, allowed for the generation of a SAR, leading to the synthesis of several pyrone and pyridinone derivatives based on the initial fragment hits. Ultimately, five small molecules with in vitro IC50 values of <200 nM, and two with IC50 values of <50 nM, were obtained. The most active compound (71) was found to inhibit endonuclease activity with an IC50 of 14 nM. Compound 71 showed minimal cytotoxicity with a CC50 of 280 μM and could rescue cells from a lethal challenge of H1N1 influenza virus with a viral EC50 of 2.1 μM. As opposed to other reported screening methods, which generally involve much larger libraries and more costly methods (e.g., crystallography), the FBDD approach described here readily produced ligand efficient fragments that targeted the metal active site. This targeted library screening method shows the potential to be a more time- and cost-effective route than those employed by traditional FBDD and HTS campaigns, yielding a similar number of active hits and ultimately resulting in the development of effective lead molecules.
Experimental Section
General Experimental Details
All reagents and solvents were obtained from commercial sources and used without further purification. All reactions, unless otherwise stated, were performed under a nitrogen atmosphere. Reactions were monitored using either aluminum or glass-backed silica TLC plates impregnated with a fluorescent indicator, absorbing at 254 nm. Silica gel column chromatography was performed on a CombiFlash Rf Teledyne ISCO system using hexane, ethyl acetate, methylene chloride, or methanol as eluent. Reverse phase column chromatography (C18 column) was performed on the same instrument using 0.1% formic acid in methanol, acetonitrile, or water as eluent. Separations were monitored by mass spectrometry via a Teledyne ISCO RF+ PurIon ESI-MS or APCI-MS detector with 1 Da resolution. The purity of all compounds used in assays was determined to be ≥95% by 1H NMR spectroscopy and confirmed by high-resolution mass spectrometry (HRMS) experiments using an Agilent 6230 Accurate-Mass LC-TOFMS located in the University of California, San Diego, Molecular Mass Spectrometry Facility (MMSF). Standard resolution MS was performed at either the aforementioned MMSF or Teledyne ISCO RF+ PurIon MS. Microwave reactions were performed using a CEM Discover series S-class microwave reactor in pressure-sealed vessels. Docking simulations were performed using MOE, version 2014.0901. Flexible receptor modeling (induced fit) was employed in these simulations, and metal-binding atoms were fixed.
Synthesis of 5-hydroxy-2-methyl-4-oxo-4H-pyran-3-carboxamide derivatives is outlined in Scheme 1. Bromopyruvic acid was transformed to bromodiethoxypropanoic acid by treatment with triethyl orthoformate in the presence of catalytic sulfuric acid. The acetal-protected propanoic acid was then activated as the p-nitrophenyl ester 74. Pyrone ring formation was achieved over two steps by the slow addition of 74 to a solution of ethyl or tert-butyl acetoacetate that was previously deprotonated by sodium hydride. After the initial nucleophilic attack of the activated ester by the acetoacetate, ring closing was accomplished via nucleophilic addition by heating the reaction mixture to reflux for 4–6 h. The pyrone-acetal 75a was deesterified by stirring briefly with trifluoroacetic acid (TFA) in CH2Cl2. The tert-butyl ester was removed selectively by controlling the time of the reaction, as the ester is more labile in the absence of water than the acetal. Key intermediate 76 was used to prepare various amides using analogous conditions; specifically, 76 was activated with HATU and triethylamine in DMF. Addition of the amine was followed by heating and stirring for ∼18 h at 60 °C. After isolation of the formed amide the acetal was deprotected in water and acid to reveal the 3-keto intermediate, which rapidly tautomerizes to form the desired 3-hydroxide species.
Scheme 1.

Reagents and conditions: (a) triethyl orthoformate, H2SO4 (catalytic), rt, 24 h; (b) 4-nitrophenyl trifluoroacetate, pyridine, rt, 18 h; (c) ethyl or tert-butyl acetoacetate, NaH, reflux in dry THF, 4–6 h; (d) TFA, CH2Cl2, rt, 2–4 h; (e) HATU, triethylamine, DMF, 60 °C, o/n; (f) 1:1 HCOOH/H2O, 80 °C, 2–6 h.
