

Abstract
Tumor vessels are characterized by abnormal morphology and hyper-permeability that together cause inefficient delivery of chemotherapeutic agents. Although VEGF has been established as a critical regulator of tumor angiogenesis, the role of mechanical signaling in the regulation of tumor vasculature or tumor endothelial cell (TEC) function is not known. Here, we show that the mechanosensitive ion channel TRPV4 regulates tumor angiogenesis and tumor vessel maturation via modulation of TEC mechanosensitivity. We found that TEC exhibit reduced TRPV4 expression and function, which is correlated with aberrant mechanosensitivity towards ECM stiffness, increased migration and abnormal angiogenesis by TEC. Further, syngeneic tumor experiments revealed that the absence of TRPV4 induced increased vascular density, vessel diameter and reduced pericyte coverage resulting in enhanced tumor growth in TRPV4 KO mice. Importantly, overexpression or pharmacological activation of TRPV4 restored aberrant TEC mechanosensitivity, migration and normalized abnormal angiogenesis in vitro by modulating Rho activity. Finally, a small molecule activator of TRPV4, GSK1016790A, in combination with anti-cancer drug Cisplatin, significantly reduced tumor growth in WT mice by inducing vessel maturation. Our findings demonstrate TRPV4 channels to be critical regulators of tumor angiogenesis and represent a novel target for anti-angiogenic and vascular normalization therapies.
Keywords: Angiogenesis, Calcium, Endothelial, Extracellular Matrix, Mechanotransduction, Tumor, TRPV4, Vascular normalization
Introduction
Angiogenesis, the formation of new blood vessels from pre-existing capillaries, is critical for solid tumor maintenance, growth and progression as it ensures proper oxygen and nutrient delivery to the tumor. However, the tumor vasculature is structurally and functionally abnormal, as characterized by its high tortuosity, non-uniform pericyte coverage and hyper-permeability1, 2. These abnormal vessels cause irregular blood flow and distribution, impaired oxygen delivery and impede immune cell function, which together lead to inefficient delivery of anti-cancer agents causing tumor cell resistance to radiation and chemotherapies3-5. Conventional anti-angiogenic therapies focus on either neutralizing the effect of soluble angiogenic factors such as vascular endothelial growth factor (VEGF) using specific antibodies or inhibiting VEGFR kinase activity6-8. These conventional anti-angiogenic strategies showed only modest success in clinical trials due to the development of resistance (evasive or intrinsic) as tumor endothelial cells (TEC) became refractory to anti-VEGF therapy over time3-5. These findings led to the emergence of a new concept called “vascular normalization”, i.e. transient inhibition of tumor angiogenesis leading to normalization of tumor vasculature which improves the efficacy of chemo- and radio therapies 1, 2. Although vascular normalization combined with chemotherapy have shown transient benefits, clinically they failed to exhibit long term beneficial effects owing to these strategies still being focused onVEGF-targeted therapies, despite their shortcomings 2, 9, 10. Infact, recent positron emission tomograpy (PET) imaging results have demonstrated a rapid decrease in the delivery of chemotherapeutic drugs to tumors post anti-VEGF therapy, in non-small cell lung cancer (NSCLC) patients 11.Therefore, an urgent need arises for the development of novel vascular normalization strategies.
In addition to soluble stimuli, such as VEGF and PDGF (platelet derived growth factor) local mechanical cues conveyed by the extracellular matrix (ECM), due to cyclic deformation of blood vessels and hemodynamic forces, are also potent inducers of directional capillary growth and vascular remodeling in vitro and in vivo 12-16. Endothelial cells (ECs) sense mechanical forces associated with tissue distortion through integrin receptors that mediate their adhesion to the surrounding ECM12, 14, 15, 17. Unlike normal ECM, the tumor ECM becomes stiffer as a result of continuous remodeling of matrix components by tumor cells and stromal fibroblasts. Further, the tumor vasculature is hyper-permeable due to irregular basement membrane and poor pericyte coverage and therefore releases plasma components into the surrounding extracellular space, eventually leading to increased ECM stiffness. Cells sense changes in ECM stiffness through integrin receptors which transduce these mechanical signals into the cell through the actin cytoskeleton. As shown previously in tumor epithelial cells18, 19 and TEC20, this increase in ECM stiffness also feeds back to enhance integrin-mediated Rho/ROCK (Rho-associated kinase) activity and contraction that can lead to aberrant mechanosensitivity of TEC.
One of the earliest responses of cells to mechanical force is the influx of calcium through the activation of mechanosensitive ion channels21-23. Although activation of these channels is thought to be independent of integrin mechanosensing, several lines of evidence indicate that both integrins and mechanosensitive ion channels are well connected22, 24-28. We have previously shown that the application of cyclic strain to normal endothelial cells (NEC) induces cell reorientation through activation of mechanosensitive Transient Receptor Potential Vanilloid 4 (TRPV4) ion channel-dependent calcium influx, which in turn activates additional integrins and causes down-stream cytoskeletal reorganization16. Importantly, siRNA knockdown of TRPV4 channels inhibited mechanical strain-induced reorientation of NEC, thus confirming their key role in endothelial cell mechanosensitivity. Notably, endothelial cell mechanosensitivity strongly influences vascular formation and patterning20, and thus, tumor vessel malformations could arise from deregulation of TEC mechanosensing. Indeed, we have previously shown that TEC fail to reorient in response to cyclic strain, exhibit aberrant mechanosensitivity to ECM stiffness and undergo abnormal angiogenesis in vitro. Importantly, this abnormal TEC behavior resulted, at least in part, from abnormally high basal Rho activity20. However, the upstream signaling molecule or the molecular mechanism(s) governing high Rho-mediated aberrant mechanosensitivity and angiogenesis by TEC remain unknown. In the present study, we explored whether the mechanosensitive TRPV4 ion channel is a key determinant of TEC dysfunction leading to tumor angiogenesis and abnormal vasculature.
Results
Tumor endothelial cells express lower levels of TRPV4 and TRPV4-dependent calcium influx
We and others have previously shown that TRPV4 channels are functionally expressed in endothelial cells and act as mechanosensor of cyclic stretch and flow16, 29. We also demonstrated that TRPV4 channels induce mechanical force-dependent calcium influx in integrin specific manner in endothelial cells and this TRPV4-dependent mechanotransduction regulates cyclic strain–induced endothelial cell reorientation16. Interestingly, we found that tumor derived endothelial cells (TEC) failed to reorient in response to cyclic strain similar to that of TRPV4 knockdown endothelial cells suggesting TEC may mimic TRPV4 knockdown phenotype 20. Because mechanical force (cyclic strain)-induced reorientation in endothelial cells is dependent on TRPV4 channels16, we investigated whether these channels contribute to the abnormal mechanosensitivity of TEC20. TEC and normal EC (NEC) used in the present study have been isolated and characterized for EC markers, EC function and mechanosensing at different passages20, 30. We first compared TRPV4 expression levels between NEC and TEC using Western blot analysis. While NEC exhibited strong expression of TRPV4,16, 31 it was significantly lower in TEC (Fig.1A, B). To determine the functional implications of lower TRPV4 expression in TEC, we measured calcium influx in Fluo-4/AM loaded cells, in response to the addition of specific TRPV4 activators, GSK1016790A or 4-α-PDD. By using calcium imaging, patch-clamp, siRNA knockdown and TRPV4 null cells, we have previously demonstrated that these compounds specifically activates TRPV4-mediated calcium influx in endothelial cells16, 31. GSK1016790A (100 nM) induced calcium influx in both NEC and TEC but the influx in TEC was decreased by almost 40-50% compared to NEC (p≦0.001) (Fig.1C, D). Another TRPV4 agonist, 4-α-PDD (10 μM) also induced a rapid calcium influx in NEC, which was again significantly reduced by ∼40% (p≦ 0.01) in TEC (Supplementary Fig.S1 A, B). These results demonstrate that TRPV4 expression and function (calcium influx) are impaired in TEC.
