

Abstract
Over 90% of HIV/AIDS positive individuals in sub-Saharan Africa are infected with highly heterogeneous HIV-1 subtype C (HIV-1C) viruses. One of the best ways to reduce the burden of this disease is the development of an affordable and effective prophylactic vaccine. Mosaic immunogens are computationally designed to overcome the hurdle of HIV diversity by maximizing the expression of potential T cell epitopes. Mycobacterium bovis BCG ΔpanCD auxotroph and modified vaccinia Ankara (MVA) vaccines expressing HIV-1C mosaic Gag (GagM) were tested in a prime-boost regimen to demonstrate immunogenicity in a mouse study. The BCG-GagM vaccine was stable and persisted 11.5 weeks post vaccination in BALB/c mice. Priming with BCG-GagM and boosting with MVA-GagM elicited higher Gag-specific IFN-γ ELISPOT responses than the BCG-GagM only and MVA-GagM only homologous vaccination regimens. The heterologous vaccination also generated a more balanced and persistent CD4+ and CD8+ T cell Gag-specific IFN-γ ELISPOT response with a predominant effector memory phenotype. A Th1 bias was induced by the vaccines as determined by the predominant secretion of IFN-γ, TNF-α, and IL-2. This study shows that a low dose of MVA (104 pfu) can effectively boost a BCG prime expressing the same mosaic immunogen, generating strong, cellular immune responses against Gag in mice. Our data warrants further evaluation in non-human primates. A low dose vaccine would be an advantage in the resource limited countries of sub-Saharan Africa and India (where the predominating virus is HIV-1 subtype C).
Introduction
HIV-1 subtype C (HIV-1C) is the most prevalent subtype in the world, accounting for over 50% of all global infections [1,2]. It is the dominant subtype in southern Africa where it accounts for more than 95% of all HIV-1 infections in the region [3–6] and in India (which accounts for 6% of the HIV-1 global prevalence of HIV-1C) [2,7]. In addition, HIV-1C infections have been shown to be on the increase in Wales, England [8], and South America [9,10].
A prophylactic HIV-1 vaccine is thought to be the most effective means of controlling the spread of this epidemic. Furthermore, there is general agreement that a successful vaccine will need to induce strong humoral and cellular immune responses [11–13]. Developing such a vaccine is particularly challenging in the face of the diversity of HIV-1. In HIV-infected elite controllers and long-term non-progressors lower viral loads are significantly correlated with Gag-specific CD4+ and CD8+ T cell responses [14–16]. Therefore HIV-1 Gag is a critical immunogen for inclusion in the T-cell inducing component of an HIV-1 vaccine. One of the methods used to address the diversity of HIV-1, is the generation of mosaic and conserved immunogens which can be designed computationally (reviewed in [17,18]). Mosaic immunogens are optimised in silico to increase the coverage of both CD8+ and CD4+ T cell epitopes from natural sequences with the hope of reducing the HIV-1 escape pathways [19–26]. This strategy provides a higher level of diversity coverage when compared to natural sequence vaccine candidates.
In this paper Mycobacterium bovis bacillus Calmette-Guérin (BCG) and modified vaccinia Ankara (MVA) are investigated as HIV vaccine vectors to deliver an HIV-1C Gag mosaic antigen targeted at inducing T cell responses. Since its development as a safe tuberculosis vaccine, BCG has been administered to over 4 billion infants and is accessible to 80% of infants globally [27–29]. The results from animal model studies indicate that recombinant BCG (rBCG) can be used as a vaccine vector that can induce both humoral and antigen-specific cellular immune responses to HIV-1 (reviewed in [30,31]). In the event that HIV-1-infected and immunocompromised individuals unintentionally get enrolled in future BCG-vectored HIV-1 vaccine campaigns, it is essential to use the safer BCG strains as vectors [32]. Strategies have been developed for the production of safer BCG strains, in particular auxotrophic BCG strains. These strains have mutations in genes required for the production of essential growth compounds, and so cannot replicate unless supplemented with the necessary growth compounds [33–36]. We used a ΔpanCD auxotroph which is unable to synthesize pantothenic acid, a key precursor of coenzyme A, which is essential for several mycobacterial intracellular processes [32,37,38]. Furthermore, since safer strains of BCG are being developed against childhood TB, it is important to use these strains as HIV-1 vaccine vectors for use as dual vaccines.
The only effective HIV vaccine trial (RV144) tested a combination of a canary poxvirus vector and protein as candidate vaccines encouraging further exploration of poxviruses as HIV vaccine vectors. MVA is a highly attenuated strain of the smallpox virus vaccine, vaccinia virus (VACV), that is unable to complete its replication cycle in human cells due to large deletions [39]. Furthermore, deletion of immune-modulatory genes results in a rapid local immune response at the point of infection [39,40]. MVA has adjuvant properties and potential to induce long-lasting immunity. However, viral and foreign genes can still be efficiently expressed. Since smallpox was eradicated and vaccination ceased in 1979, people up to 37 years of age would not have been exposed to either Variola virus or VACV and therefore would not have been immunized with MVA. MVA provides the safety of an attenuated or killed virus vaccine, yet provides the immunogenicity of a live virus vaccine with its ability to express foreign antigen. Added to their safety, both rBCG and rMVA are affordable to produce [41–43].
In an effort to make affordable vaccines suitable for the regions most affected by HIV-1, we developed stable HIV-1 vaccines—based on BCG and MVA vectors—expressing an HIV-1C mosaic Gag immunogen and assessed the vaccine immunogenicity in mice.
Materials and Methods
Construction of rBCG
The pantothenic acid auxotroph strain derived from BCG Pasteur, BCGΔpanCD, was kindly provided by Professor William R Jacobs and cultured as previously described [37].
The BCG shuttle vector pTJBCG3 was constructed as follows: the full length HIV-1C mosaic gag gene (gagM [19]) was codon optimised for use in BCG and cloned into the ClaI and HpaI sites of pHS400 [37]. A control plasmid that had no insert in it (pEM19) was included in the study. To generate plasmid pEM19, pHS400 was digested with SnaBI and HpaI, to remove gagM, and religated.
Recombinant BCGΔpanCD vaccine stocks resuspended in 8.5% w/v NaCl; 10%glycerol; 10% Tyloxapol were made as previously described [37] and stored at -80°C until required.