To acquire SAR at the 6-position, a library of amine derivatives was prepared from commercially available kojic acid (Scheme 2). The phenolic oxygen of 8 was selectively protected as a benzyl ether by treatment with benzyl bromide in the presence of potassium carbonate in DMF at 80 °C. Compound 78 was prepared quantitatively by reacting 77 with thionyl chloride. Nucleophilic addition of various primary and secondary amines followed by selective hydrolysis of the benzyl ether in a mixture of TFA, concentrated HCl, and glacial acetic acid afforded aminomethylpyrones in good yields. Amide and sulfonamide derivatives were generated by the nucleophilic addition of sodium azide to 78, followed by reduction of 79 with triphenylphosphine to afford 80 as a key intermediate. Compound 80 was reacted with various acid chlorides and sulfonyl chlorides to generate 28–31, after deprotection of the phenol by boron trichloride.
Scheme 2.

Reagents and conditions: (a) benzyl bromide, K2CO3, DMF, 80 °C, 8–12 h; (b) thionyl chloride, CH2Cl2, rt, 8 h; (c) R1R2NH, triethylamine, DMF, 75 °C, o/n; (d) 5:5:1 HOAc/HCl/TFA, rt to 40 °C, 24–48 h; (e) sodium azide, DMF, rt, o/n; (f) triphenylphosphine, THF, rt, 30–60 min; (g) acid chloride or sulfonyl chloride, CH2Cl2, rt, o/n; (h) BCl3, CH2Cl2, 0 °C, 30 min, rt, 30 min.
Preparation of pyridinone derivatives of allomaltol (2) possessing N-aryl and -alkyl substituents is described generally in Scheme 3. Kojic chloride (81) was derived from 8 and reduced to 2 using metallic zinc and strong acid. While dehydration of the hydroxypyrone ring to afford hydroxypyridinone derivatives was shown to be possible, benzyl protection of the hydroxyl group prior to dehydration greatly improved yields and suppressed formation of side products. Key intermediate 82 was irradiated in a microwave reactor in the presence of excess amine and acetic acid to produce a wide variety of pyridinone derivatives. Microwave heating under increased pressure greatly accelerated reaction rates; dehydration using conventional heating could be accomplished but required refluxing reactants in ethanol for a period of days. Removal of the benzyl ether after dehydration was shown to be accomplished quickly and efficiently employing boron trichloride as a dealkylating agent or less efficiently using a 5:5:1 mixture of concentrated hydrochloric acid, glacial acetic acid, and TFA, as a milder alternative to boron trihalides (Supporting Information).
Scheme 3.

Reagents and conditions: (a) thionyl chloride, CH2Cl2, rt, 4–6 h; (b) zinc dust, HCl, water, 70 °C, 4–6 h; (c) benzyl bromide, K2CO3, DMF, 80 °C, 8–12 h; (d) aryl- or alkylamine, HOAc, 3:1 EtOH/H2O, microwave 125 °C, 90–120 min; (e) BCl3, CH2Cl2, 0 °C, 30 min, rt, 30 min; (f) 5:5:1 HOAc/HCl/TFA, rt to 40 °C, 24–48 h.
