

Abstract
The x-ray structure of LeuT, a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporter, provides a great opportunity to better understand the molecular basis of monovalent cation selectivity in ion-coupled transporters. LeuT possesses two ion-binding sites, NA1 and NA2, which are highly selective for Na+. Extensive all-atom free energy molecular dynamics simulations of LeuT embedded in an explicit membrane are performed at different temperatures and various occupancy states of the binding sites to dissect the molecular mechanism of ion selectivity. The results show that the two binding sites display robust selectivity for Na+ over K+ or Li+, the competing ions of most similar radii. Of particular interest, the mechanism primarily responsible for selectivity for each of the two binding sites appears to be different. In site NA1, selectivity for Na+ over K+ arises predominantly from the strong electrostatic field arising from the negatively charged carboxylate group of the leucine substrate coordinating the ion directly. In site NA2, which comprises only neutral ligands, selectivity for Na+ is enforced by the local structural restraints arising from the hydrogen-bonding network and the covalent connectivity of the poly-peptide chain surrounding the ion according to a snug-fit mechanism.
INTRODUCTION
In the central nervous system, ion-coupled membrane transporters regulate signaling by the transport of neurotransmitter molecules (glycine, serotonin, dopamine and many others) from the synapse back to the synaptic neuron, thus terminating the the synaptic transmission. More generally, these transporters belong to a highly-specialized class of membrane proteins that utilize the electro-chemical potential of specific ions to pump organic substrates and amino acids (glycine, leucine, aspartate, etc…) as well as biogenic amines (serotonine, dopamine) across the cell membrane.1–3 Many of these proteins were discovered decades ago,3–5 and substantial progress has been made towards the characterization of their transport properties and functional activities.2 The determination of the x-ray crystallographic structure of LeuT (at 1.65 Å resolution), a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporter, gives a fresh impetus to efforts directed at better understanding the molecular basis of ion-coupled transporters.6 Prior to the LeuT structure, it was not possible to understand the mechanism of ion-substrate co-transport at the molecular level.
The x-ray structure shows that one protomer of LeuT is composed of 12 transmembrane helical regions, TM1 to TM12. The TM1 and TM6 helices maintain the highest density of conserved residues among entire NSS family and form the majority of the LeuT core. LeuT possesses two highly selective Na+ binding sites, NA1 and NA2, that are well-resolved in the x-ray structure. Both the TM1 and TM6 helices display breaks in the helical structure around the middle of lipid bilayer, resulting in the exposure of carbonyl backbone oxygens that are essential for the formation of the Na+ binding sites and substrate coordination.6 The Na+ ion in site NA1 is coordinated by 6 ligands, while the ion in site NA2 is coordinated by 5 ligands. The helical break resulting in the formation of the neutral site NA2 is reminiscent of the GxxxG helix-break-helix motif reported for other membrane proteins.7 Two residues (G20 and V23) from the helix-break-helix motif in the LeuT transporter formed by the G20XXXG24 sequence in the TM1 helix are involved in the Na+ coordination in the binding site NA2 and are conserved among most members of the NSS family. (G20 is conserved in 98% of reported sequences).8 Additional residues from TM3 and TM8 also contribute to form the binding sites for ions and substrate. The two binding sites are directly coupled because the two Na+ are coordinated by main-chain carbonyl oxygens from Ala22 and Val23, respectively. Interestingly, the crystal structure of LeuT revealed that the leucine provides a negatively-charged carboxylate group coordinating the Na+ in site NA1, thus revealing a direct interaction between the leucine substrate and one of the two bound Na+ ions. Examination of the hydrogen bonding network in vicinity of the binding pocket suggests that that the leucine substrate is in the zwitterionic form, a conclusion that is also supported by free energy simulations.9
The existence of two Na+-selective binding sites—both structurally coupled to the transported leucine substrate—is a distinctive structural feature of LeuT revealed by the x-ray structure. A recent study by Beuming et al.8 points to the functional importance of the two binding sites and suggests that LeuT represents a reliable template for future studies of the neurotransmitter transport in eukaryotes that can be generalized to the broader number of proteins from the NSS-family. Sequence alignments analysis between different transporters from the NSS family indicates that 4 out of 5 residues providing coordination for the site NA2 are preserved in the eukaryotic Na+–coupled transporters. Interestingly, side-chains from Thr354 and Ser355 involved in the coordination of the Na+ ion in the NA2 binding site are also well conserved. For instance, Thr354 of the LeuT transporter is conserved in 77% of NSS members and replaced by Ser in TnaT and TyT1 transporters.8 It was proposed that binding and dissociation of ions and subtrate to the helix-break-helix motif catalyzes conformational changes during transport cycle.10
Strict selectivity of the two binding sites is essential to realize the strong coupling between the transport of the leucine substrate and the Na+ influx.6;11 For example, loss of selectivity for Na+ over K+, which is abundant inside the cell, could result in the substrate being pumped outward. The underlying mechanism responsible for selectivity is thus of great biological interest. Upon determination of the crystal structure,6 it was proposed that the binding sites are precisely adapted to Na+, and are structurally constrained by the protein structure from expanding or contracting to accommodate an ion of a different size. Such a structural explanation of selectivity draws naturally from the ion-ligand distances observed in high-resolution crystal structures. While the concept of a precise snug-fit may capture the dominant factor controlling selectivity in the case of fairly rigid chelating agents, the conditions required for supporting this mechanism in the case of flexible protein structures needs to be assessed quantitatively, on a case-by-case basis. As the atomic radius of Na+ and K+ differs only by 0.38 Å, a purely structural view of selectivity relies on the ability of the binding sites to retain their geometry with sub-angstrom precision. Selectivity in LeuT appears to be, nonetheless, particularly robust. For example, measurements reported by Gouaux and co-workers6;12 indicate that the transporter displays high selectivity—around 1000 folds—for Na+ over K+,12 even at elevated temperatures (90–95°C). This is significantly more selective than Na+- or K+-dependent soluble enzymes (around 10–100 folds).13; 14 At such high temperature, the atomic thermal fluctuations of most proteins functioning at ambient conditions would be significantly larger than the size difference between Na+ and K+. However, it is important to recall that LeuT was isolated from Aquifex aeolicus, a thermophilic bacterial organism living around 90–95°C, hence it may correspond to a three-dimensional structure of particularly high stability. In addition, one may note that strict size-selectivity can sometimes be circumvented, as shown by the recent crystal structure of the Na+-coupled aspartate transporter GltPh,11 where the larger Tl+ cations were observed occupying the Na+-selective binding sites. This suggests that the structural forces preventing the binding sites from expanding to accommodate a larger cation may not be always a critical factor in the formation of selective binding sites.11 With the availability of a high resolution x-ray structure, LeuT represents an ideal testing ground for computational studies of monovalent ion selectivity in membrane co-transporters.