In Vitro and In Vivo Genetic Integrity of rBCG Vaccine Stocks
Crude cell lysates prepared from BCGΔpanCD vaccine stocks were electroporated into E. coli DH5α electro-competent cells which were plated on Luria agar [44]. To determine the in vitro plasmid stability, single colonies from the electroporated E. coli cells were used to inoculate liquid media, plasmid DNA was isolated and mapped using restriction enzyme digestion. In vivo shuttle vector integrity was determined on spleens and lymph nodes isolated from mice 11.5 weeks after vaccination by polymerase chain reaction (PCR) using crude cell lysates of BCG colonies obtained after plating homogenised splenocytes and lymph nodes from previously vaccinated mice on Middlebrook 7H10 agar. The PCR components were 5 μl of BCG crude cell lysate, 25μl PCR ImmoMix Red (Bioline, USA), 10μM each primer (pCB119F: 5'–CAT ATG AAG CGT GGA CTG AC– 3' and pEMRev: 5'–AGC AGA CAG TTT TAT TGT TC—3') and 10μl distilled water. The PCR reaction conditions were initial denaturation at 95°C for 10 minutes, followed by 30 cycles of DNA denaturation at 95°C for 30 seconds, annealing of primers at 56°C for 30 seconds, and DNA extension at 72°C for one minute with a 5 second increment per cycle. A final extension step at 72°C for 4 minutes completed the reaction. PCR products were confirmed for lack of mutations by sequence analysis.
Construction of Recombinant MVA (rMVA)
The transfer vector, pTJMVA2, was designed to insert the gagM gene, under the control of the VACV mH5 promoter, between the A11R and A12L transcriptionally convergent open reading frames (ORFs) (Fig 1). The genes for green fluorescent protein (gfp) and blasticidin resistance (bsd) were used as marker and selection genes respectively and expressed as a fusion protein under the transcriptional control of the pSS promoter. The gfp-bsd ORF was placed outside of the A12L flank, so that it could be recombined out at a later stage and non-fluorescing foci could be screened for the final recombinant containing gagM alone between the A11R and A12L ORFs.
Fig 1. Recombinant MVA construction.
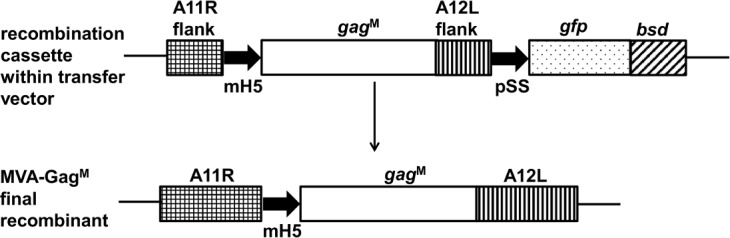
The transfer vector pTJMVA2 was designed to insert the gagM gene under the transcriptional control of the mH5 promoter, between the MVA A11R and A12L ORFs. The selection (bsd) and marker (gfp) genes were expressed as a fused protein under the transcriptional control of the pSS MVA synthetic promoter.
Adherent BHK cell monolayers were infected with wild type (wt) MVA (multiplicity of infection (MOI) = 0.1) diluted in DMEM-0 (DMEM supplemented with 1000U/ml penicillin (Lonza, Belgium), 1000U/ml streptomycin (Lonza, Belgium), and 10μg/ml fungin (Invivogen, USA)) and transfected 2 hours later with a 1:1 ratio of 4μg of the recombinant plasmid DNA, pTJMVA2: 4μl X-treme Gene HP® transfection reagent (Roche, Switzerland) according to the manufacturer’s instructions. Cells were incubated at 37°C in a 5% CO2 humidified incubator for 48 hours. Total virus was harvested and used to re-infect a fresh BHK monolayer. Virus was passaged 4 times in the presence of blasticidin and an intermediate rMVA expressing GFP was purified. Virus was then passaged in the absence of blasticidin and non-fluorescing foci were plaque purified 7 times. PCR was carried out to screen for the presence of the intermediate and final MVA recombinants using cell lysates of infected cells as template. The presence of the intermediate recombinant MVA was confirmed by PCR amplification using the primers A11Rfor (5'-ACAAACACCATCCTTGGGAGTA-3') and gfprev (5'-AAAGTTCTTCACCCTTAGACGCC-3') that bind to A11R and gfp respectively, or colE1for (5'-GCGTGAGCTATGAGAAAGCGCCAAAT-3') and A12Lrev (5'-CGGTGGAGATGCAGCCGTCAA-3') that bind to the E. coli ORI and A12L respectively. The presence of the final recombinant was confirmed using a gag-specific primer in combination with a primer which bound on either side of the A12L or A11R flanking site in the MVA genome (gagfor: 5'-CCCTAGAAAGAAAGGCTGCTGGAA-3' and A12Lrev: 5'-AATCGGTGGAGATGCAGCCGTCAA-3' or A11Rfor: 5'-ACAAACACCATCCTTGGGAGTATTC-3' and gagrev: 5'-TTTCCGCCAGGCCTCAGT-3') and using primers A11Rfor and A12Lrev alone. Cell lysates for PCR were obtained using a method described by David Tscharke (personal communication). Briefly, when plaques were visible, the cells were gently washed with 1ml PBS/well. A volume of 250μl 1 X PCR buffer (10mM TrisCl, 2.5mM MgCl2, 50mM KCl, pH 8.3) with proteinase K (10μg/ml; Sigma-Aldrich, USA) was added to the cells, incubated at -80°C until frozen, and then thawed at 37°C. Cell lysates were incubated at 56°C for 20 minutes, and then at 85°C for 10 minutes. Cell debris was removed by centrifugation for 10 minutes at 2000rpm (Eppendorf Centrifuge 5417C, Germany). A 5–10μl aliquot of the supernatant was used for PCR using an ImmoMix Red PCR mix (Bioline, USA) according to the manufacturer’s instructions. The PCR product for the final recombinant was confirmed by sequencing using the ABI Prism® BigDye™ Terminator Cycle Sequencing kit (Applied Biosystems, USA) according to the manufacturer’s instructions. This was done as a service offered by the Central Analytical Facilities laboratory (Stellenbosch University, South Africa). MVA-GagM virus stock was grown in BHK cells and purified on a 36% sucrose cushion (36% in PBS) at 4°C and 15 000rpm (Sorvall RCSC Plus, USA) for an hour. The virus stock was further purified on a sucrose-dextran gradient (36% sucrose; 10% dextran) under the same conditions described above, and stored in PBS at −80°C until required. MVA-GagM virus titration was done on BHK-21 cells as previously described by Chapman et al., 2012 [37]. Western blot analysis to determine GagM protein expression was carried out as previously described [42,45].
Mice Vaccinations
Groups of four to five 6–8 weeks old female BALB/c mice were used for each experiment. The BCG vaccine was administered intraperitoneally at a dose of 2 x 107cfu per mouse. MVA vaccines were administered bilaterally into the tibialis muscle of each hind leg (50μl each) at a total dose of 102, 104 or 106 pfu MVA. Mice were sacrificed by cervical dislocation. The vaccination schedule (see Table 1) and all the animal procedures were approved by the University of Cape Town Animal Research Ethics Committee (reference UCT AEC 12–059) and performed by a trained animal technologist.