Fragment merging was accomplished as shown in Scheme 4. Merging of N-arylpyridinones with 5-position carboxylates began with advanced intermediate 76a. Hydrolysis of the acetal followed by tautomerization yielded 83. After benzyl protection of the phenol, dehydration was accomplished with microwave heating using dry ethanol as a solvent. Hydrolysis of only the benzyl ether using strong acid afforded compounds 67–69 in moderate yields; hydrolysis of both ester and ether was not observed. Further base catalyzed hydrolysis of the ethyl ester proved difficult and ultimately resulted in the decomposition of the ring system. As an alternative route, compound 86 was hydrolyzed to the free carboxylic acid by stirring in 4% KOH and methanol for several hours. This reaction proceeded almost quantitatively with no apparent decomposition of the starting material. Compound 86 was found to be stable to strongly acidic conditions and was hydrolyzed to 70 using a mixture of strong acids with good yields. Synthesis of compound 71 began with chloride compound 78. Nucleophilic substitution with 4-chloro-N-methylaniline afforded pyrone 87. Dehydration, as previously described, yielded 88 in moderate to low yields. Hydrolysis in the presence of strong acid afforded 71.
Scheme 4.

Reagents and conditions: (a) 1:1 HCOOH/H2O, 80 °C, 2–6 h; (b) benzyl bromide, K2CO3, DMF, 80 °C, 8–12 h; (c) arylamine, HOAc, 3:1 EtOH/H2O, microwave 130 °C, 3–5 h; (d) 5:5:1 HOAc/HCl/TFA, rt to 40 °C, 24–48 h; (e) 4% KOH, 1:2:1 THF/MeOH/water, rt, o/n; (f) RCH3NH, triethylamine, DMF, 75 °C, o/n.
Acknowledgments
The authors acknowledge Dr. Yongxuan Su (University of California, San Diego, Molecular Mass Spectrometry Facility) for aid with HR-MS and HPLC fragment purity analysis. The authors thank Eric Chen (University of California, San Diego) for providing the recombinant expression plasmid for PA endonuclease, suggestions for protein purification, and assay protocols. The authors thank Dr. David T. Puerta (University of California, San Diego) for helpful discussions. This work was supported by grants from the U.S. National Institutes of Health (Grant R01 GM098435) and the California HIV/AIDS Research Program (Grant ID12-SD-231). S.M.C. has an equity interest in Cleave Biosciences and Forge Therapeutics, a companies that may potentially benefit from the research results, and also serves on the Scientific Advisory Board for these companies. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Glossary
Abbreviations Used
- ATCC
American Type Culture Collection
- FBDD
fragment-based drug discovery
- FRET
Forster resonance energy transfer
- HTS
high-throughput screening
- LE
ligand efficiency
- MBP
metal-binding pharmacophore
- MDCK cells
Madin–Darby canine kidney epithelial cells
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00628.
The authors declare no competing financial interest.
Supplementary Material
References
- Dunning J.; Baillie J. K.; Cao B.; Hayden F. G. International Severe Acute, R.; Emerging Infection, C., Antiviral combinations for severe influenza. Lancet Infect. Dis. 2014, 14 (12), 1259–1270. 10.1016/S1473-3099(14)70821-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills C. E.; Robins J. M.; Lipsitch M. Transmissibility of 1918 pandemic influenza. Nature 2004, 432 (7019), 904–906. 10.1038/nature03063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjusted vaccine effectiveness estimates for influenza seasons from 2005 to 2015. http://www.cdc.gov/flu/professionals/vaccination/effectiveness-studies.htm (accessed February 26, 2016).
- Monod A.; Swale C.; Tarus B.; Tissot A.; Delmas B.; Ruigrok R. W.; Crepin T.; Slama-Schwok A. Learning from structure-based drug design and new antivirals targeting the ribonucleoprotein complex for the treatment of influenza. Expert Opin. Drug Discovery 2015, 10 (4), 345–371. 10.1517/17460441.2015.1019859. [DOI] [PubMed] [Google Scholar]
- Stiver G. The treatment of influenza with antiviral drugs. Can. Med. Assoc. J. 2003, 168 (1), 49–57. [PMC free article] [PubMed] [Google Scholar]
- Administration, U. S. F. a. D. Tamiflu pediatric adverse events: questions and answers. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm107840.htm (accessed February 26, 2016).