To examine the fundamental basis for the cation selectivity in LeuT, we used all-atom free energy perturbation (FEP) molecular dynamics (MD) simulations. The simulations are carried out at three different temperatures and various states of occupancy of the binding sites are considered. On the basis of those FEP/MD computations, the role of the substrate in the formation of the binding site and in the coupling between the two binding sites is clarified and an analysis of the relative enthalpic and entropic contributions to ion selectivity is presented. To broaden the present analysis, the FEP/MD computations are complemented by a survey of the coordination structure of bound Na+ and K+ from an ensemble of proteins taken from the Protein Data Base (PDB). The results are discussed in the context of previous studies of the KcsA15–18 and NaK19; 20 channels. It is concluded that selectivity of the binding site NA1 for Na+ is primarily governed by the electrostatic nature of the coordinating ligands (according to a “field-strength” mechanism), while the selectivity of the site NA2 is explained by a the local structure (according to a “snug-fit” mechanism). A unified view of the microscopic factors governing Na+ selectivity in biological channels and transporters emerges from the FEP/MD computations and the survey of the PDB. The inclusion of a negatively charged “high-field” ligand is a key factor for the emergence of robust selectivity for Na+. But binding sites comprising only neutral ligands can also display Na+ selectivity, typically by exploiting the local stiffness arising from the covalent connectivity along the poly-peptide chain according to a snug-fit mechanism.
RESULTS
a) All-atom FEP simulations of Na+/K+ selectivity in LeuT
We first carry out a series of FEP/MD simulations based on an all-atom model of the LeuT embedded into an explicit solvated lipid membrane. In these simulations, no constraints are applied to the system and all the atoms of the protein, lipid membrane, and solvent are allowed to freely fluctuate. FEP/MD calculations are performed for the two binding sites NA1 and NA2. Their local structure is displayed in Figure 2. The binding site NA1 of LeuT comprises 6 ligands, including the the carbonyl oxygens (O) from Ala22, Thr254, the hydroxyl oxygen (Oγ) from Thr254, side chain amide oxygens Oδ from Asn27 and Asn286 as well as a negatively charged carboxylate group from the leucine substrate (Figure 2A). The binding site NA2 comprises 5 ligands, including three main chain carbonyl oxygens (O) from G20, V23 and A351 and hydroxyl oxygens (Oγ) from T354 and S355. (Figure 2B). The two binding sites are organized into the binding pocket (Figure 2C), which plays an important role in the substrate binding to LeuT.9
Figure 2.
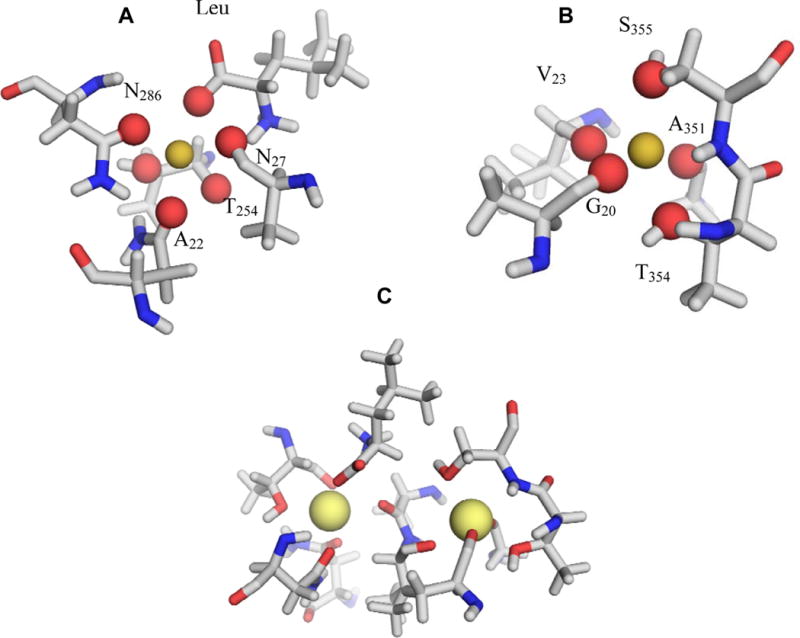
Binding site organization in the LeuT. (A) Site NA1: TM1: O—Ala22, Oδ—Asn27, TM6: O—Thr254, Oγ—Thr254, TM7: Oδ—Asn286, OT1—L-Leu. (B) Site NA2: TM1: O—Gly20, O—Val23, TM8: O—Ala351, Oγ—Thr354, Oδ—Ser355. (C) Organization of the double NA1-NA2 binding site.
The results of FEP/MD simulations carried out for the sites NA1 and NA2 are reported in Table 1. These results provide the most realistic representation of the system that is available computationally and, thus, play an important role in the validation of the computational methodology. Based on previous computations17; 19; 21 with different crystal structures of the KcsA15; 16 and the NaK channels,20 the variations between the CHARMM PARAM2722 and AMBER23 force fields19 indicate that the accuracy and overall significance of the calculated free energy differences is roughly on the order of 1.0 to 1.5 kcal/mol. We performed block-average computations of the selectivity free energies for the Na+/K+ selectivity of the LeuTAa transporter as function of different temperatures. The computed relative free energies are accurate on the order of 0.5–0.8 kcal/mol consistent with previously reported estimates. Though different force-fields provides slightly different estimates for the selectivity, previous studies indicate that the computed relative free energies are quite robust for all major force fields.18; 19
Table 1.