Fig.1. TRPV4 channel expression and function in normal and tumor endothelial cells.
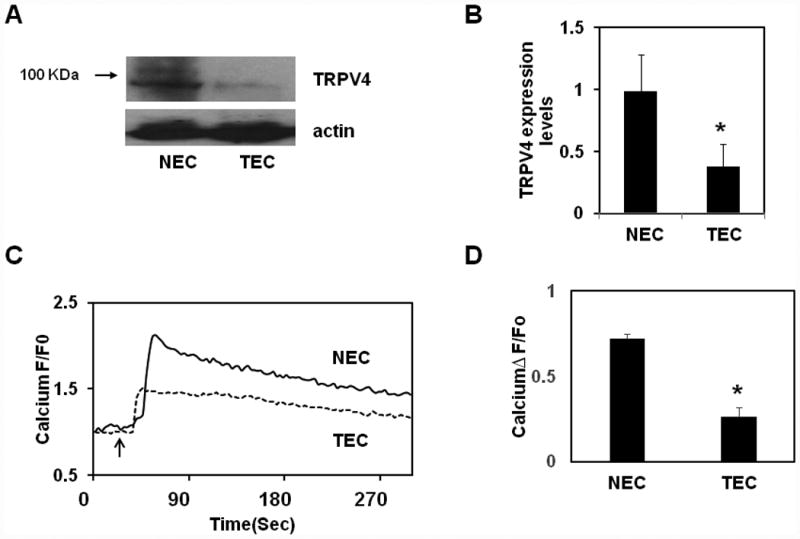
A) Western blot analysis of TRPV4 expression in normal (NEC) and tumor-derived endothelial cells (TEC). B) Quantitative analysis of the Western blots showing significant (p≦0.05) reduction in TRPV4 expression in TEC. C) Representative traces showing relative changes in cytosolic calcium in response to a selective TRPV4 agonist, GSK1016790A (100 nM) in Fluo-4 loaded normal and tumor endothelial cells (n=300). Arrow denotes the time when the cells were stimulated with the TRPV4 agonist D) Quantitative analysis of cytosolic calcium influx induced by GSK1016790A in NEC and TEC. (F/F0 = ratio of normalized Fluo-4 fluorescence intensity relative to time 0). The results shown are mean ± SEM from 3 independent experiments. The significance was set at p≦ 0.05.
Tumor Angiogenesis and tumor growth are enhanced in TRPV4 null mice
Next, to confirm if TRPV4 expression level contributes to tumor angiogenesis in vivo, we induced tumors in TRPV4 KO and WT mice (C57BL/6) by subcutaneously injecting mouse Lewis lung carcinoma cells (LLC). We found that tumor growth was 2-3 times greater in TRPV4 KO mice compared to WT mice at day 21 (Fig.2A). Importantly, immunohistochemical analysis revealed that tumors in TRPV4 KO mice exhibited a greater fraction of hyper-dilated (malformed) vessels with significantly larger vessel diameters (p≦0.05) compared to WT mice (Fig. 2, B, C). In contrast to tumor vessels in WT mice, those in TRPV4 KO mice exhibited poor pericyte coverage, as determined by very weak α-SMA staining, indicating the immature nature of these vessels. (Fig. 2D, E). Further, the tumors from TRPV4 KO mice exhibited increased microvessel density compared to tumors from WT mice (Supplementary Fig.S2 A, B). Next, we assessed the leakiness of TRPV4 KO vessels by performing permeability assays using TRITC-dextran (3000 MW) perfusion, via tail vein injections. Immunohistochemical analysis revealed increased TRITC staining within the tumor tissue, surrounding the vessels, in TRPV4 KO mice compared to WT tumors suggesting that TRPV4KO vessels are indeed hyperpermeable (Suppl Fig.S3). Taken together, these results clearly suggest that TRPV4 plays a critical role in modulating tumor angiogenesis and the absence of TRPV4 can lead to abnormal tumor angiogenesis (immature (leaky) vessels), possibly through altered mechanotransduction exhibited by endothelial cells.
Fig.2. Vessel malformations and tumor growth are enhanced in TRPV4 knockout mice.
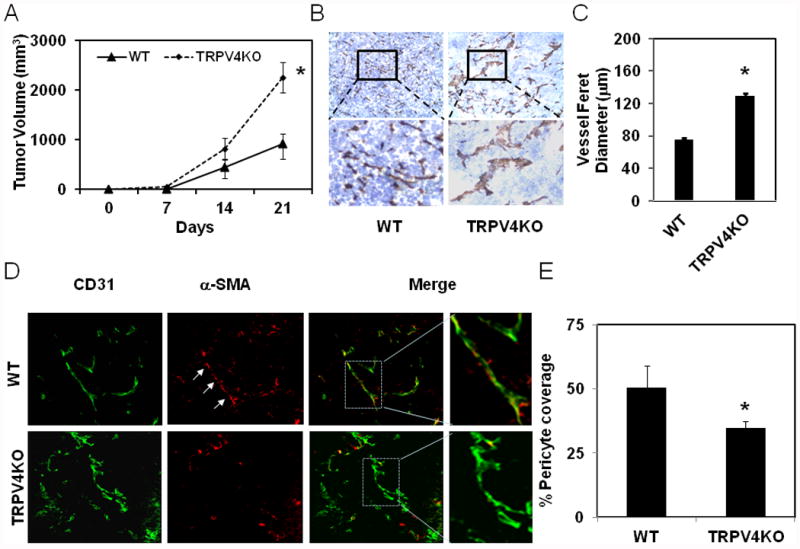
A) Time-dependent growth of the tumors in WT and TRPV4KO mice. Mouse Lewis lung carcinoma (LLC) cells (2 × 106) were subcutaneously injected in to wild type C57BL/6 mice (WT) or TRPV4 knockout mice in C57BL/6 background (TRPV4KO) and tumor growth was measured using calipers at indicated days. The data shown are ± SEM of three independent experiments (n=8-10 mice for each group). B) Immunohistochemical analysis showing increased vessel diameter (feret) in tumors (21 days) from TRPV4 knockout mice (TRPV4KO) compared to wild type mice (WT). C) Quantitative analysis of microvessel diameter in tumors from WT and TRPV4KO mice. D) Frozen sections of tumors (10 μm thickness) were stained with CD31 (green) and α-SMA (red) to measure pericyte coverage (matured vessels). E) Quantitative analysis of pericyte covered microvessels in tumors from WT and TRPV4KO mice. The results shown are mean ± SEM from 3 independent experiments. The significance was set at p≦ 0.05.