Table 1. Mouse immunization regimen.
| Group | Prime | Boost | Sacrifice |
|---|---|---|---|
| 1 | Day 0: 2x107 cfu BCG-GagM | Day 70: 102 pfu MVA-GagM | Day 82 |
| 2 | Day 0: 2x107 cfu BCG-GagM | Day 70: 104 pfu MVA-GagM | Day 82 |
| 3 | Day 0: 2x107 cfu BCG-GagM | Day 70: 106 pfu MVA-GagM | Day 82 |
| 4 | Day 0: 2x107 cfu BCGE | Day 70: 102 pfu MVA-GagM | Day 82 |
| 5 | Day 0: 2x107 cfu BCGE | Day 70: 104 pfu MVA-GagM | Day 82 |
| 6 | Day 0: 2x107 cfu BCGE | Day 70: 106 pfu MVA-GagM | Day 82 |
| 16 | Day 0: 2x107 cfu BCG-GagM | Day 70: 2x107 cfu BCG-GagM | Day 82 |
| 17 | Day 0: 104 pfu MVA-GagM | Day 12 | |
| 18 | Day 0: 104 pfu MVA-GagM | Day 28: 104 pfu MVA-GagM | Day 40 |
Isolation of Splenocytes and Lymph Nodes
At the experimental endpoint, spleens and mesenteric lymph nodes were harvested. Organs of the same type in each group were pooled before processing. A single cell suspension from the spleens was prepared as previously described [37,46] for use in the IFN-γ ELISPOT assay, intracellular cytokine staining and for the cytometric bead array assay. Mesenteric lymph nodes and left over splenocytes from the BCG vaccinations were stored at -80°C in BCG resuspension buffer (8.5% w/v NaCl, 10% glycerol, 10% tyloxapol) until required for evaluating the integrity of the shuttle vectors.
Immunogenicity Assays to Evaluate Mosaic Vaccines
IFN-γ ELISPOT and cytometric bead array (CBA) assays were carried out as previously described [37] and intracellular cytokine staining and staining of cell surface molecules was carried out as previously described by Burgers et al., 2006 [47]. The following antibodies were used; anti-CD3+ Alexa 700, anti-CD4+ PE-Cy7, anti-CD8+ APC-Cy7, anti-CD62L APC, and anti-CD44 FITC). The cytokine antibodies were all PE-conjugated (0.2μg anti-TNF-PE, 0.06 μg anti-IL-2-PE, 0.06 μg anti-IFN-γ-PE) and were obtained from BD biosciences. The anti-CD4+ PE-Cy7, anti-CD8+ APC-Cy7 were obtained from BD biosciences. All the other antibodies were obtained from eBioscience.
Statistical Analysis
Data was statistically analysed using Prism version 5.0 (Graphpad Software, San Diego, CA). The t test for independent unpaired parametric comparisons was applied to assess the level of significance of comparisons between means. All tests were two-tailed. P values ≤ 0.05 were considered significant. The false discovery rate (FDR) step-down procedure described in the paper by Columb & Sagadai [48] was used to correct for multiple comparisons.
Results
Determination of rBCG Vaccine Stability
As the stability of rBCG can be compromised in vitro and in vivo (reviewed by Chapman et al., 2010; [49]), the genetic integrity of the rBCGΔpanCD vaccine stocks was assessed by restriction enzyme mapping of the shuttle vectors using XhoI (pTJBCG3) and SmaI (pEM19) (Fig 2A–2D). A single batch of each vaccine stock was tested and the same batch was then used to inoculate mice. pTJBCG3 and pEM19 plasmid DNA isolated from vaccine stocks of BCG-GagM and BCGE respectively gave DNA fragments of the expected sizes (Fig 2C and 2D).
Fig 2. Schematic representation of the BCG shuttle vectors and the determination of their genetic integrity before and after vaccination.

Schematic representation of (A) pTJBCG3, which was used to make the BCG-GagM vaccine, (B) pEM19 which was used to make the BCGE vaccine. Restriction sites used for cloning and restriction mapping analysis are indicated in black bold type. OriE–E. coli origin of replication; OriM–mycobacterial origin of replication, 19kD ss– 19kD signal sequence; KanR–kanamycin resistance gene. (C) pTJBCG3 digested with XhoI. Lane 1 is a positive control of pTJBCG3 DNA prior to transformation into BCGΔpanCD. Lanes 2–21 contain pTJBCG3 DNA obtained from recombinant BCG-GagM vaccine stocks. (D) pEM19 digested with SmaI. Lanes 1–15 contain pEM19 plasmid DNA isolated from recombinant BCGE vaccine stocks, and lane 16 pEM19 plasmid DNA isolated prior to transformation into BCGΔpanCD (positive control). PCR amplification of DNA from rBCG obtained from the spleens and lymph nodes of mice vaccinated with BCG-GagM (Group 2) (E) or BCGE (Group 5) (F) 11.5 weeks post vaccination. Lane 1 –negative control; Lane 2 –positive control; Lane 3–12 are PCR products from rBCG isolated from homogenised spleen (3–7) or lymph nodes (8–12). Lanes M in C—F contain the molecular weight marker O’GeneRulerTM 1kb DNA ladder.
The genetic integrity of the BCG shuttle vectors in vivo was determined by PCR analysis of plasmid DNA extracted from rBCG isolated from the spleens and lymph nodes of mice 11.5 weeks post vaccination. PCR products of the expected sizes of 1869bp and 344bp were obtained from pTJBCG3 and pEM19 respectively (Fig 2E and 2F). Eight of the PCR products were also sequenced to confirm genetic integrity and no mutations were observed (data not shown).
Generation of a Recombinant MVA Expressing GagM
A recombinant MVA was constructed with HIV-1C gagM inserted between ORFs A11R and A12L and under the control of the vaccinia virus mH5 promoter. GagM expression was confirmed by immunostaining using anti-HIV-1 p24 Gag antibody (ARP432) (Fig 3A). Uninfected cells and cells infected with wtMVA were used as negative controls. In vitro expression of the GagM protein in the vaccine stocks was also confirmed by Western blot analysis following SDS PAGE. Cell lysates derived from BHK-21 cells infected with MVA-GagM, when probed with a Gag-specific antibody, showed expression of a protein of the correct size of 55kD (Fig 3B; Lane 4). Lysates from uninfected cells were used as a negative control and a BHK cell lysate transfected with a plasmid known to express full length Gag was used as a positive control (Fig 3B; Lane 1) [47].
Fig 3. In vitro expression of Gag by BHK-21 cells infected with MVA-GagM.