Na+/K+ and Li+/Na+ selectivity (kcal/mol)
| Temperature | Na+/K+ | Li+/Na+ | ||
|---|---|---|---|---|
| Site NA1 | Site NA2 | Site NA1 | Site NA2 | |
| 315 K | 6.11 | 3.22 | −0.86 | 1.51 |
| 350 K | 4.84 | 2.40 | −0.80 | 1.48 |
| 369 K | 3.87 | 2.28 | 0.83 | 0.66 |
Reference bulk water values for this set of potential parameters are: At T=315K, ΔGbulk(K+ to Na+) =18.12 kcal/mol and ΔG bulk(Li+ to Na+) = 24.17 kcal/mol; at T=350K, ΔGbulk(K+ to Na+) = 18.05 kcal/mol and ΔGK bulk(Li+ to Na+) = 23.86 kcal/mol; and at T=369K, ΔGbulk(K+ to Na+) = 17.71 kcal/mol and ΔG bulk(Li+ to Na+) = 23.64 kcal/mol. By convention, a positive sign in the Table 1 reflects a binding site preference for Na+ over X+ (K+ or Li+). The bulk solvation free energies of ions at different temperatures were evaluated with free energy simulations using the following scheme: Box of 500 water molecules (TIP3P64) was equilibrated for 5 ns at a given T to follow up with free energy simulations of the ion charging/uncharging. We have used 11 perturbation windows run forward and backward with the length of 1 ns each. The resulting free energies were determined with WHAM analysis.66 See supplementary materials for the discussions of the ion parameters used in the present work.
The calculated relative free energies for Na+ relative to K+ in the two binding sites compare reasonably well with the trends expected from experimental data.6; 11 The free energy for Na+ relative to K+ is 5–6 kcal/mol in the case of site NA1, which comprises 6 ligands including the negatively charged carboxylate group of the leucine substrate, and it is 3–4 kcal/mol in the case of site NA2, which comprises only 5 neutral ligands. The theoretical data is in excellent accord with the communicated experimental data on Na+/K+ selectivity of LeuT (several thousands folds difference in the free energy of Na+ and K+ binding to LeuT).12
b) All-atom FEP simulations of Li+/Na+ selectivity
The results of free energy simulations on Li+/Na+ selectivity are summarized in the Table 1. Within statistical error (±1 kcal/mol), there is no selectivity for Na+ over Li+ in the site NA1. The presence of a negatively charged ligand in site NA1 does not confer selectivity for Na+ over the smaller Li+ ion. The site NA1 in LeuT provides six oxygens in octahedral arrangement to solvate either Na+ or Li+. In the simulations, the oxygen atoms from the carbonyl, carboxylate, and hydroxyl groups of the site NA1 collapse onto the smaller Li+. The computed thermal fluctuations for the oxygen atoms surrounding the ions are within the range of 0.6 to 0.9 Å RMS (depending on the temperature), which is larger than the size difference between Na+ and K+ or Na+ and Li+.
c) Simulations of the effects of ion-substrate coupling on the Na+/K+ selectivity of LeuT
A number of computational experiments consisting of free energy simulations in the presence or absence of the leucine substrate for different states of occupancy of the binding sites NA1 and NA2 were performed to examine the coupling between the 2 binding sites and with the transported substrate. The results are given in Table 2. In the absence of the negative charge from the substrate’s carboxylate group, the selectivity of the site NA1 for Na+ decreases substantially. Subsequent removal of the ion in the neighboring binding site NA2 essentially annihilates the selectivity of site NA1 for Na+. These results indicate that the presence of the negative (carboxylate) charge in the coordinating shell of Na+ cation contributes significantly to yield a selective site NA1. In contrast, the selectivity in the site NA2 is only slightly affected by the presence or absence of the leucine substrate, or the ion in the neighboring NA1 site. This suggests that the dominant mechanism conferring selectivity to site NA2 arise from the optimal configuration of the ligands, enforced via the local structure. The relative independence of the selectivity of the site NA2 on the presence of the leucine substrate and/or the ion in the neighboring binding site NA1 indicates that this site can exist independently. This suggests that the site NA2 with its bound Na+ may have primarily a structural role in stabilizing the binding pocket region for the leucine substrate and the ion in site NA1.
Table 2.
Effect of the substrate and binding sites occupancies on Na+/K+ selectivity
| Binding pocket composition* | Site NA1 | Site NA2 |
|---|---|---|
| Leu••NA1••NA2 | 3.87 | 2.28 |
| NA1••NA2 | 1.28 | 1.46 |
| Leu••NA1 | 1.96 | – |
| Leu••NA2 | – | 1.30 |
| NA1 | 0.57 | |
| NA2 | – | 1.36 |
Occupancy of the binding pocket in LeuT: Leucine substrate, and the Na+ ions in the binding site NA1 and NA2. All FEP/MD simulations were done at T=369 K according to the methodology described in Methods section. All setups with and without substrates were subjected to 4 ns equilibration run prior to FEP/MD computations. All units are kcal/mol. A positive number implies selectivity for Na+ over K+.
d) Computational experiments with partially constrained models
In Table 3 are given the results of additional “computational experiments” in which FEP/MD simulations similar to those described above were carried out for a partially or completely frozen protein with K+–adapted binding sites. While those partially restrained models are somewhat artificial, the computational experiments are helpful to ascertain the importance of the local geometry and dissect the underlying mechanism of ion selectivity in LeuT. The latter were produced by optimizing the x-ray structure using energy minimization after replacing the Na+ by a K+ ion in each of the binding sites. To produce a K+–adapted site, the surrounding structure has to expand slightly to accommodate the larger ion (though the atomic displacements needed to achieve this are very small). The FEP/MD simulations were then repeated using the same methodology as used in Table 1. When the entire protein is kept frozen in this K+–adapted protein configuration, then the two binding sites are essentially non-selective (2nd row of Table 3). The sites almost fully recover their selectivity for Na+ when only the nearest atoms around the ions (within 3.5 Å) are allowed to freely fluctuate while all the remaining protein atoms are kept frozen in the K+–adapted configuration (3rd row of Table 3).
Table 3.