TRPV4 overexpression restores mechanosensitivity and reduces abnormal cell migration in tumor endothelial cells
The above findings suggest that lower levels of TRPV4, a known mechanosensor in endothelial cells 16, 22, 32, 33 may contribute to the previously reported aberrant mechanosensitivity of TEC in response to ECM stiffness and cyclic strain 20. Since ECM stiffness is known to increase in tumors 19, which can also influence TEC spreading, migration and tube formation, we investigated if overexpression of TRPV4 might rescue the abnormal mechanosensitivity exhibited by TEC. To achieve this, we expressed a human TRPV4-EGFP construct in TEC; EGFP fluorescence revealed that more than 80% cells were transfected with TRPV4-EGFP (Supplementary Fig. S4A). We found that overexpression of TRPV4 increased GSK1016790A-induced calcium influx in TEC by almost 4-fold (p≦ 0.01) compared to EGFP-alone expressing cells (Supplementary Fig.S4 B, C). The other TRPV4 specific activator, 4-α-PDD, also increased calcium influx in these TRPV4-transfected TEC cells compared to EGFP-expressing controls (not shown).
To explore if TRPV4 overexpression influences TEC mechanosensitivity towards ECM stiffness, we next cultured TRPV4-overexpressing TEC on transglutaminase linked gelatin gels of varying stiffness (370 and 2280 Pa; representing intermediate and high stiffness, respectively) which mimic the stiffness of tumor ECM 19 for 6 h and compared their degree of spreading20 with that of control EGFP-expressing cells (Fig. 3A, B). In past studies, we found that both NEC and TEC spread similarly on low stiffness (98 Pa) gels20. However, in that study, NEC exhibited increased spreading on intermediate stiffness (370 Pa) which reached plateau at high stiffness (2280 Pa) while TEC continue to increase their spreading with increased stiffness. Therefore, in the present study, we focused on intermediate (370 Pa) and high (2280 Pa) stiffness gels. We found that NEC (expressing only EGFP) spread to similar extent on both intermediate and high stiffness gels (370 and 2280 Pa) further confirming that the spreading of NEC reached plateau at 370 Pa (Fig.3 A, B), similar to our previous study20. On the other hand, spreading of TEC (expressing only EGFP) was significantly more on high stiffness gels compared to intermediate stiffness gels (Fig.3 A,B). In contrast, TEC-TRPV4-EGFP exhibited reduced spreading on high stiffness gels compared to TEC-EGFP alone, with no significant difference between intermediate and high stiffness substrates (Fig. 3A, B) suggesting that TRPV4 overexpression restored substrate mechanosensitivity in these cells. We did not find any change in NEC spreading when TRPV4-EGFP was overexpressed (data not shown). To further confirm that TRPV4 overexpression restores mechanosensitivity, we exposed TEC-EGFP and TEC-TRPV4-EGFP cells to cyclic strain, which is widely used to test endothelial cell mechanosensitivity, as previously described16. As shown earlier20, we found that TEC-EGFP failed to reorient while TEC overexpressing TRPV4-EGFP significantly oriented in response to cyclic strain (data not shown).
Fig.3. TRPV4 overexpression restores mechanosensitivity towards ECM stiffness, and reduces migration in TEC.
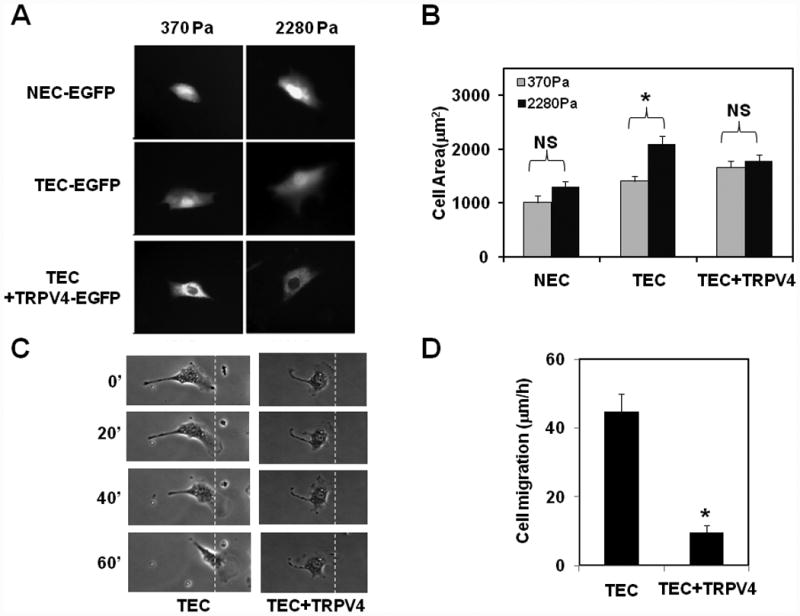
A) Representative images showing normal (NEC) and tumor (TEC) endothelial cell spreading (indicative of mechanosensitivity) on intermediate (370 Pa) and high (2280 Pa) stiffness ECM gels. Cells were transfected with either EGFP alone or TRPV4-EGFP B) Quantitative analysis of projected cell areas of NEC and TEC (expressing EGFP alone) and TEC+TRPV4 (expressing TRPV4-EGFP) on ECM gels of intermediate and high stiffness (370 and 2280 Pa). The results shown are mean ± SEM from 3 independent experiments. The significance was set at p≦ 0.05. NS= non-significant. C) Time lapse phase contrast micrographs showing migration of TEC-EGFP (TEC) and TEC+TRPV4-EGFP (TEC+TRPV4) cells plated on the surface of ECM gels of 370 Pa stiffness. Cells were allowed to spread for 4 h at 37°C and later shifted on to a microscope stage and random cell migration was recorded every 10 min using time lapse microscopy. Dashed line denotes the border of the leading edge at time 0. D) Quantification of cell migration as measured by marking the centroid of migrating cells overtime. Note: NEC migrated with a speed of 10 μm/h (Supplementary Fig.S5) suggesting that overexpression of TRPV4 normalized TEC migration. The results shown are mean ± SEM from 3 independent experiments. The significance was set at p≦ 0.05.
Since cell migration is dependent on mechanosensing of matrix stiffness and is a critical component of angiogenesis, we next explored if TRPV4 overexpression which normalized mechanosensitivity also influence TEC migration. To demonstrate this, we chose substrates of intermediary stiffness, as they supported optimal TEC cell spreading with or without TRPV4 overexpression. Consistent with their abnormal mechanosensitivity, TEC exhibited high cell migration speed (40 μm/h; (Fig.3 C, D), which was significantly suppressed (10 μm/h) when cells were transfected with TRPV4 (Fig.3 C, D). Further, we found that NEC migrated at a speed of 10 μm/h (Supplementary Fig.S5) suggesting that overexpression of TRPV4 normalized TEC cell migration to that of NEC. Taken together, all the above findings clearly demonstrate that TRPV4 expression restored mechanosensitivity in TEC and normalized migration.