- Furuse Y.; Suzuki A.; Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrob. Agents Chemother. 2009, 53 (10), 4457–4463. 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore A. E.; Fry A.; Shay D.; Gubareva L.; Bresee J. S.; Uyeki T. M. Antiviral agents for the treatment and chemoprophylaxis of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP). Morbidity Mortality Wkly. Rep. 2011, 60 (1), 1–24. [PubMed] [Google Scholar]
- Huang T. S.; Palese P.; Krystal M. Determination of influenza virus proteins required for genome replication. J. Virol. 1990, 64 (11), 5669–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J. W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (9), 4171–4175. 10.1073/pnas.90.9.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor E. The RNA polymerase of influenza a virus: mechanisms of viral transcription and replication. Acta. Virol. 2013, 57 (2), 113–122. 10.4149/av_2013_02_113. [DOI] [PubMed] [Google Scholar]
- Plotch S. J.; Bouloy M.; Ulmanen I.; Krug R. M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23 (3), 847–858. 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- Dias A.; Bouvier D.; Crepin T.; McCarthy A. A.; Hart D. J.; Baudin F.; Cusack S.; Ruigrok R. W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458 (7240), 914–918. 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- Beaton A. R.; Krug R. M. Selected host cell capped RNA fragments prime influenza viral RNA transcription in vivo. Nucleic Acids Res. 1981, 9 (17), 4423–4436. 10.1093/nar/9.17.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.; Wartchow C.; Shia S.; Uehara K.; Steffek M.; Warne R.; Sutton J.; Muiru G. T.; Leonard V. H.; Bussiere D. E.; Ma X. Molecular basis of mRNA cap recognition by Influenza B polymerase PB2 subunit. J. Biol. Chem. 2016, 291, 363–370. 10.1074/jbc.M115.693051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama D.; Shoji M.; Yamayoshi S.; Hirota T.; Nagae M.; Yanagisawa S.; Nakano M.; Ohmi N.; Noda T.; Kawaoka Y.; Kuzuhara T. A novel functional site in the PB2 subunit of influenza A virus essential for acetyl-CoA interaction, RNA polymerase activity, and viral replication. J. Biol. Chem. 2014, 289 (36), 24980–24994. 10.1074/jbc.M114.559708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok V.; Cianci C.; Tibbles K. W.; Inglis S. C.; Krystal M.; Digard P. Inhibition of the influenza virus RNA-dependent RNA polymerase by antisera directed against the carboxy-terminal region of the PB2 subunit. J. Gen. Virol. 1996, 77 (Part 5), 1025–1033. 10.1099/0022-1317-77-5-1025. [DOI] [PubMed] [Google Scholar]
- DuBois R. M.; Slavish P. J.; Baughman B. M.; Yun M. K.; Bao J.; Webby R. J.; Webb T. R.; White S. W. Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog. 2012, 8 (8), e1002830. 10.1371/journal.ppat.1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa M.; Kadowaki S. E.; Watanabe I.; Kadowaki Y.; Takei M.; Fukuda H. PA subunit of RNA polymerase as a promising target for anti-influenza virus agents. Antiviral Res. 2008, 78 (3), 194–201. 10.1016/j.antiviral.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Tomassini J.; Selnick H.; Davies M. E.; Armstrong M. E.; Baldwin J.; Bourgeois M.; Hastings J.; Hazuda D.; Lewis J.; McClements W.; et al. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 1994, 38 (12), 2827–2837. 10.1128/AAC.38.12.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J. C.; Selnick H.; Wolanski B.; Tomassini J. E. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob. Agents Chemother. 1996, 40 (5), 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci C.; Chung T. D. Y.; Meanwell N.; Putz H.; Hagen M.; Colonno R. J.; Krystal M. Identification of N-hydroxamic acid and N-hydroxyimide compounds that inhibit the influenza virus polymerase. Antiviral Chem. Chemother. 1996, 7 (6), 353–360. 10.1177/095632029600700609. [DOI] [Google Scholar]