(A) BHK-21 cells were infected with MVA-GagM, wild type MVA (wtMVA), or left uninfected for 72 hours. HIV-1 Gag was detected with anti-Gag antibody ARP432 (α-Gag; top panels) as well as an anti-VACV antibody (α-MVA; bottom panels), followed by an anti-rabbit HRP-conjugated antibody. (B) Cell lysates were prepared from BHK-21 cells infected with MVA-GagM (lane 4), wild type MVA (lane 3), or left uninfected (lane 2). BHK cells transfected with a plasmid known to express full length Gag was used as a positive control (lane 1). Western blots were probed with a rabbit anti-HIV-1-p24 Gag antibody (ARP432), followed by an anti-rabbit antibody conjugated to alkaline phosphatase (Sigma-Aldrich, USA). A Precision Plus Protein Kaleidoscope pre-stained standard (lane M; Biorad, USA) was used and the sizes are indicated on the left.
Determination of the Optimal Dose of MVA-GagM to Boost a BCG-GagM Prime
To determine the optimal MVA dose required to effectively boost the BCG prime, mice were primed with 2 x 107 cfu of either the recombinant BCG-GagM or the control BCGE and boosted on day 70 with 102, 104, or 106 pfu of MVA-GagM (see table insert in Fig 4). The BCG vaccine priming dose of 2 x 107 cfu was previously determined as optimal in our lab (Dr Ros Chapman, personal communication).
Fig 4. Determination of the optimal dosage of MVA-GagM to boost a BCG-GagM prime.

(A) Mice were primed on day 0 with 2 x 107 cfu BCG-GagM (Group 1–3) or BCGE (Group 4–6) and boosted on day 70 with 102 (Group 1 and 4), 104 (Group 2 and 5), or 106 (Group 3 and 6) pfu MVA-GagM. (B) Cumulative IFN-γ ELISPOT CD8+ and CD4+ responses of vaccinated mice to HIV-1 Gag peptides. The ELISPOT assay was carried out using three Gag-specific peptides for stimulation of pooled splenocytes that were isolated 12 days post the MVA-GagM boost. Bars represent the magnitude of net responses to individual peptides, expressed as sfu/106 splenocytes after subtracting the background. (C) and (D) Total frequency of T cells producing IFN-γ, IL-2, and/or TNF-α, after subtracting the background, in response to HIV-1 Gag peptide stimulation following a rBCG prime and an MVA-GagM boost at different doses. Cells were positive for cytokine production if the proportion was ≥0.05% after subtracting the background. These results are from a single experiment using pooled splenocytes.
Priming with the BCG-GagM or BCGE and boosting with 102 pfu MVA-GagM elicited no detectable Gag-specific IFN-γ ELISPOT responses in mice (Fig 4B; Groups 1 and 4 respectively). However, high cumulative HIV-1 Gag-specific IFN-γ ELISPOT responses were induced in mice primed with BCG-GagM and boosted with 104 pfu (1273 sfu/106 splenocytes) and 106 pfu (2023 sfu/106 splenocytes) MVA-GagM. These responses targeted both Gag CD4 and CD8 peptides in almost equal magnitudes (Fig 4B, Groups 2 and 3 respectively). The magnitudes of these responses were 3.2 and 4.1 fold higher, than those induced in mice primed with the control BCG (BCGE) and boosted with a 104 or 106 pfu of MVA-GagM respectively (Groups 5 & 6). Thus, MVA-GagM efficiently boosts a BCG-GagM prime at a dose of 104 pfu or 106 pfu but not at a dose of 102 pfu.
To further characterise the immune responses induced by boosting BALB/c mice with different doses of MVA-GagM following a BCG prime, intra-cellular cytokine staining followed by flow cytometry was carried out to determine the combined frequency of IFN-γ-, TNF-α-, and IL-2- producing cells (Fig 4C and 4D).
Priming with BCG-GagM or with the control BCGE, and boosting with 102 pfu MVA-GagM did not elicit any detectable HIV-1 Gag-specific cytokine-producing CD8+ T cells (Fig 4D, Groups 1 and 4). Mice primed with BCG-GagM and boosted with 104 pfu (Group 2) or 106 pfu MVA-GagM (Group 3) elicited almost double the frequency of HIV-1 Gag-specific cytokine-producing CD8+ T cells as compared to control-primed mice that were similarly boosted (Groups 5 &6), suggesting an effective BCG-GagM prime.
Immune Responses in BALB/c Mice Elicited by BCG Prime-MVA Boost Vaccines Expressing a GagM Immunogen
While the greatest cumulative immune response to the Gag peptides was detected from mice boosted with 106 pfu MVA-GagM (Fig 4B; Group 3), the amount of background responses in the IFN-γ ELISPOT assay were unacceptably high (up to 420 sfu/106 splenocytes). An MVA-GagM boost of 104 pfu for rBCGΔpanCD-primed mice was therefore chosen as the optimal dose to compare immune responses to different rBCGΔpanCD prime vaccinations. As controls, mice vaccinated with a single dose of MVA-GagM as well as those vaccinated with two homologous doses of BCG-GagM or MVA-GagM vaccines were included. The immunisation schedules were chosen to give the peak immune response for each vaccine vector or combinations of vaccine vectors. Data from our group (unpublished) has indicated immune responses are improved if the MVA boost is given at 10 weeks rather than 4 weeks post the BCG prime.
Mean cumulative responses for Group 2 mice (BCG-GagM/MVA-GagM) reached a magnitude of 1143 ± 117 sfu/106 splenocytes with almost equally balanced responses targeted to Gag CD8 (475 ± 55 sfu/106 splenocytes) and Gag CD4 (668 ± 32.7 sfu/106 splenocytes; Fig 5B) peptides. There was a 2.8-fold difference between the magnitudes of cumulative responses of mice primed with BCG-GagM (Group 2) and those that received a control BCGE prime (Group 5–410 ± 98 sfu/106 splenocytes). Thus, BCG-GagM efficiently primes the adaptive immune system.
Fig 5. Evaluation of a BCG-GagM prime/ MVA-GagM boost in BALB/c mice.

(A) Vaccination schedule used. (B) Cumulative IFN-γ ELISPOT CD8+ and CD4+ responses of vaccinated mice to HIV-1 Gag peptides. The ELISPOT assay was done in triplicate using pooled spleens on the day of sacrifice using three Gag-specific peptides for stimulation. Bars are the mean and standard deviation of the mean responses for the indicated individual peptides from 3 independent experiments. Responses are expressed as sfu/106 splenocytes after background subtraction. Horizontal bars with asterisks indicate statistical significance of the mean responses between the indicated groups. **p<0.01, ***p<0.001; Student t-test of unpaired data followed by FDR for multiple comparisons. (C) and (D) Total frequency of T cells producing IFN-γ, IL-2, and/or TNF-α in response to HIV-1 Gag peptide stimulation. Cell surface staining and flow cytometry were carried out in duplicate on pooled spleens per group using three Gag-specific peptides for stimulation. The memory distribution of the cytokine producing T- cells in the central and effector memory compartment (TCM and TEM) are represented as pie charts above each corresponding bar per group. Cells were positive for cytokine production if the proportion was ≥ 0.05% after subtracting the background. The cellular phenotype was positive if there were ≥ 10 cells per test. (E) The levels of cytokines in the culture supernatants were quantified using a Th1/Th2 cytokine bead array assay followed by flow cytometry. The recorded results were obtained after subtracting the background. The distance from the centre of the plot indicates a log10-fold change (ranging from 1 to 10 000) and cytokine levels were expressed as pg/ml.