Na+/K+ selectivity in altered binding sites (kcal/mol)
| Site NA1 | Site NA2 | |
|---|---|---|
| Fully Flexible Proteina | 6.11 | 3.22 |
| Frozen protein in a K+ adapted conformationb | −0.11 | 0.81 |
| Partially frozen protein in a K+ adapted conformationc | 4.27 | 2.11 |
No constrains were applied on the system during molecular dynamics simulation (T=315 K).
Na+ ions were replaced by potassium in binding sites followed by energy minimization to insure optimal K+ coordination in binding sites NA1 and NA2, minimized system was fixed during free energy simulations.
Same as b, but ligands in the first coordination shell of the ion were free to move.
e) Results of PDB survey for Na+ and K+ binding sites
The main results of the statistical survey are summarized in Table 4. The overarching aim of the PDB survey was to characterize the nature of ion coordination at protein sites where a K+ or a Na+ is bound. An important caveat in interpreting the results of such a survey is that observation of an x-ray structure with a bound Na+ or K+ does not imply that a particular binding site is actually selective for the bound ion, though it does mean that the ion binds with sufficient affinity to be observed in the electronic density. In a number of cases, multiple structures with Na+ or K+ bound at a particular are present in the protein database.24 This is consistent with the fact that some enzymes are functionally active with either cation, suggesting that relatively non-specific metal-protein interactions may primarily have a structural role in those cases. For many proteins such as thrombin, the reported measurements are indicative of relatively modest Na+/K+ selectivity (10–100 folds).14 In spite of these limitations, the survey provides valuable information on the various molecular contexts able to bind K+ or Na+ in a stable fashion.
Table 4.
Summary of the PDB survey results for Na+ and K+ binding sites in proteins
| Na+ sites | K+ sites | |
|---|---|---|
| Nstructures | 132 | 61 |
| Average Ion-Ligand distance | 2.35 ± 0.15 Å | 2.77 ± 0.25 Å |
| Maximum coordination number (Nmax) | 7 | 9 |
| Minimum coordination number (Nmin) | 3 | 4 |
| Average coordination number (Nave) | ~5 | ~6 |
| Nave in neutral binding sites | ~4.4 | ~5.3 |
| Nave in charged binding sites | ~5.4 | ~6.1 |
| Number of charged ligands per binding site | ~1.12 | ~0.7 |
| Fraction of structures with charged ligands | 0.50 | 0.41 |
| Number of coordinating water molecule per site | 1.17 | 1.01 |
| Fraction of structures with water coordination | 0.67 | 0.59 |
According to the statistical analysis of the database, the average ion-ligand distances are 2.35 ± 0.15 Å and 2.79 ± 0.25 Å for Na+ and K+, respectively. Approximately 50% of Na+ binding sites comprise at least one or more negatively charged ligand, as observed for site NA1 of LeuT. Only 41% of analyzed K+ binding sites contains one or more charged ligands. The average number of charges in the coordination sphere of Na+ is 1.16 compared to 0.7 for K+. This observation is broadly consistent with the classical concepts introduced by Eisenman,25 predicting that binding sites with a high field strength ligand tend to be selective for Na+ over K+.18;19 In good agreement with previous surveys of the PDB,24;26–29 the coordination number of Na+ is ~4.89 ± 0.95 on average, close to the estimated coordination number of Na+ in bulk water.30 Similarly, the coordination number of K+ is 5.65± 1.15 on average (6 as reported by Page et al.24). However, the coordination number may vary from 3 up to 7 for Na+, and from 4 up to 9 K+. For an extensive discussion of the particular coordination geometry of Na+ and K+ binding sites, see exhaustive review by Page and Di Cera.24
DISCUSSION
a) Na+/K+ selectivity and functional implications
According to all-atom FEP/MD simulations, the site NA1 comprising a charged ligand is the most selective site of LeuT. By comparison, the site NA2 comprising only neutral ligands is moderately selective for Na+ over the larger K+. Both binding sites remain selective for Na+ over K+ at elevated temperatures (T~369 K), though the site NA1 remaining more selective than site NA2. Intuitively, such robust selectivity should confer a clear advantage in the case of a transporter that must undergo relatively large conformational changes—on the order of 6–10 Å according to experiments11—to perform its normal function. The selective binding of a Na+ ion coordinated by the negatively charged carboxylate group from the transported leucine substrate is a key feature of LeuT. The latter is especially important for this transporter protein expressed from the thermophile organism living at a temperature of ~350 K. As described previously,19 coordination of the ion by a negatively charged ligand, as realized in site NA1, provides the most effective and robust mechanism to discriminates for Na+ over K+. The high-field ligand favors the smaller ion and provides a strong ion-protein interaction serving to offset the dehydration cost.
Two Na+ are bound in the X-ray structure of LeuT, although the true stoichiometry for the Na+ and substrate co-transport function is not known.31 The fact that one negatively charged coordinating ligand in site NA1 is actually provided by the transported substrate itself suggests a tight coupling, and perhaps cooperativity in Na+ and leucine binding. More generally, the binding stoichiometry of ions and substrate in NSS transporters and whether 1 or 2 ion are absolutely required for substrate transport has become a controversial issue.8; 31; 32 Some transporters in the family such as GAT-133 and GlyT1b34 are known to transport 2 Na+ ions per each substrate molecule. Similarly, Na+ stoichiometry for the homologous transporter hSERT has been determined to be one Na+ ion per every substrate molecule (5-HT or serotonin).32 To address issues of coupling and cooperativity, additional FEP/MD simulations with and without leucine substrate and Na+ were carried out (Table 2). Selectivity of the site NA1 is reduced from 3.87 kcal/mol to 2.41 kcal/mol in the absence of an ion in NA2 (this value is similar to that of a fully flexible reduced model, see below). The presence of a Na+ cation in NA2 thus serves to enhance the selectivity of NA1. More dramatically, the free energy decreases to 0.57 kcal/mol in the absence of substrate. Selectivity of the site NA1 is abolished without the carboxylate ligands provided by the leucine substrate.