- Tomassini J. E.; Davies M. E.; Hastings J. C.; Lingham R.; Mojena M.; Raghoobar S. L.; Singh S. B.; Tkacz J. S.; Goetz M. A. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996, 40 (5), 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuhara T.; Iwai Y.; Takahashi H.; Hatakeyama D.; Echigo N. Green tea catechins inhibit the endonuclease activity of influenza A virus RNA polymerase. PLoS Curr. 2009, 1, RRN1052. 10.1371/currents.RRN1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E.; Zubieta C.; Wolkerstorfer A.; Szolar O. H.; Ruigrok R. W.; Cusack S. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS Pathog. 2012, 8 (8), e1002831. 10.1371/journal.ppat.1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong H. Y.; Bauman J. D.; Patel D.; Das K.; Arnold E.; LaVoie E. J. Phenyl substituted 4-hydroxypyridazin-3(2H)-ones and 5-hydroxypyrimidin-4(3H)-ones: inhibitors of influenza A endonuclease. J. Med. Chem. 2014, 57 (19), 8086–8098. 10.1021/jm500958x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhi A. K.; Xiang A.; Bauman J. D.; Patel D.; Vijayan R. S.; Das K.; Arnold E.; Lavoie E. J. Phenyl substituted 3-hydroxypyridin-2(1H)-ones: inhibitors of influenza A endonuclease. Bioorg. Med. Chem. 2013, 21 (21), 6435–6446. 10.1016/j.bmc.2013.08.053. [DOI] [PubMed] [Google Scholar]
- Bauman J. D.; Patel D.; Baker S. F.; Vijayan R. S.; Xiang A.; Parhi A. K.; Martinez-Sobrido L.; LaVoie E. J.; Das K.; Arnold E. Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem. Biol. 2013, 8 (11), 2501–2508. 10.1021/cb400400j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman B. M.; Jake Slavish P.; DuBois R. M.; Boyd V. A.; White S. W.; Webb T. R. Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS Chem. Biol. 2012, 7 (3), 526–534. 10.1021/cb200439z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen J. A.; Fullagar J. L.; Miller M. T.; Cohen S. M. Identifying chelators for metalloprotein inhibitors using a fragment-based approach. J. Med. Chem. 2011, 54 (2), 591–602. 10.1021/jm101266s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9 (10), 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Sanna D.; Buglyo P.; Biro L.; Micera G.; Garribba E. Coordinating properties of pyrone and pyridinone derivatives, tropolone and catechol toward the VO2+ ion: an experimental and computational approach. Eur. J. Inorg. Chem. 2012, 2012 (7), 1079–1092. 10.1002/ejic.201101249. [DOI] [Google Scholar]
- Raha K.; Merz K. M. Jr. A quantum mechanics-based scoring function: study of zinc ion-mediated ligand binding. J. Am. Chem. Soc. 2004, 126 (4), 1020–1021. 10.1021/ja038496i. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang W.; Piquemal J. P.; Ren P. Modeling structural coordination and ligand binding in zinc proteins with a polarizable potential. J. Chem. Theory Comput. 2012, 8 (4), 1314–1324. 10.1021/ct200812y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaert A.; Dallocchio R.; Dessi A.; Pala N.; Rogolino D.; Sechi M.; Naesens L. Mutational analysis of the binding pockets of the diketo acid inhibitor L-742,001 in the influenza virus PA endonuclease. J. Virol. 2013, 87 (19), 10524–10538. 10.1128/JVI.00832-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong H. Y.; Parhi A.; Bauman J. D.; Patel D.; Vijayan R. S.; Das K.; Arnold E.; LaVoie E. J. 3-Hydroxyquinolin-2(1H)-ones as inhibitors of influenza A endonuclease. ACS Med. Chem. Lett. 2013, 4 (6), 547–550. 10.1021/ml4001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm H. J.; Flohr A.; Stahl M. Scaffold hopping. Drug Discovery Today: Technol. 2004, 1 (3), 217–224. 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.