Mean cumulative IFN-γ ELISPOT HIV-1 Gag responses of mice vaccinated with the BCG-GagM/MVA-GagM heterologous prime-boost regimen (Group 2) were 3- and 1.7-fold greater than the BCG-GagM/BCG-GagM homologous prime-boost (Group 16; 380 ± 64.7 sfu/106 splenocytes) and MVA-GagM/MVA-GagM homologous prime-boost; 656.7 ± 8.5 sfu/106 (Group 18) mice respectively. The BCG-GagM/MVA-GagM heterologous prime-boost was therefore significantly more efficient in generating an immune response than the BCG-GagM/BCG-GagM (p<0.001) and MVA-GagM/MVA-GagM (p<0.01) homologous prime-boost vaccinations. Interestingly, responses to the CD8 Gag peptide were similar for Groups 17 (MVA-GagM) and 18 (MVA-GagM/ MVA-GagM; Fig 5B). The second MVA-GagM vaccination, however boosted CD4+ T cell responses to Gag (Fig 5B).
As shown in Fig 5C and 5D, the BCG-GagM/MVA-GagM heterologous prime-boost regimen (Group 2) resulted in CD8+ T cells with a higher frequency of effector memory phenotype (91.6%) than those in the control group (Group 5–66.5%). A BCG-GagM homologous prime-boost resulted in cytokine positive CD8+ T cells that were predominantly of a central memory phenotype (58%; Group 16). There were fewer cytokine-producing CD4+ T cells than there were cytokine-producing CD8+ T cells for all vaccination regimens except for the Group 18 mice that received an MVA-GagM homologous prime-boost vaccination (Fig 5C and 5D). A single MVA-GagM vaccination resulted in cytokine-positive CD4+ T cells with a predominant effector memory phenotype (Group 17–76%), and a homologous boost increased the proportion of effector memory CD4+ T cells to 99% (Group 18). Cytokine-positive CD4+ T cells following a BCG-GagM homologous prime-boost all had an effector memory phenotype (Group 16).
To assess the Th1/Th2 bias of the immune response to the vaccines used, Th1 and Th2 cytokines were quantified in culture media of splenocytes stimulated with Gag CD4 and CD8 peptides in a cytokine bead array assay (Fig 5E). As anticipated, IFN-γ, TNF-α, and IL-2 (Th1 cytokines) had the highest cumulative levels in all groups of mice. IFN-γ, TNF-α, and IL-2 were 2.4-, 4.9-, and 4.7-fold higher, respectively, in Group 2 (BCG-GagM/MVA-GagM) compared to Group 5 mice (BCGE/MVA-GagM) confirming an efficient prime with the BCG-GagM vaccine. The MVA-GagM vaccine also potently boosted the BCG-GagM prime. Cumulative IFN-γ, TNF-α, and IL-2 levels were higher in mice that received the heterologous BCG-GagM/MVA-GagM prime-boost vaccination (Group 2) compared to Group 16 mice that received a BCG-GagM/BCG-GagM homologous vaccination. Splenocytes from the heterologous prime-boost vaccination (Group 2) produced high levels of IFN-γ, TNF-α, and IL-2 compared to any of the homologous prime-boost vaccinations (Group 16 and 18; BCG-GagM/BCG-GagM and MVA-GagM/MVA-GagM respectively).
Discussion
In this study, rBCGΔpanCD and rMVA vaccines expressing an HIV-1C mosaic Gag (BCG-GagM and MVA-GagM, respectively) were made and evaluated in mice. Studies done by others have shown HIV-1 Group M mosaic vaccines to induce broader T cell and higher magnitude responses than vaccines expressing antigens derived from natural or consensus HIV-1 sequences [21,26,50–52]. Here we have shown BCG-GagM and MVA-GagM vaccines expressing HIV-1C mosaic Gag to be immunogenic in mice, particularly when administered as a heterologous prime-boost regimen. BALB/c mice have a limited number of HIV-1 epitopes that can be used to evaluate the breadth of candidate vaccines thus this could not be evaluated in this study. Further studies using bi- and tri-valent HIV-1C mosaic immunogens to increase breadth, which is essential for clearing diverse strains of HIV-1 in infected individuals, will be carried out in non-human primates.
The stability and expression of transgenes is critical in recombinant BCG vaccine development. This is essential for memory cells to elicit a correct and potent immune response to the antigen in the event of an infection. The rBCGΔpanCD vaccines made in this study were stable in vitro and in vivo. The shuttle vectors in the BCG-GagM and BCGE vaccines were detectable in peripheral lymphoid organs (spleen and lymph nodes) of vaccinated mice 11.5 weeks post vaccination. Furthermore, the gagM DNA sequence obtained from BCG-GagM in the peripheral lymphoid organs was unaltered as determined by PCR and sequencing. This was encouraging, as these are sites where adaptive immune responses are initiated [53].
In order to make a stable rMVA an insertion site was selected that would be stable. In the past the del II and del III regions which lie within the variable terminal regions were used as sites of insertion in rMVA vaccine development. These regions are often prone to deletions and other mutational changes. Inserting a foreign gene into the variable terminal region makes it prone to such deletion mutations. Therefore to increase transgene stability, foreign genes have been inserted between transcriptionally convergent conserved genes where no possible transcriptional promoters could be disrupted [54,55]. In this study gagM was inserted between the A11R and A12L genes. A GagM protein of the correct size (55kD) was shown to be expressed from MVA-GagM after vaccine scale up confirming the stability of the vaccine.
Live attenuated SIV and CMV vaccines that elicit persistent CD8+ T cell responses, have been shown to control viral load in macaques [56–58]. In this study, we demonstrated that BCG-GagM persists in the tissues of vaccinated mice as determined by the presence of BCG-GagM colonies in the peripheral lymphoid organs (spleen and lymph nodes) of these mice 11.5 weeks post vaccination (Fig 2). Our group and others have also shown that rBCG persists in vivo up to 20 weeks post vaccination [28,59–61]. Future studies of BCG-GagM should include experiments to confirm the long term persistence of HIV-specific T cell responses.
The replication of BCG in vivo is slow and rBCG persistence subsequently results in low antigen expression and low levels of antigen presentation [62]. Low T cell immune responses to the antigen are induced and differentiate into memory phenotype, and get stimulated when boosted with a matching antigen [63]. Vaccination with a BCG-GagM prime MVA-GagM boost generated predominantly effector memory cytokine-positive T cells, a T cell subset shown to play a role in the control of viral load after vaccination with CMV-based vaccines [56]. Effector memory cells act as the first line of defence at the site of HIV infection. Hansen and colleagues have shown that the protection of vaccinated non-human primates from SIV challenge was due to both CD4+ and CD8+ effector memory T cell responses [56–58].