These results suggest a plausible mechanistic scenario: Na+ ion binds first to the site NA2, where it structurally stabilizes the helix-break-helix motif. Subsequently, a second Na+ ion would either bind to NA1, or would translocate into the site while being associated with the leucine substrate. This scenario provides a possible explanation for the different stoichiometries observed in radiolabeled uptake experiments31; 35 and electrophysiology.36; 37 Perhaps, the two-ion motif is required to assure selective binding to the site NA1, but the actual transport of the amino acid relies only on a single cation.
b) Li+ binding to LeuT
Interestingly, while the sites NA1 and NA2 display strong selectivity for Na+ over the larger K+ ion, such size specificity is not reciprocated for the smaller Li+ ion (see Table 1). The site NA2 is slightly more selective for Na+ over the smaller Li+ ion than the site NA1, but the selectivity is relatively modest. This may be rationalized from a biological standpoint. Selectivity for Na+ over the abundant K+ ion is required for the normal biological function of the transporter, but a high selectivity for Na+ over Li+, which is present only to very small concentration under normal physiological conditions, is not essential for the function. Nonetheless, understanding the mechanism for the relative affinity of the binding site for Na+ and Li+ is of great importance for the pharmacology of secondary amino-acid transporters. It was reported that use of Li+-based pharmaceuticals inhibit glutamate uptake in mice and monkeys,38 so that it can be used as an antimanic agent and treatment for depressive phases of bipolar disorder.
Our simulations indicates that Li+ ion interacts more strongly with the ligand of the site NA1 than the larger Na+ ion, though the gain in favorable interaction is counterbalanced by the large difference in the dehydration energies. Distortions of the binding site upon Li+ binding resulting in steric penalties further reduce the favorable gain ion-protein interaction energy. Such effect is magnified in the neutral binding site NA2 lacking the presence of the negatively charged high-field ligand. The modest Na+/Li+ selectivity displayed by the FEP/MD simulations is consistent with experiments indicating that Li+, but not K+, supports ligand uptake in the absence of Na+6 in Na+-coupled amino-acid transporters. For example, reported Kd of substrate-to-transporter binding for Glt measured in presence of Li+ are about 100-fold higher than those measured in presence of Na+.11 Similar data were obtained for transporters from the NSS family homologous to LeuT (NET, DAT39 and the hSERT transporters40). A recent crystal structure of the aspartate transporter Glt suggests that Na+ can be substituted by Li+ in the site analogous to NA1 of LeuT.9 Recent experimental studies of the flux/current ratio in the EAAC1 transporter as function of the substitute cation41 lend additional support for the possibility of Li+ binding to the Na+-binding site in Na+-selective secondary transporters. All these observations together with moderate Na+/Li+ selectivity of LeuT in the computations led us to the conclusion that Li+ can bind to the site NA1 without inducing a major conformational change.
c) Mechanism of ion selectivity of the sites NA1 and NA2
The results of the all-atom FEP/MD computations are broadly in accord with the trends expected from experiments. This means that, though the computational methodology could be improved, it is sufficiently accurate in its present state to yield meaningful insight into the mechanism controlling the selectivity of the Na+ binding sites in a NSS protein. In the following, we turn to the task of examining the outcome of a number of computer experiments, each designed to highlight an aspect of the mechanism yielding selectivity.
The results of FEP/MD simulations from partially constrained all-atom models of LeuT with K+–adapted binding sites, shown in Table 3, suggest that selectivity depends sensitively on some local property of the binding sites rather than some long-range cooperative feature of the entire structure. For instance, when the entire protein is kept frozen in this K+–adapted protein configuration, the two binding sites display only a slight selectivity. It is noteworthy that such partially constrained model does not yield binding sites that are selective for K+, despite the perfect K+–adapted frozen geometry. As a comparison, a similar experiment performed under the same conditions with the selectivity filter of the KcsA channel yielded binding sites that were highly favorable for K+ (about 9.7 kcal/mol for the site S2 in the selectivity filter).17 Remarkably, the sites almost fully recover their selectivity for Na+ when only the nearest atoms around the ions (within 3.5 Å) are allowed to freely fluctuate while all the remaining protein atoms are kept frozen in the K+–adapted configuration (3rd row of Table 3).
An alternative and useful strategy in seeking to further highlight the dominant microscopic factors responsible for the selectivity of the two binding sites and complement the all-atom FEP/MD computations is to construct reduced simulation systems (minimal “toy models”) that are able to retain some of the properties of the complete system. Such a strategy previously helped clarify some of the basic principles controlling the K+/Na+ selectivity in the KcsA and NaK channels.18; 19 The minimal models used here are illustrated in Figure 3A and 3B, together with the results of FEP/MD simulations. Obviously, these reduced models are exceedingly simplified caricatures of a complex reality, and are intended to illustrate the role of local forces on ion selectivity in site NA1 and NA2. First, we constructed models of NA1 and NA2 including only the functional groups in the first coordination shell of the ion (Figure 3A and 3B). The reduced model of NA1 is composed of 18 atoms formed by the carbonyl group of Ala22, side-chain amide groups providing the oxygens Oδ from Asn27 and Asn286 and the whole Thr254 residue and charged carboxylate group donated by the substrate (Figure 3A). The reduced model of NA2 includes 30 atoms from the covalently bound motif T354-S355 that provides two side-chain hydroxyls to coordinate the ion, main chain carbonyl groups of G20, V23 and A351. In the reduced models, these functional groups are allowed to fluctuate freely by confining only the oxygen atoms of the ligands within a sphere of 3.5 Å radius using a flat-bottom harmonic restraining potential. This simple restraint is meant to represents the confinement of the binding pocket enforced by the protein fold, without assuming detailed structural features arising from the configuration of the ligands.
Figure 3.

Simplified models considered for analysis. (A and B). Minimal model of the binding site NA1 (A) and NA2 (B), where all ligands are allowed to move freely within a sphere of 3.5 Å radius (indicated by dashed line). The calculated free energies ΔΔG is 2.41 kcal/mol for NA1 (A) and −0.3 kcal/mol for NA2 (B). (C) Na+-selective toy-model of the binding site NA2 described in the text. The calculated free energy is 1.49 kcal/mol when all functional groups within the 3.5 radii are allowed to fluctuate freely, while the outside groups were frozen.