TRPV4 overexpression or pharmacological activation normalizes abnormal tube formation in vitro via modulation of Rho activity
Since TRPV4 overexpression restored TEC mechanosensitivity to ECM stiffness which reduced abnormal cell migration, we explored whether TRPV4 expression normalizes vessel formation and if so, further determine the molecular mechanisms that underlie this process. First, TEC expressing TRPV4-EGFP or EGFP alone were tested for their ability to form capillary networks using a Matrigel® based in vitro 2D angiogenesis assays. We have previously shown that TEC form robust tubes when plated at low density (2 × 104 cells/ well) on Matrigel (2D) but at high density (8 × 104 cells/ well), these cells form tubes and then undergo multicellular retraction with disruption of tubular networks20. Importantly, TEC cultured within (rather than on top of) Matrigel formed tubular structures that were abnormally dilated and non-uniform, an abnormal morphology 20; (Fig. 4A) reminiscent of that observed within the cancer microvasculature in vivo, whereas NEC reorganized into tubular structures of relatively uniform size under similar culture conditions. These findings suggest that TEC are capable of tube formation in both 2D and 3D Matrigels. However, they collapse on 2D Matrigel due to high contraction mediated by cumulative high Rho activity 34, but form abnormal tubes in 3D. Therefore, we used both 2D and 3D Matrigel assays to determine if TRPV4 overexpression could normalize abnormal tube formation by TEC. To achieve this, first, we plated TEC expressing EGFP alone or overexpressing TRPV4 (expressing TRPV4-EGFP) at high density (8 × 104 cells/ well) on Matrigel for 2D tube formation. As expected, TEC expressing EGFP alone, formed tubes at high density (8 × 104 cells/ well) but these formed tubes underwent multicellular retraction and collapsed (Fig.4A). In contrast, TEC overexpressing TRPV4 formed a robust tubular network, with quantitative analysis revealing a significant increase (almost 10 fold) in tube length in TRPV4-expressing TEC compared to EGFP-expressing counterparts (Supplementary Fig.S6). Interestingly, we did not find any change in tube formation by NEC either expressing EGFP alone (Fig.4A) or overexpressing TRPV4-EGFP (not shown). We also found that TRPV4 overexpression normalized tube formation in 3D Matrigels (Fig4A).
Fig.4. TRPV4 overexpression normalizes abnormal angiogenesis by tumor EC through the inhibition of abnormal Rho activity.
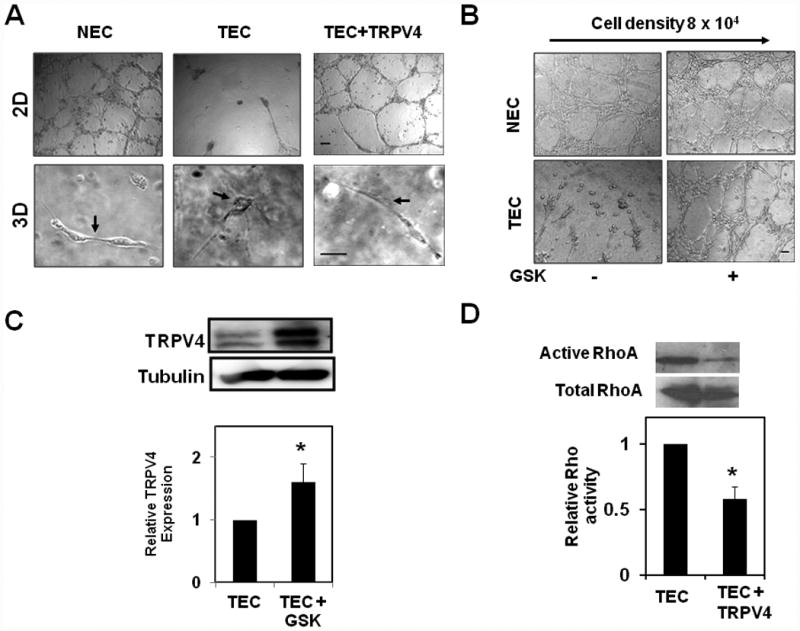
A) Phase contrast micrographs showing the normalizing effects of TRPV4 overexpression on TEC angiogenic behavior when plated on 2D Matrigels (at high densities; 8 × 104 well that cause collapse of tubular networks) and 3D Matrigels. NEC-EGFP (NEC), TEC-EGFP (TEC) and TEC-TRPV4-EGFP (TEC+TRPV4) cells were plated and cultured on the surface of Matrigel for 18 h (2D) or mixed in Matrigel and cultured for 14 days (3D). Note that the overexpression of TRPV4 in TEC cells restored tube formation on 2D Matrigel and normalized abnormal tubes in 3D Matrigel. Scale bar= 10 μm. B) Phase contrast micrographs showing the normalizing effects of pharmacological activation of TRPV4 with GSK1016790A (100 nM) on TEC angiogenic behavior when plated on 2D (at high densities; 8 × 104/ well that cause collapse of tubular networks). Scale bar= 10 μm. C) Representative Western blot showing TRPV4 expression in TEC untreated or treated with GSK1016790A (100 nM) for 24 h. Densitometry analysis of relative changes in TRPV4 expression measured by normalizing the levels of TRPV4 with that of tubulin. D) Representative Western blot showing the levels of active-Rho and total Rho for TEC and TEC+TRPV4 cells. Rho activity was analyzed in TEC (EGFP) and TEC+TRPV4 cells cultured under regular growth conditions using the Rhotekin-RBD binding assay. Densitometry analysis of relative changes in Rho activity. Rho activity levels were measured by normalizing the levels of active Rho with that of total Rho.
Next, we examined if pharmacological activation of TRPV4 normalizes abnormal tube formation in vitro. For this, we activated TRPV4 in TEC using the small molecule activator of TRPV4, GSK1016790A (GSK; 100 nM). We found that, similar to TRPV4 overexpression, TEC, but not NEC, treated with GSK formed robust tubes at high plating density (8 × 104 cells/ well) (Fig.4B). Since activity-dependent regulation of ion channels was shown in various cancers35, 36, we then asked if activation of TRPV4 with GSK modulates TRPV4 expression in TEC. We found that treatment of TEC with GSK for 24 h significantly increased TRPV4 protein expression (Fig. 4C). These results demonstrate that TRPV4 overexpression or pharmacological activation by GSK normalizes abnormal angiogenesis exhibited by TEC through the restoration of mechanosensitivity towards ECM stiffness and thus identifies TRPV4 as a critical mediator of angiogenesis.