Antigens derived from mycobacteria are processed and presented by macrophages. Antigens delivered into the phagolysosome usually get processed by the HLA class II pathway. Such antigens would induce CD4+ T cell responses (reviewed by Hess et al., 2000; [64]. In our study, the BCG-GagM prime and MVA-GagM boost resulted in the frequency of cytokine-secreting CD8+ T cells being greater than that of CD4+ T cells (Fig 5C and 5D). The GagM antigen in our study was linked to the 19kD signal sequence in the BCG shuttle vector. This targets the antigen to the cell wall, making it accessible for processing by the HLA class I pathway and inducing CD8+ T cell responses [65]. Previous studies carried out by our group also showed that the BCGΔpan strain induced predominant CD8+ T cell responses to HIV-1 Gag [32,37,60]. CD4+ cells are essential for providing help to CD8+ T cells [66–69]. However, CD4+ T cells are also the target of HIV-1 infection (reviewed by Grossman et al., 2006 [70]). There is therefore a fine balance between inducing enough of a CD4+ response to provide help to CD8+ cells and inducing too many CD4+ cells, which will increase the pool of target cells for HIV-1 infection.
The GagM vaccines in our study induced a Th1 bias when administered in a heterologous or homologous prime-boost regimen (Fig 5E). A Th1 immune response has been shown to be important for protection against viral challenge in mice and humans as reported by Someya and colleagues (2004; [71]) and by Betts and colleagues respectively [72,73]. Both IFN-γ and TNF-α are important mediators of antiviral activity by CD8+ T cells in HIV-1 infection whilst increased production of IL-2 is associated with reduced viral loads in elite controllers [73–75]. It is therefore desirable for candidate vaccines to induce these cytokines as potential correlates of protection.
This study shows that subtype-specific monovalent HIV-1 GagM vaccines are highly immunogenic in mice. Furthermore, a low dose MVA-GagM boosted a BCG-GagM prime. This is very attractive for dose sparing and reduced costs for the targeted resource-limited regions should the vaccine get to clinical trials, licencing, and large scale distribution. This promising immunogenicity data warrants further evaluation in non-human primates.
Acknowledgments
We thank Rodney Lucas for technical assistance with the mouse work and Desiree Bowers and Shireen Gallant for assistance with the mouse work and immunological assays. We also appreciate the technical assistance provided by Tracy Meiring and Craig Adams. Thanks to Bette Korber and Bart Haynes for giving us permission to use their HIV-1 subtype C Gag mosaic sequence.
Data Availability
All relevant data are within the paper.
Funding Statement
This work is also based upon research supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation. Tsungai Jongwe received bursaries from the Schlumberger Foundation, the Oppenheimer Memorial Trust and the Poliomyelitis Research Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Osmanov S, Pattou C, Walker N, Schwardlander B, Esparza J, WHO-UNAIDS Network for HIV Isolation and Characterization. Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000. J Acquir Immune Defic Syndr 2002. February 1;29(2):184–190. [DOI] [PubMed] [Google Scholar]
- 2.UNAIDS. Global fact sheet July 2014. Available: http://www.unaids.org/sites/default/files/en/media/unaids/contentassets/documents/factsheet/2014/20140716_FactSheet_en.pdf. Accessed November 2014.
- 3.Department of Health. Republic of South Africa. Available: www.health-e.org.za/. Accessed November 2014.
- 4.Van Harmelen JH, Van der Ryst E, Loubser AS, York D, Madurai S, Lyons S, et al. A predominantly HIV type 1 subtype C-restricted epidemic in South African urban populations. AIDS Res Hum Retroviruses 1999. March 1;15(4):395–398. [DOI] [PubMed] [Google Scholar]
- 5.Guevara H, Johnston E, Zijenah L, Tobaiwa O, Mason P, Contag C, et al. Prenatal transmission of subtype C HIV-1 in Zimbabwe: HIV-1 RNA and DNA in maternal and cord blood. J Acquir Immune Defic Syndr 2000. December 15;25(5):390–397. [DOI] [PubMed] [Google Scholar]
- 6.Bredell H, Martin DP, Van Harmelen J, Varsani A, Sheppard HW, Donovan R, et al. HIV type 1 subtype C gag and nef diversity in Southern Africa. AIDS Res Hum Retroviruses 2007. March;23(3):477–481. [DOI] [PubMed] [Google Scholar]
- 7.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 1999. January;73(1):152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tatt ID, Barlow KL, Clewley JP, Gill ON, Parry JV. Surveillance of HIV-1 subtypes among heterosexuals in England and Wales, 1997–2000. J Acquir Immune Defic Syndr 2004. August 15;36(5):1092–1099. [DOI] [PubMed] [Google Scholar]
- 9.Gräf T, Pinto AR. The increasing prevalence of HIV-1 subtype C in Southern Brazil and its dispersion through the continent. Virology 2013. 1/5;435(1):170–178. 10.1016/j.virol.2012.08.048 [DOI] [PubMed] [Google Scholar]
- 10.Alcalde R, Guimaraes ML, Duarte AJ, Casseb J. Clinical, epidemiological and molecular features of the HIV-1 subtype C and recombinant forms that are circulating in the city of Sao Paulo, Brazil. Virol J 2012. August 9;9:156-422X-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Excler JL, Robb ML, Kim JH. Prospects for a globally effective HIV-1 vaccine. Vaccine 2015. June 20. [DOI] [PubMed] [Google Scholar]
- 12.Haynes BF. New approaches to HIV vaccine development. Curr Opin Immunol 2015. June 4;35:39–47. 10.1016/j.coi.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esparza J. A New Scientific Paradigm may be Needed to Finally Develop an HIV Vaccine. Front Immunol 2015. March 18;6:124 10.3389/fimmu.2015.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 2007. January;13(1):46–53. [DOI] [PubMed] [Google Scholar]
- 15.Prendergast A, Goodliffe H, Clapson M, Cross R, Tudor-Williams G, Riddell A, et al. Gag-specific CD4+ T-cell responses are associated with virological control of paediatric HIV-1 infection. AIDS 2011. June 19;25(10):1329–1331. 10.1097/QAD.0b013e3283478575 [DOI] [PubMed] [Google Scholar]
- 16.Ranasinghe S, Flanders M, Cutler S, Soghoian DZ, Ghebremichael M, Davis I, et al. HIV-specific CD4 T cell responses to different viral proteins have discordant associations with viral load and clinical outcome. J Virol 2012. January;86(1):277–283. 10.1128/JVI.05577-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tongo M, Burgers WA. Challenges in the design of a T cell vaccine in the context of HIV-1 diversity. Viruses 2014. October 23;6(10):3968–3990. 10.3390/v6103968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann JK, Ndung'u T. HIV-1 vaccine immunogen design strategies. Virol J 2015. January 24;12:3-014-0221-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer W, Perkins S, Theiler J, Bhattacharya T, Yusim K, Funkhouser R, et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat Med 2007. January;13(1):100–106. [DOI] [PubMed] [Google Scholar]