The fully flexible toy model representing the site NA1 spontaneously displays a selectivity for Na+ over K+ of 2.41 kcal/mol, even in the absence of any precise structural restraint (other than the featureless confinement of the ligands). The calculated ΔΔG is comparable to that from all-atom simulations (3.87 kcal/mol, Table 1). This result is entirely expected according to the classical concept of field strength, originally introduced by Eisenman.25 In contrast, the reduced model for site NA2 yields only a Na+/K+ selectivity of −0.30 kcal/mol, and thus fails to reproduce the results observed in the FEP/MD simulations of the complete system (2.28 kcal/mol, Table 1). Such failure to recover Na+ selectivity for the reduced model of site NA2 implies that the interplay of the freely fluctuating ion-ligand and ligand-ligand interactions confined in a small region is not enough to reproduce the correct behavior without further structural information communicated by the protein. From this point of view, the ligand composition of site NA2 (3 carbonyl and 2 hydroxyl) is essentially nonselective; i.e., it does not favor nor disfavor Na+ or K+. Parenthetically, this computer experiment also demonstrates that the number of coordinating ligands in NA2, which matches more closely the hydration number of Na+ in bulk water than that of K+, is by itself unable to generate Na+ selectivity. This observation appears to be incompatible with recent suggestions that selectivity of protein binding sites could be topologically controlled by the coordination numbers.42 Therefore, some additional ingredient must be involved to recover the selectivity for Na+ over K+ that is observed in the all-atom simulations.
The most obvious ingredient that is left out from the simplified toy model of site NA2 corresponds to the structural forces constraining the ligands in a configuration to optimally coordinate Na+ but not K+. Such structural forces could arise, as proposed in the context of ion channels,20; 43 from the long-range delocalized packing of the amino acids surrounding a binding site. However, the results from computational experiments with a partially frozen protein structure with a K+–adapted binding site argue against this in the case of LeuT because releasing only the atoms nearest to the ion is actually sufficient to restore the proper selectivity observed in all-atom simulations. This indicates that the structural forces conferring selectivity to the site NA2 are local. To test this idea, we consider a different reduced model including the complete amino acids involved in the formation of the site NA2. Figure 3C is illustrating this artificial construct, where all functional groups within the 3.5 radii were allowed to fluctuate freely, whereas the outside groups were frozen). This reduced model is augmented to include the effect of molecular stiffness. Accounting for the local stiffness results in a moderately Na+ selective binding site with ΔΔG of 1.49 kcal/mol. This result compares reasonably well with the ΔΔG of 2.28 kcal/mol estimated from all-atom MD simulations of LeuT generated at the same temperature. While the functional groups around Na+ in this reduced model are allowed to move freely in any direction if their oxygens ligands are within 3.5 Å from the ion and no restraining force is applied to the coordinating ligands, all bonds, angles and dihedrals belonging to groups outside the coordination sphere of the ion are constrained to maintain the local organization. This indicates that only a reduced model of NA2 incorporating aspects of molecular stiffness is selective for Na+. This view is in accord with the structural explanation initially proposed by Yamashita et al.6 It is, however, interesting to observe that, in large part, the microscopic forces constraining the ligands in a proper configuration to optimally coordinate Na+ originate from local features (e.g., chemical bonds, angles, and hydrogen bonding). This is in accord with the results of a survey of the coordination structure of Na+ ions bound to proteins in the PDB (see below).
d) Mechanisms of Na+ selectivity in protein binding sites: Lessons from PDB survey and simulations
The presence of a charged high-field ligand, as in site NA1, is obviously a potent mechanism to compensate for the large dehydration free energy of monovalent cation and favors for smaller Na+. A binding site comprising a negatively charged ligand, such as the carboxylate group of the leucine substrate, or the side chain of Asp or Glu acidic amino acids, can competitively dehydrate a small cation such as Na+ ion better than a larger cation such as K+. High field ligands are as found more frequently coordinating Na+ and K+ in the PDB (50% for Na+ binding sites compared to 44% for K+), results that are consistent with the previously reported data from the Harding’s Lab.26 On average, Na+ site comprise ~1.1 charged ligands, compared to ~0.7 for K+ sites. These observations along with the results of minimal models of site NA1 suggest that the most common mechanistic features of the transporters might be constructed with a strong coupling to the carboxylate residue presented by the substrate or protein (with a high field ligand). Nevertheless, as in the case of site NA2 in LeuT, the presence of a negative charge to coordinate Na+ is not an absolute requirement. Bound Na+ without any negatively charged ligands are also observed in the PDB. A large number of such “neutral” Na+ binding sites appear to be located at the protein surface and the coordination frequently comprises a relatively small number of protein ligands (~3) as well as some water molecules. About 20% of all binding sites for Na+ with more than 3 protein ligands are formed by backbone and side-chain atoms, from either serine or threonine.
The organizations of four Na+ binding sites (3 with only neutral ligand and 1 with a single charged ligand) are illustrated in Figure 5. Different geometries of the binding site can occur. Most of the neutral sites display either distorted tetrahedral, bi-pyramidal or octahedral coordination. Binding sites formed by 6 or more coordinating ligands comprises at least one negative charge and display a high connectivity. Interestingly, Na+ binding sites appear to be frequently formed by backbone and side-chain from the same (i with i) or nearest amino acid neighbor along the sequence (i with i−1 or i+1). This is illustrated in Figure 5e for the Na+-coupled aspartate transporter Glt,11 where Na+ coordination is achieved by the presence of a 4-ligand motif involving main-chain oxygens from residues Ser349, to Thr352. The hydroxyl group of the Thr352 side-chain forms a stable hydrogen bond with main chain nitrogen of the Ser349. The site NA2 in LeuT, with its Ser354–Thr355 motif and overall high connectivity, thus displays an organization that is very typical of the neutral binding sites found in the database (See also supplementary results). In the case of K+, such near sequence-neighbor motifs are virtually absent in the protein structures of the PDB. While coordination of K+ in enzymes often occurs through a main chain carbonyl oxygen24; 26 it is more frequently achieved by distant residues in the protein sequence (Supplementary Materials). Actually, the absence of well defined motifs in known K+ enzymes renders bioinformatics detection of K+ binding sites in proteins virtually impossible without structural data.24
Figure 5.