To understand the molecular mechanism downstream of TRPV4 that mediates normalization of angiogenesis, we focused on Rho which regulates endothelial contraction that is required for partial rounding of endothelial cells during tube formation (34). We have previously demonstrated that TEC (expressing only EGFP) showed high basal Rho activity 20 which is the reason for abnormal collapse of TEC on 2D Matrigel, when plated at high densities. However, we found that overexpression of TRPV4-EGFP significantly reduced this high basal Rho activity exhibited by TEC (approximately ½-fold lower; p≦0.001) (Fig.4 D). We found that TRPV4 activator GSK1016790A (100 nM) also inhibited high basal Rho activity in TEC (Supplementary Fig.S7) suggesting that TRPV4 is a critical modulator of Rho activity in TEC.
TRPV4 specific small molecule activator GSK1016790A normalizes tumor vasculature in vivo and reduces tumor growth in combination with Cisplatin
Finally, we explored if pharmacological activation of TRPV4 induces tumor vascular normalization/maturation in vivo and improves efficacy of chemotherapeutic drug (Cisplatin). To achieve this, we injected TRPV4 activator GSK1016790A intraperitonially every day for 14 days in WT mice that had developed palpable tumors (around 100 mm3, after 7 days). Cisplatin was given once per week starting 2-4 days after GSK treatment and tumor growth was monitored every week until day 21. First, we examined vessel maturity in tumors by staining for pericyte coverage. We observed that the vessels in GSK and GSK-Cisiplatin-treated tumors, but not in control or Cisplatin treated ones, showed increased pericyte coverage (Fig.5A, B) suggesting that TRPV4 activation normalized tumor angiogenesis and induced vessel maturation which may help efficient delivery of Cisplatin. Consistent with this observation, we found that tumor growth was markedly reduced in GSK-Cisplatin treated animals (Fig.5C), but not in control or Cisplatin treated mice, suggesting the improved delivery of Cisplatin due to the normalization of vessels by TRPV4 activation.
Fig.5. TRPV4 activation with a small molecule activator together with Cisplatin reduces tumor growth in WT mice.
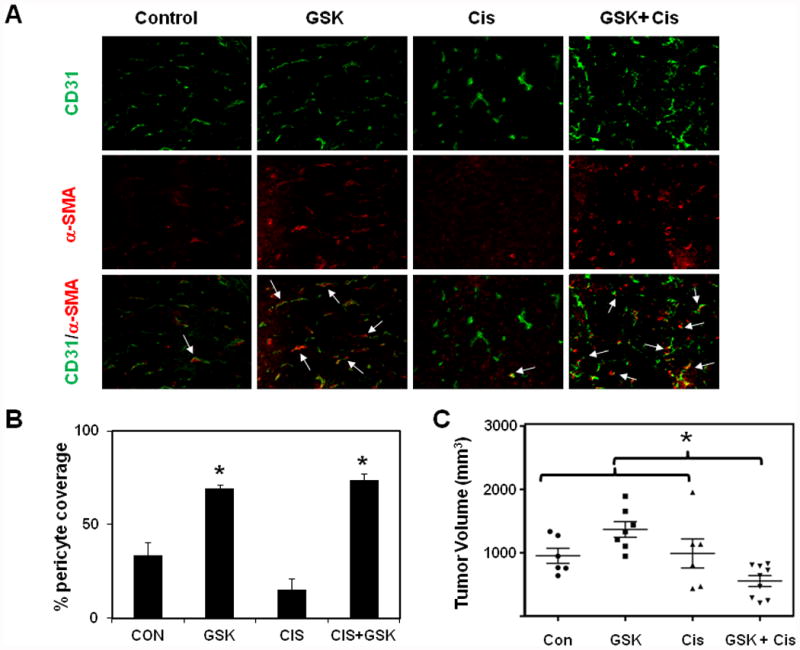
A) Syngeneic tumors (LLC) were injected in the back of WT (C57BL/6) mice and tumor growth was monitored for 21 days. TRPV4 activator, GSK1016790A (GSK) was injected i.p. everyday starting from day 7 (after palpable tumors were observed) until 21 days. Cisplatin was injected i.p. (once/week) 3 days after the injection of GSK1016790A. Frozen sections of tumors (10 μm thickness; from 21 day) were stained with CD31 (green) and α-SMA (red) to measure pericyte coverage (matured vessels). B) Quantitative analysis of pericyte covered microvessels in tumors from control, GSK, Cisplatin, and GSK + Cisplatin treated mice. The results shown are mean ± SEM from 3 independent experiments. The significance was set at p≦ 0.05. C) Tumor volumes among mice groups. Note that tumor growth was reduced in GSK + Cisplatin (*) treated mice. However, treatment with either of the drug alone did not inhibit tumor growth, indicating that GSK treatment improved Cisplatin delivery through the normalization of the abnormal tumor vasculature.
Discussion
Tumor angiogenesis has been widely shown to be regulated by soluble growth factors such as VEGF and fibroblast growth factor (FGF). However, regulation of tumor angiogenesis by mechanical forces is not well known. In the present study, we found that tumor endothelial cells express lower levels of TRPV4, a mechanosensitive ion channel, which is a key upstream signaling molecule that regulates tumor endothelial cell mechanosensitivity, tumor angiogenesis and tumor vessel maturation. Importantly, we demonstrate that overexpression of TRPV4 normalizes aberrant TEC mechanosensitivity, migration and angiogenesis through modulation of Rho activity. We further confirmed these findings in vivo by demonstrating that TRPV4 KO mice lacking this mechanosensing molecule exhibit severe tumor vessel malformations, characterized by increased vessel diameter, length and density, and enhanced tumor growth. Finally, we demonstrated that administration of the specific small molecule TRPV4 activator, GSK1016790A induced vessel maturation and, in combination with a chemotherapeutic drug, Cisplatin, reduced tumor growth in WT mice. To the best of our knowledge, this is the first report to demonstrate a role for TRPV4 in angiogenesis in vitro or in vivo.
TRPV4 channels are non-selective calcium ion channels ubiquitously expressed in endothelial cells that act as a mechanosensor of cyclic strain and shear stress in endothelial cells 16, 22, 29, 32, 33, 37. We have previously shown that endothelial cells express functional TRPV4 channels (Fluo-4 imaging, patch-clamp) and that these channels mediate mechanical signals from integrin to integrin that are critical for reorientation of NEC in response to cyclic strain 16. The fact that EC isolated from tumors fail to reorient in response to cyclic strain and exhibit aberrant mechanosensitivity towards substrate stiffness20, suggested that TRPV4 channel signaling might be altered in these cells. Indeed, we found that TEC express low levels of functionally active TRPV4 channels compared to NEC. TRPV4 protein usually exhibits two bands that are below and above of 100 kDa in Western blots16,29,53-56. Although the upper band has been demonstrated to be the glycosylated form of TRPV453 which influences its membrane translocation and activity, we did not find any significant difference in terms of the relative amount of each band in NEC or TEC, suggesting that TRPV4 expression is down-regulated. Consistently, TRPV4 expression has been shown to be modulated by microRNA -20357. However, it is not clear how tumor cells or tumor stroma may influence TRPV4 expression in TEC. Importantly, we demonstrate that TRPV4 overexpression or pharmacological activation by a small molecular activator, restored mechanosensitivity to matrix stiffness and cyclic strain, reduced migration and normalized the abnormal angiogenesis exhibited by TEC in vitro.