- 20.Korber BT, Letvin NL, Haynes BF. T-cell vaccine strategies for human immunodeficiency virus, the virus with a thousand faces. J Virol 2009. September;83(17):8300–8314. 10.1128/JVI.00114-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santra S, Muldoon M, Watson S, Buzby A, Balachandran H, Carlson KR, et al. Breadth of cellular and humoral immune responses elicited in rhesus monkeys by multi-valent mosaic and consensus immunogens. Virology 2012. July 5;428(2):121–127. 10.1016/j.virol.2012.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fenimore PW, Muhammad MA, Fischer WM, Foley BT, Bakken RR, Thurmond JR, et al. Designing and testing broadly-protective filoviral vaccines optimized for cytotoxic T-lymphocyte epitope coverage. PLoS One 2012;7(10):e44769 10.1371/journal.pone.0044769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shedlock DJ, Aviles J, Talbott KT, Wong G, Wu SJ, Villarreal DO, et al. Induction of broad cytotoxic T cells by protective DNA vaccination against Marburg and Ebola. Mol Ther 2013. July;21(7):1432–1444. 10.1038/mt.2013.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goulder PJ, Sewell AK, Lalloo DG, Price DA, Whelan JA, Evans J, et al. Patterns of immunodominance in HIV-1-specific cytotoxic T lymphocyte responses in two human histocompatibility leukocyte antigens (HLA)-identical siblings with HLA-A*0201 are influenced by epitope mutation. J Exp Med 1997. April 21;185(8):1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nkolola JP, Bricault CA, Cheung A, Shields J, Perry J, Kovacs JM, et al. Characterization and immunogenicity of a novel mosaic M HIV-1 gp140 trimer. J Virol 2014. September 1;88(17):9538–9552. 10.1128/JVI.01739-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hulot SL, Korber B, Giorgi EE, Vandergrift N, Saunders KO, Balachandran H, et al. Comparison of Immunogenicity in Rhesus Macaques of Transmitted-Founder, HIV-1 Group M Consensus, and Trivalent Mosaic Envelope Vaccines Formulated as a DNA Prime, NYVAC, and Envelope Protein Boost. J Virol 2015. June 15;89(12):6462–6480. 10.1128/JVI.00383-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatherill M. Prospects for elimination of childhood tuberculosis: the role of new vaccines. Arch Dis Child 2011. September;96(9):851–856. 10.1136/adc.2011.214494 [DOI] [PubMed] [Google Scholar]
- 28.McShane H. Tuberculosis vaccines: beyond bacille Calmette-Guerin. Philos Trans R Soc Lond B Biol Sci 2011. October 12;366(1579):2782–2789. 10.1098/rstb.2011.0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ottenhoff TH, Kaufmann SH. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog 2012;8(5):e1002607 10.1371/journal.ppat.1002607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuo K, Yasutomi Y. Mycobacterium bovis Bacille Calmette-Guerin as a Vaccine Vector for Global Infectious Disease Control. Tuberc Res Treat 2011;2011:574591 10.1155/2011/574591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng YQ, Naguib YW, Dong Y, Shi YC, Bou S, Cui Z. Applications of bacillus Calmette-Guerin and recombinant bacillus Calmette-Guerin in vaccine development and tumor immunotherapy. Expert Rev Vaccines 2015;14(9):1255–1275. [PMC free article] [PubMed] [Google Scholar]
- 32.Chege GK, Burgers WA, Stutz H, Meyers AE, Chapman R, Kiravu A, et al. Robust immunity to an auxotrophic Mycobacterium bovis BCG-VLP prime-boost HIV vaccine candidate in a nonhuman primate model. J Virol 2013. May;87(9):5151–5160. 10.1128/JVI.03178-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampson SL, Dascher CC, Sambandamurthy VK, Russell RG, Jacobs WR Jr, Bloom BR, et al. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect Immun 2004. May;72(5):3031–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Im EJ, Saubi N, Virgili G, Sander C, Teoh D, Gatell JM, et al. Vaccine platform for prevention of tuberculosis and mother-to-child transmission of human immunodeficiency virus type 1 through breastfeeding. J Virol 2007. September;81(17):9408–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McAdam RA, Weisbrod TR, Martin J, Scuderi JD, Brown AM, Cirillo JD, et al. In vivo growth characteristics of leucine and methionine auxotrophic mutants of Mycobacterium bovis BCG generated by transposon mutagenesis. Infect Immun 1995. March;63(3):1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borsuk S, Mendum TA, Fagundes MQ, Michelon M, Cunha CW, McFadden J, et al. Auxotrophic complementation as a selectable marker for stable expression of foreign antigens in Mycobacterium bovis BCG. Tuberculosis (Edinb) 2007. November;87(6):474–480. [DOI] [PubMed] [Google Scholar]
- 37.Chapman R, Shephard E, Stutz H, Douglass N, Sambandamurthy V, Garcia I, et al. Priming with a recombinant pantothenate auxotroph of Mycobacterium bovis BCG and boosting with MVA elicits HIV-1 Gag specific CD8+ T cells. PLoS One 2012;7(3):e32769 10.1371/journal.pone.0032769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, et al. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med 2002. October;8(10):1171–1174. [DOI] [PubMed] [Google Scholar]
- 39.Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 1998. May 10;244(2):365–396. [DOI] [PubMed] [Google Scholar]
- 40.Price PJ, Torres-Dominguez LE, Brandmuller C, Sutter G, Lehmann MH. Modified Vaccinia virus Ankara: innate immune activation and induction of cellular signalling. Vaccine 2013. September 6;31(39):4231–4234. 10.1016/j.vaccine.2013.03.017 [DOI] [PubMed] [Google Scholar]
- 41.Mercer AA, Schmidt A, Weber O editors. Poxviruses.; 2007.
- 42.Carroll MW, Moss B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 1997. November 24;238(2):198–211. [DOI] [PubMed] [Google Scholar]
- 43.Gheorghiu M, Lagrange PH, Fillastre C. The stability and immunogenicity of a dispersed-grown freeze-dried Pasteur BCG vaccine. J Biol Stand 1988. January;16(1):15–26. [DOI] [PubMed] [Google Scholar]
- 44.Parish T, Stolker NG editors. Mycobacteria protocols: Springer protocols; 2001. [Google Scholar]
- 45.Burgers WA, Shephard E, Monroe JE, Greenhalgh T, Binder A, Hurter E, et al. Construction, characterization, and immunogenicity of a multigene modified vaccinia Ankara (MVA) vaccine based on HIV type 1 subtype C. AIDS Res Hum Retroviruses 2008. February;24(2):195–206. 10.1089/aid.2007.0205 [DOI] [PubMed] [Google Scholar]
- 46.Y. J. Shen. An investigation into the use of lumpy skin disease virus as a vaccine vector for a potential HIV-1 vaccineUniversity of Cape Town; 2010.