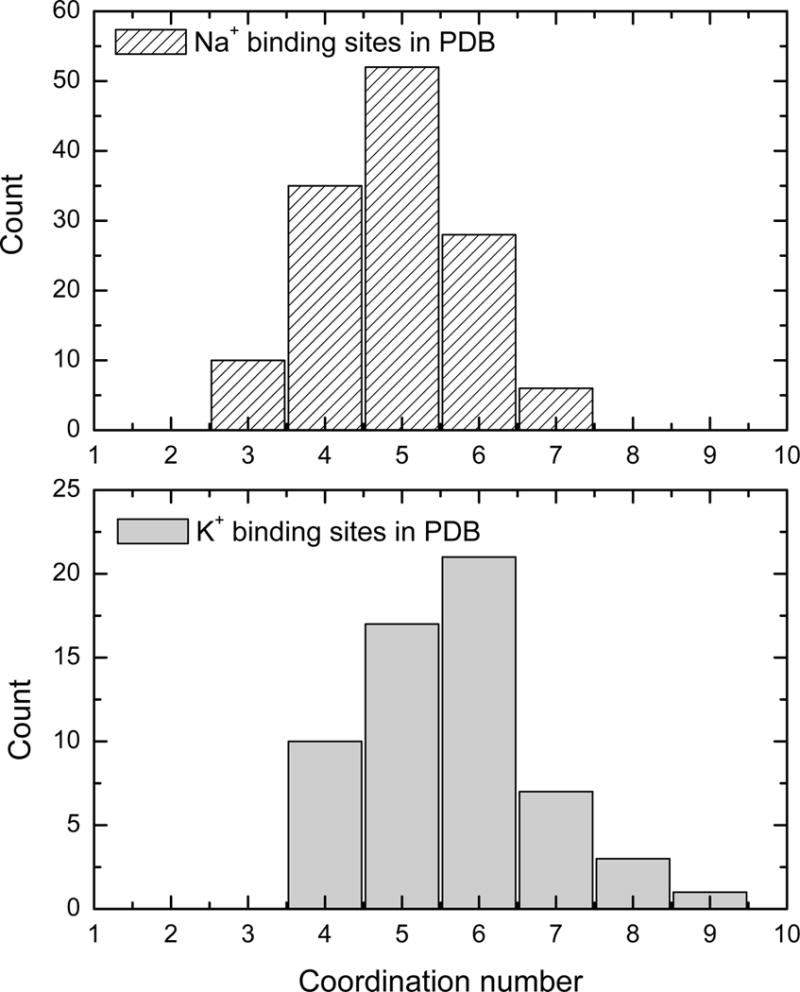
Statistics on the average coordination of Na+ and K+ from the PDB survey.
In structural biology, ion selectivity of protein binding sites is traditionally explained by invoking the concept of snug-fit, whereby only a specific ion has the optimal radius to occupy the binding cavity.44 Such an explanation, familiar in host-guest chemistry, is often appropriate in the case of small and rigid chelating molecules, though its validity must be assessed carefully in the case of flexible protein structures. For instance, the mechanism requires that there is sufficient molecular stiffness in the binding site to give rise to an energy penalty for ions or the “wrong” size.18; 45 A good example of the snug-fit mechanism is provided by the small cyclic membrane carrier valinomycin, as confirmed by numerous computational studies.19; 46; 47 In this case, the needed molecular stiffness of the binding site is achieved locally, via the connectivity of the cyclic covalent structure and the hydrogen bonding network. The binding sites in LeuT are not, of course, formed by a cyclic covalent structure, although similar elements of covalent connectivity are undeniably present. In particular, coordination of the ion by backbone and side chain atoms of near-neighbor residues along the sequence (i, i+1, i−1) exploits the covalent connectivity to form binding sites that possess sufficient local molecular stiffness.
Hydrogen bonding also contributes to build-up the local molecular stiffness. To explore their importance, we have performed a statistical analysis of the hydrogen bonding of the residues forming binding sites NA1 and NA2. The difference in hydrogen bonding connectivity between the two binding sites in LeuT is striking (Table 5). The binding site NA1 contains only a single long-lived hydrogen bond between the non-coordinating backbone nitrogen group of the Thr254 and the carbonyl oxygen of Gln250. In contrast, there are two stable hydrogen-bonds in site NA2, with Gly20–Ala19-Val23 and Ser355–Gly352-A351, forming ring-like motifs coordinating the ion. In addition, there are at least 3 bifurcated bonds. The backbone atoms of Ser355 and Thr354 residues coordinating the ion in the binding site NA2 are covalently bound. With its high connectivity, the NA2 binding site is reminiscent of the organization of cyclic ionophores such as valinomycin.19; 46; 47
Table 5.
Hydrogen-Bond Statistics for the Binding Sites*
| Binding Site | Donor | Acceptor | Life Time (ps) | Occupancy |
|---|---|---|---|---|
| NA1 | HN Glu290 | O Asn286 | 100 | 0.71 |
| HN Thr254 | O Gln250 | 2500 | 0.9 | |
| NA2 | HN Gly20 | O Leu16 | 9.2 | 0.87 |
| HN Val23 | O Ala19 | 2500 | 0.98 | |
| HN Ala351 | O Leu348 | 9.8 | 0.91 | |
| HG1 Thr354 | O Phe350 | 9.6 | 0.92 | |
| HN Ser355 | O Gly352 | 2500 | 0.98 |
The geometrical criteria (distance between donor and acceptor less than 2.7 A and heavy–heavy atoms distance less than 3.5 Å was used to determine hydrogen bond). Life-time column represent the average life-time of a certain hydrogen bond as seen in the simulation. Occupancy represents the probability of a certain H-bond in the trajectory.