The aberrant mechanosensitivity of TEC is known to be mediated by high basal Rho activity20; however, the molecular mechanism up-stream of Rho responsible for this abnormal behavior is not yet known. Here, we show that overexpression of TRPV4 significantly inhibited high basal Rho activity in TEC. This reduction in basal Rho activity could possibly inhibit the high basal contraction of TEC 34 which then may be responsible for the restored mechanosensitivity of these cells towards substrate stiffness and normalization of angiogenesis. In fact, transient inhibition of Rho kinase with Y-27632 can normalize TEC responses to cyclic strain20. Although the exact molecular mechanism through which TRPV4 regulates Rho activity is not known, it is plausible that TRPV4 exerts its effects by modulating integrin activation, which has been shown to be sensitive to mechanical force-induced TRPV4-dependent calcium influx16. Importantly, binding of integrins to ECM transiently inhibit Rho activity38, 39 and facilitate cell shape changes, leading to cell spreading and reorientation. Interference with binding of additional integrins using function blocking antibodies also significantly inhibits TRPV4-dependent cyclic strain-induced CE reorientation16.
Although TRPV4 role in endothelial migration was shown 40, the role of TRPV4 in physiological or pathological angiogenesis is not demonstrated. Our results demonstrating TRPV4 overexpression normalized tube/vessel formation in 2D Matrigels, coupled with increased vessel malformations and enhanced tumor growth in TRPV4 KO mice, confirms that TRPV4 plays a critical role in tumor angiogenesis. Further, combination of GSK and Cisplatin, but not each them alone, inhibited tumor growth suggesting that TRPV4 activation-induced vascular maturation improved the efficacy of Cisplatin. These findings have significant value in vascular normalization therapies directed towards the treatment of cancer. Since the initial proposal that tumors rely on neovascularization for their survival and growth41, 42, angiogenesis has become a potential target for cancer therapy. In fact, multiple angiogenesis inhibitors have now been identified, and some clinically approved (e.g., Avastin), that inhibit the growth of a wide variety of experimental tumors in many animal models albeit with limited success in patients. Although VEGF is required for normal proliferation and migration of EC, it is important to emphasize that the tumor vasculature in certain cancers, such as in renal cell carcinoma, is largely refractory to therapies designed to alter VEGF signaling, as growth factors are redundant for angiogenesis 3-6, 43. Nevertheless, current vascular normalization therapies still focus on targeting soluble growth factors such as VEGF and PDGF 2, 10, 44-51. Notably, recently it was shown NSCLC patients treated with humanized VEGF antibody, bevacizumab reduced both perfusion and net influx rate of [(11)C] docetaxel within 5 h as measured by PET. Further, it was demonstrated that these effects persisted even after 4 days11. Importantly, these findings show no evidence for a substantial improvement in drug delivery to tumors by anti-VEGF treatment and highlight the importance of drug scheduling and supports further studies to optimize scheduling or the use of anti-angiogenic drugs.
Thus, our results showing that a mechanosensitive ion channel, TRPV4 regulates tumor angiogenesis through the modulation of Rho-dependent mechanosensitivity of TEC and, a small molecule activator of TRPV4, GSK1016790A together with Cisplatin, inhibits tumor growth, suggests TRPV4 as a target for developing a novel mechanotransduction-based therapy for vascular normalization. To our knowledge, TRPV4 channels have not previously been studied as potential therapeutic targets in angiogenesis, therefore we believe that elucidation of TRPV4-dependent mechanotransduction mechanisms and their role in angiogenesis may open entirely new avenues for developmental therapeutics for cancer as well as other angiogenic disorders, such as age-related macular degeneration and diabetic retinopathy.
Materials and Methods
Cell culture
Normal (NEC) and Tumor (TEC) endothelial cells were obtained from a transgenic adenocarcinoma mouse prostate (TRAMP) model, as previously described20, 30. Cells were plated on fibronectin (FN) or gelatin-coated tissue culture dishes and grown in a defined medium composed of low glucose DMEM, 10% fetal bovine serum, 10% Nu Serum IV, VEGF(1ng/ml) basic fibroblast growth factor (3 ng/ml), heparin salt (0.1 mg/ml), 1% insulin-transferrin-selenium and antibiotic/mycotic mix. Cells were cultured in a 37°C, 5%CO2 incubator, split at ∼90-95% confluence, and used between passages 11-22. These cells (NEC and TEC) were characterized for the presence of endothelial markers and function. We found that both of these cells expressed endothelial markers including CD31(PECAM-1), VE-cadherin, Von Wildebrand Factor (vWF), endothelial nitric oxide synthase (eNOS) and bind to isolectin-IB4 as measured by FACS, Western blotting, RT-PCR and immunocytochemistry. In contrast, these cells do not express mesenchymal markers α-SMA and PDGFR-β receptor20, 30. NEC and TEC formed robust tubular structures/sprouts in 2D and 3D Matrigel/Fibrin gel angiogenesis assays further confirming that these are functional EC 20, 30. We have shown these EC retained their endothelial phenotype and TRPV4 expression (not shown) even up to 22 passages20.
Transfection
Cells were transfected with TRPV4-EGFP (kind gift of Dr. Jendrach, Germany) or EGFP constructs using targefect (targetingsystems)31. The transfection efficiency was found to be 80-90%.
Cell spreading on flexible substrates
Transglutaminase-crosslinked gelatin hydrogels of increasing stiffness were prepared using 3, 5 and 10% (w/v) final gelatin concentration and incubated at 4°C overnight to stabilize crosslinking (19). Cells in regular culture medium were plated at low density (to minimize cell-cell interactions) and allowed to spread for 6 h.
Rho activation assay
Rho activity was determined using the Rhotekin-RBD affinity precipitation assay as described previously20, 52. Briefly, TEC overexpressing TRPV4-EGFP or EGFP alone or TEC treated with GSK1016709A (Sigma-Aldrich; 100 nM for 15 min) were lysed in 1% Triton X-100 buffer and centrifuged at 12000× g for 15 min. Equal volumes (compensated for equal protein concentration) of clarified lysate were incubated with GST-Rhotekin-RBD beads (Cytoskeleton Inc.), for 1 h at 4°C. The beads were collected by centrifugation and washed 3 times with wash buffer. The bound GTP-Rho was extracted with SDS-sample buffer and was detected using Rho mAb (SantaCruz) on a Western blot. GTP-Rho levels were calculated from the densitometric analyses of Western blot and normalized to the levels of total-Rho and presented as relative Rho activity with that of TEC transfected with EGFP alone or TRPV4-EGFP.