- 47.Burgers WA, van Harmelen JH, Shephard E, Adams C, Mgwebi T, Bourn W, et al. Design and preclinical evaluation of a multigene human immunodeficiency virus type 1 subtype C DNA vaccine for clinical trial. Journal of General Virology 2006;87(2):399–410. [DOI] [PubMed] [Google Scholar]
- 48.Columb MO, Sgadai S. Multiple comparisons. Current Anaesthesia & Critical Care 2006;17:233–236. [Google Scholar]
- 49.Chapman R, Chege G, Shephard E, Stutz H, Williamson AL. Recombinant Mycobacterium bovis BCG as an HIV vaccine vector. Curr HIV Res 2010. June;8(4):282–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santra S, Liao HX, Zhang R, Muldoon M, Watson S, Fischer W, et al. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat Med 2010. March;16(3):324–328. 10.1038/nm.2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barouch DH, O'Brien KL, Simmons NL, King SL, Abbink P, Maxfield LF, et al. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat Med 2010. March;16(3):319–323. 10.1038/nm.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong WP, Wu L, Wallstrom TC, Fischer W, Yang ZY, Ko SY, et al. Expanded breadth of the T-cell response to mosaic human immunodeficiency virus type 1 envelope DNA vaccination. J Virol 2009. March;83(5):2201–2215. 10.1128/JVI.02256-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy K, P. editor. Janeway's Immunobiology.; 2011.
- 54.Wyatt LS, Earl PL, Xiao W, Americo JL, Cotter CA, Vogt J, et al. Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J Virol 2009. July;83(14):7176–7184. 10.1128/JVI.00687-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong YC, Lin LC, Melo-Silva CR, Smith SA, Tscharke DC. Engineering recombinant poxviruses using a compact GFP-blasticidin resistance fusion gene for selection. J Virol Methods 2011. January;171(1):295–298. 10.1016/j.jviromet.2010.11.003 [DOI] [PubMed] [Google Scholar]
- 56.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 2009. March;15(3):293–299. 10.1038/nm.1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011. May 26;473(7348):523–527. 10.1038/nature10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen SG, Piatak M Jr, Ventura AB, Hughes CM, Gilbride RM, Ford JC, et al. Immune clearance of highly pathogenic SIV infection. Nature 2013. October 3;502(7469):100–104. 10.1038/nature12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saubi N, Mbewe-Mvula A, Gea-Mallorqui E, Rosario M, Gatell JM, Hanke T, et al. Pre-clinical development of BCG.HIVA(CAT), an antibiotic-free selection strain, for HIV-TB pediatric vaccine vectored by lysine auxotroph of BCG. PLoS One 2012;7(8):e42559 10.1371/journal.pone.0042559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chapman R, Stutz H, Jacobs W Jr, Shephard E, Williamson AL. Priming with recombinant auxotrophic BCG expressing HIV-1 Gag, RT and Gp120 and boosting with recombinant MVA induces a robust T cell response in mice. PLoS One 2013. August 20;8(8):e71601 10.1371/journal.pone.0071601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chapman R, Bourn WR, Shephard E, Stutz H, Douglass N, Mgwebi T, et al. The use of directed evolution to create a stable and immunogenic recombinant BCG expressing a modified HIV-1 Gag antigen. PLoS One 2014. July 25;9(7):e103314 10.1371/journal.pone.0103314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Faassen H, Dudani R, Krishnan L, Sad S. Prolonged antigen presentation, APC-, and CD8+ T cell turnover during mycobacterial infection: comparison with Listeria monocytogenes. J Immunol 2004. March 15;172(6):3491–3500. [DOI] [PubMed] [Google Scholar]
- 63.van Faassen H, Saldanha M, Gilbertson D, Dudani R, Krishnan L, Sad S. Reducing the stimulation of CD8+ T cells during infection with intracellular bacteria promotes differentiation primarily into a central (CD62LhighCD44high) subset. J Immunol 2005. May 1;174(9):5341–5350. [DOI] [PubMed] [Google Scholar]
- 64.Hess J, Schaible U, Raupach B, Kaufmann SH. Exploiting the immune system: toward new vaccines against intracellular bacteria. Adv Immunol 2000;75:1–88. [DOI] [PubMed] [Google Scholar]
- 65.Al-Zarouni M, Dale JW. Expression of foreign genes in Mycobacterium bovis BCG strains using different promoters reveals instability of the hsp60 promoter for expression of foreign genes in Mycobacterium bovis BCG strains. Tuberculosis (Edinb) 2002;82(6):283–291. [DOI] [PubMed] [Google Scholar]
- 66.Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol 2011. January;85(2):733–741. 10.1128/JVI.02030-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johansen P, Stamou P, Tascon RE, Lowrie DB, Stockinger B. CD4 T cells guarantee optimal competitive fitness of CD8 memory T cells. Eur J Immunol 2004. January;34(1):91–97. [DOI] [PubMed] [Google Scholar]
- 68.Kemball CC, Pack CD, Guay HM, Li ZN, Steinhauer DA, Szomolanyi-Tsuda E, et al. The antiviral CD8+ T cell response is differentially dependent on CD4+ T cell help over the course of persistent infection. J Immunol 2007. July 15;179(2):1113–1121. [DOI] [PubMed] [Google Scholar]
- 69.Ramsburg EA, Publicover JM, Coppock D, Rose JK. Requirement for CD4 T cell help in maintenance of memory CD8 T cell responses is epitope dependent. J Immunol 2007. May 15;178(10):6350–6358. [DOI] [PubMed] [Google Scholar]
- 70.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med 2006. March;12(3):289–295. [DOI] [PubMed] [Google Scholar]
- 71.Someya K, Xin KQ, Matsuo K, Okuda K, Yamamoto N, Honda M. A consecutive priming-boosting vaccination of mice with simian immunodeficiency virus (SIV) gag/pol DNA and recombinant vaccinia virus strain DIs elicits effective anti-SIV immunity. J Virol 2004. September;78(18):9842–9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Betts MR, Exley B, Price DA, Bansal A, Camacho ZT, Teaberry V, et al. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc Natl Acad Sci U S A 2005. March 22;102(12):4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 2006. June 15;107(12):4781–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zimmerli SC, Harari A, Cellerai C, Vallelian F, Bart PA, Pantaleo G. HIV-1-specific IFN-gamma/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc Natl Acad Sci U S A 2005. May 17;102(20):7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, et al. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog 2010. May 27;6(5):e1000917 10.1371/journal.ppat.1000917 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.