Calcium Imaging
TEC overexpressing TRPV4-EGFP or EGFP alone or NEC were cultured on MatTek glass bottom dishes and loaded with Fluo-4/AM (1-4 μM) for 30 min, washed 3 times in calcium medium (136 mM NaCl, 4.7 mM KCl,1.2 mM MgSO4, 1.1 mM CaCl2, 1.2 mM KH2PO4, 5 mM NaHCO3, 5.5mM glucose, and 20 mM Hepes. pH 7.4). Cells were stimulated with TRPV4 activators 4-α-PDD (Sigma-Aldrich; 10 μM) or GSK1016790A (100 nM) in calcium medium16, 22, 31. Calcium imaging was performed on Leica SP2 Confocal Microscope or Olympus FluoView 300 confocal microscope and analyzed using Leica/Olympus software and Microsoft Excel. We have previously confirmed that TRPV4 channels are expressed in WT and NEC and that TRPV4 activators 4-α-PDD (10 μM) or GSK1016790A (100 nM) specifically induce calcium influx in TRPV4 expressing but not TRPV4 null endothelial cells in the calcium containing media31.
SDS-PAGE and Western blot analysis
Cells were lysed in TritonX-100 with protease and phosphatase inhibitor cocktail (Boston Bioproducts). Cell lysates were separated by electrophoresis on 8% SDS- polyacrylamide gels and transferred to Immobilon ® polyvinylidene difluoride membrane. The membrane was blocked in 5% milk in TBS with 0.1% Tween-20 (TBS-Tw) for 1 h. The blot was then incubated with the following primary antibodies anti-TRPV4 (1:300) (Alomone), anti-Actin (1:1000). The ECL (Pierce West Pico) method was used with anti-rabbit (Jackson Laboratories) at a dilution of 1:10,000 and developed using Kodak X-ray film or Protein Simple. Results were quantified using Image J software.
In vitro angiogenesis assays
Growth factor-reduced Matrigel® (BD Biosciences) was plated on 48 well plates and kept at 37°C in an incubator for 30 min. Cells overexpressing TRPV4-EGFP or EGFP alone (2-8 × 104 cells/well) were plated on Matrigel and incubated at 37°C for 16-18 h20. For 2D assays performed in the presence or absence of TRPV4-specific agonist, cells were treated with GSK1016790A (100nM) prior to plating. Tube formation was quantified by obtaining images as described below.
Microscopy, image analysis and statistics
The expression of EGFP-TRPV4 in EC was visualized using a Nikon Eclipse TE 2000-E microscope (Nikon, Japan) fitted with a CoolSnap HQ digital camera (Photometrics) or Olympus IX72 fluorescence microscope (Olympus, Japan). The cells spread on flexible gelatin hydrogels were fixed with 4% paraformaldehyde while cells forming tubular structures were left untreated, and samples from both studies were imaged using a Nikon Diaphot 300 phase contrast microscope (Nikon, Japan) fitted with a Hamamatsu digital camera (Hamamatsu Photonics, Japan) or Olympus IX72 fluorescence microscope (Olympus, Japan). Image analyses were performed using ImageJ software (NIH). For cell spreading studies, TEC cell areas were measured by tracing cell perimeter and normalized as described previously20. At least 30 cells were evaluated. For cell migration experiments, live cell images were recorded with a CCD camera (Hamamatsu, Tokyo, Japan) on a Nikon Eclipse TE 2000-E microscope (Nikon, Japan) or Olympus IX81 fluorescence microscope (Olympus, Japan) equipped with phase contrast optics and processed using the Image J. The microscope was also equipped with an on-stage heater that maintained the temperature at 37°C at all times, and the culture medium was covered with a thin layer of mineral oil to prevent evaporation. Cell migration was measured by marking the centroids of the migrating cell recorded at 20 min intervals over 2 h and then used to calculate the speed of cell migration. All data are expressed as mean ± SEM and evaluated for differences using student's t-test and/or one-way ANOVA.
Syngeneic tumor model in mice and analysis of tumor growth, vascular malformation and angiogenesis
All the experiments were performed according to approved protocol by Northeast Ohio Medical University, IACUC. Mouse Lewis lung carcinoma (LLC) cells (2 × 106) were subcutaneously injected in the flank region of wild type C57BL/6 mice (WT) or TRPV4 knockout mice in C57BL/6 background (KO). Tumor size was measured using calipers at 7, 14 and 21 days and tumor volume was calculated according to the formula V= 4/3*Pi*Length/2*(width/2)2. At day 21, mice were euthanized and tumor tissues were collected and fixed for immunohistochemistry or stored at -80°C. To measure tumor angiogenesis, tumor tissue sections of 10 μm thicknesses were stained with anti-CD31 (PECAM-1) to visualize the microvessels, α-SMA to stain pericytes and DAPI to label the nuclei. Images were acquired using Olympus IX72 microscope and the microvessels density, diameter (Feret) and length were calculated using Image J software. For the in vivo drug experiments, six to eight mice/group were used and the animals were divided in to four groups: 1) WT (control) 2) WT + TRPV4 activator 3) WT + Cisplatin and 4) WT + TRPV4 activator + Cisplatin. Once the tumors were palpable (after 7 days), the mice were daily given an intraperitonial (i.p) injection of TRPV4 agonist GSK1016790A (10 μg/kg) to groups 2 and 4 until day 21. The anti-cancer drug Cisplatin (3 mg/kg/week) was administered i.p. once/week to groups 3, and 4, 3 days post treatment with TRPV4 activator, until day 21. The WT control received saline as a vehicle.
Supplementary Material
Acknowledgments
This study was supported by the start-up funds from NEOMED (CKT) and NIH grants CA55833 and CA45548 (DI).
Sources of Funding: Supported by the start-up funds from NEOMED (CKT) and NIH grants CA55833 and CA45548 (DI).
Footnotes
Conflict of Interest: CKT and DI have rights in a patent based on some of the results presented in this manuscript. The remaining authors have no conflict of interest.
References
- 1.Fukumura D, Jain RK. Imaging angiogenesis and the microenvironment. Apmis. 2008;116:695–715. doi: 10.1111/j.1600-0463.2008.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain RK. Taming vessels to treat cancer. Sci Am. 2008;298:56–63. doi: 10.1038/scientificamerican0108-56. [DOI] [PubMed] [Google Scholar]
- 3.Abdollahi A, Folkman J. Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist Updat. 2010;13:16–28. doi: 10.1016/j.drup.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duda DG, Batchelor TT, Willett CG, Jain RK. VEGF-targeted cancer therapy strategies: current progress, hurdles and future prospects. Trends Mol Med. 2007;13:223–230. doi: 10.1016/j.molmed.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folkman J. Angiogenesis inhibitors: a new class of drugs. Cancer Biol Ther. 2003;2:S127–133. [PubMed] [Google Scholar]
- 9.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain RK, Carmeliet P. SnapShot: Tumor Angiogenesis. Cell. 2012;149:1408–1408 e1401. doi: 10.1016/j.cell.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 11.Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21:82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 12.Ingber DE. Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ Res. 2002;91:877–887. doi: 10.1161/01.res.0000039537.73816.e5. [DOI] [PubMed] [Google Scholar]
- 13.Ingber DE, Prusty D, Sun Z, Betensky H, Wang N. Cell shape, cytoskeletal mechanics, and cell cycle control in angiogenesis. J Biomech. 1995;28:1471–1484. doi: 10.1016/0021-9290(95)00095-x. [DOI] [PubMed] [Google Scholar]
- 14.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457:1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mammoto A, Mammoto T, Ingber DE. Rho signaling and mechanical control of vascular development. Curr Opin Hematol. 2008;15:228–234. doi: 10.1097/MOH.0b013e3282fa7445. [DOI] [PubMed] [Google Scholar]
- 16.Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, et al. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res. 2009;104:1123–1130. doi: 10.1161/CIRCRESAHA.108.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingber DE. Integrins, tensegrity, and mechanotransduction. Gravit Space Biol Bull. 1997;10:49–55. [PubMed] [Google Scholar]
- 18.Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 19.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh K, Thodeti CK, Dudley AC, Mammoto A, Klagsbrun M, Ingber DE. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci U S A. 2008;105:11305–11310. doi: 10.1073/pnas.0800835105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinac B. Mechanosensitive ion channels: molecules of mechanotransduction. J Cell Sci. 2004;117:2449–2460. doi: 10.1242/jcs.01232. [DOI] [PubMed] [Google Scholar]
- 22.Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2:435–442. doi: 10.1039/c0ib00034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sukharev S, Corey DP. Mechanosensitive channels: multiplicity of families and gating paradigms. Sci STKE. 2004;2004:re4. doi: 10.1126/stke.2192004re4. [DOI] [PubMed] [Google Scholar]
- 24.Alessandri-Haber N, Dina OA, Joseph EK, Reichling DB, Levine JD. Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J Neurosci. 2008;28:1046–1057. doi: 10.1523/JNEUROSCI.4497-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci. 2004;24:4444–4452. doi: 10.1523/JNEUROSCI.0242-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HS, Millward-Sadler SJ, Wright MO, Nuki G, Salter DM. Integrin and mechanosensitive ion channel-dependent tyrosine phosphorylation of focal adhesion proteins and beta-catenin in human articular chondrocytes after mechanical stimulation. J Bone Miner Res. 2000;15:1501–1509. doi: 10.1359/jbmr.2000.15.8.1501. [DOI] [PubMed] [Google Scholar]
- 27.Shakibaei M, Mobasheri A. Beta1-integrins co-localize with Na, K-ATPase, epithelial sodium channels (ENaC) and voltage activated calcium channels (VACC) in mechanoreceptor complexes of mouse limb-bud chondrocytes. Histology and histopathology. 2003;18:343–351. doi: 10.14670/HH-18.343. [DOI] [PubMed] [Google Scholar]
- 28.Wilson PD, Geng L, Li X, Burrow CR. The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest. 1999;79:1311–1323. [PubMed] [Google Scholar]
- 29.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, et al. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One. 2007;2:e827. doi: 10.1371/journal.pone.0000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudley AC, Khan ZA, Shih SC, Kang SY, Zwaans BM, Bischoff J, et al. Calcification of multipotent prostate tumor endothelium. Cancer Cell. 2008;14:201–211. doi: 10.1016/j.ccr.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, et al. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301:H757–765. doi: 10.1152/ajpheart.00142.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liedtke W. TRPV4 plays an evolutionary conserved role in the transduction of osmotic and mechanical stimuli in live animals. J Physiol. 2005;567:53–58. doi: 10.1113/jphysiol.2005.088963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, et al. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol. 2010;298:H466–476. doi: 10.1152/ajpheart.00854.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ingber DE, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol. 1989;109:317–330. doi: 10.1083/jcb.109.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brackenbury WJ, Djamgoz MB. Activity-dependent regulation of voltage-gated Na+ channel expression in Mat-LyLu rat prostate cancer cell line. J Physiol. 2006;573:343–356. doi: 10.1113/jphysiol.2006.106906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fraser SP, Ozerlat-Gunduz I, Brackenbury WJ, Fitzgerald EM, Campbell TM, Coombes RC, et al. Regulation of voltage-gated sodium channel expression in cancer: hormones, growth factors and auto-regulation. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2014;369:20130105. doi: 10.1098/rstb.2013.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baylie RL, Brayden JE. TRPV channels and vascular function. Acta Physiol (Oxf) 2011;203:99–116. doi: 10.1111/j.1748-1716.2010.02217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. Embo J. 2001;20:4639–4647. doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fiorio Pla A, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, et al. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene. 2012;31:200–212. doi: 10.1038/onc.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175:409–416. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. 1971;133:275–288. doi: 10.1084/jem.133.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuevas I, Boudreau N. Managing tumor angiogenesis: lessons from VEGF-resistant tumors and wounds. Adv Cancer Res. 2009;103:25–42. doi: 10.1016/S0065-230X(09)03002-4. [DOI] [PubMed] [Google Scholar]
- 44.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 45.Chae SS, Kamoun WS, Farrar CT, Kirkpatrick ND, Niemeyer E, de Graaf AM, et al. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin Cancer Res. 2010;16:3618–3627. doi: 10.1158/1078-0432.CCR-09-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383–388. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.di Tomaso E, London N, Fuja D, Logie J, Tyrrell JA, Kamoun W, et al. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS One. 2009;4:e5123. doi: 10.1371/journal.pone.0005123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goel S, Wong AH, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med. 2012;2:a006486. doi: 10.1101/cshperspect.a006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Liao S, Huang Y, Samuel R, Shi T, Naxerova K, et al. PDGF-D improves drug delivery and efficacy via vascular normalization, but promotes lymphatic metastasis by activating CXCR4 in breast cancer. Clin Cancer Res. 2011;17:3638–3648. doi: 10.1158/1078-0432.CCR-10-2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thodeti CK, Massoumi R, Bindslev L, Sjolander A. Leukotriene D4 induces association of active RhoA with phospholipase C-gamma1 in intestinal epithelial cells. Biochem J. 2002;365:157–163. doi: 10.1042/BJ20020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu H, Fu Y, Tian W, Cohen DM. Glycosylation of the osmoresponsive transient receptor potential channel TRPV4 on Asn-651 influences membrane trafficking. Am J Physiol Renal Physiol. 2006;290:F1103–1119. doi: 10.1152/ajprenal.00245.2005. [DOI] [PubMed] [Google Scholar]
- 54.Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, et al. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. doi: 10.1016/j.yjmcc.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, et al. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci U S A. 2007;104:19138–143. doi: 10.1073/pnas.0705964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Troidl C, Troidl K, Schierling W, Cai WJ, Nef H, et al. Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J Cell Mol Med. 2009;13:2613–2621. doi: 10.1111/j.1582-4934.2008.00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu F, Zhu W, Wang L. MicroRNA-203 up-regulates nitric oxide expression in temporomandibular joint chondrocytes via targeting TRPV4. Arch Oral Biol. 2013;58:192–199. doi: 10.1016/j.archoralbio.2012.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.