Analysis of the database suggests that, on average, “neutral” binding sites tend to possess a smaller number of ligands. The average coordination number for Na+ binding sites with charged ligands is ~5.4, whereas neutral sites comprise only ~4.4 coordinating ligands on average. The site NA1 comprises 6 oxygen atoms including a negatively charged ligand (the carboxylate from the leucine substrate). The site NA2 comprises only 5 neutral coordinating oxygen atoms. The homolog of site NA2 in the Glt transporter including also only neutral ligands comprises 4 carbonyl oxygen atoms. It is likely that the reduction in the number of ligands contributes to shift the selectivity towards smaller cations by reducing the ligand-ligand steric repulsion. However, a reduced coordination number can be effective only if it is accompanied by sufficient local structural stiffness in the binding site. In a flexible site, the interaction provided by the smaller number of ligands is actually unable to effectively compensate for the large difference in hydration free energy between Na+ and K+, and as a result, such site is either non-selective, or selective for K+ “by default”. As indicated by the minimal model of site NA2 shown in Figure 3, a site with 5 ligands is not selective for Na+ over K+. Sufficient structural stiffness is required to achieve selectivity with those ligands. Hence, in binding sites without charged ligands, the requirement for stiffness from high local covalent connectivity precedes coordination numbers as a key determinant of selectivity.
The above analysis shows that the coordination number, by itself, is not the key determinant of size selectivity. This observation is consistent with a survey of ion binding sites from the PDB and reported bioinformatics data,26 which shows that both Na+ and K+ can be accommodated by a wide range of coordination numbers (see also Figure 5). For example, Na+ binding sites comprising 3 to 7 ligands, and K+ binding sites comprising 4 to 9 ligands are observed. In LeuT, the selectivity of the site NA2, with its 5 ligands, is not enhanced relative to the site NA1, with its 6 ligands. These observations indicate that selectivity of the Na+ binding sites in LeuT is not controlled primarily via coordination numbers. Obviously, a number of other factors such as the chemical nature of the ligands,17–19 and the structural stiffness enforced by the local covalent connectivity of the protein play essential roles.19;46;47
CONCLUSIONS
Many of the key concepts used to explain ion selectivity were suggested several decades ago. The need to compensate for the large hydration free energy upon entering a pore was taken into consideration already in the work of Mullins,48;49 Eisenman,25;50 Bezanilla and Armstrong,51 and Hille.52 Over the years, MD simulation studies based on detailed atomic models have contributed to refine our understanding of the microscopic factors governing ion selectivity. In particular, free energy simulations of K+ selectivity in the KcsA and NaK channels have highlighted the importance of dynamic fluctuations in those flexible pores.17–19; 53–55 Yet, attempts to explain ion selectivity exclusively in structural terms remain customary. Ultimately, it is important to realize that selectivity results from small free energy differences (a few kcal/mol) between large numbers (several tens of kcal/mol). Assessing the relative importance of the various microscopic factors requires strict quantitative analysis based on accurate atomic models and free energy simulations. The molecular basis of selectivity of a binding site should be ascertained, on a case-by-case basis.
The present computational study of two Na+ binding sites for a membrane transporter provides a good opportunity to broaden our perspective concerning the different microscopic factors affecting size selectivity. Combining FEP/MD simulations with PDB survey, we were able to analyze the role of different factors in the Na+/K+ selectivity in LeuT and draw general conclusions applicable for a broad variety of proteins. Of particular interest, the present study reveals that Na+ selectivity for two binding site in one membrane protein is controlled by two different mechanisms. This illustrates clearly how there is no unique and universal molecular mechanism explaining ion selectivity in proteins. Coordination of the ion by a negatively charged ligand as in site NA1 clearly provides the most effective and robust mechanism that discriminates for Na+ over K+. Intuitively, the robustness should confer a clear advantage in the case of a transporter that must undergo large conformational changes to function. This would be consistent with the highly conserved residues DEKA in the selectivity locus of the Na+ channels,1; 56 suggesting that Na+ may encounter negatively charged ligands along the permeation pathway of these channels. But selectivity for Na+ over K+ can also be achieved, as in site NA2, without directly implicating a charged ligand. In such neutral sites, selectivity is governed primarily by the structural constraints imposed by the ligands geometry. This is essentially the familiar snug-fit mechanism from host-guest chemistry.44 Mechanistically, such a mechanism is possible only if there is sufficient structural rigidity or stiffness to maintain the relative geometry of the coordinating ligands.18; 45 However, the presence of angstrom-scale thermal fluctuations in flexible proteins normally tend to obscure the sub-angstrom structural precision that is required to discriminate between two ions of very similar size such as Na+ and K+. In the site NA2, the protein solves this problem by exploiting the stiff degrees of freedom from the covalent structure. The structural forces involved are mainly local, and do not arise from the long-range delocalized packing of the amino acids surrounding the binding site. The coordinating ligands forming the binding site NA2 are taken from backbone and side chain atoms of nearest neighboring residues (e.g., i, i−1, and i+1) along the poly-peptide chain. This appears to be a recurrent motif, observed in numerous neutral Na+ binding sites observed in the PDB (but not as frequently for K+ binding sites). In retrospect, the covalent connectivity is an effective and simple way to confer sufficient local rigidity to a binding site by relying the natural stiffness of chemical bonds and angles degrees of freedoms. Whether a similar structural mechanism involving neutral binding sites could control the Na+ selectivity in ion channels, which must sustain a high throughput ion conduction, is unclear.
METHODS
a) Simulation Protocol
All-atom free energy MD simulations were carried out using a model of LeuT embedded in a lipid membrane with explicit solvent. The starting configuration of LeuT was taken from the x-ray coordinates deposited in the PDB (2A69). The system contains 1 LeuT, 3 bound ions (2 Na+ and 1 Cl−), 1 leucine substrate, 108 dimyristoyl-phosphatidylcholine (DMPC), solvated by a 100 mM NaCl aqueous salt solution. The simulation system comprising a total of 56399 atoms is represented in Figure 1. All the computations were carried out using the CHARMM program version c33b2.57 The simulation methodology is similar to that used previously to study ion selectivity in the KcsA K+ channel.17 Briefly, the simulation system was constructed using a membrane-building protocol.17; 58; 59 The simulations were carried at constant pressure (1 Atm) and constant temperature with periodic boundary conditions.60 Electrostatic interactions were treated using a particle mesh Ewald (PME) algorithm.61
Figure 1.
Molecular graphics representation of the Na+-coupled LeuT transporter in the explicit DMPC membrane. Chloride and Na+ ions are depicted as van der Waals spheres (green and yellow, respectively). This picture was created with PyMol.69
The free energy perturbation (FEP) calculations concern only the two Na+ binding sites of LeuT, NA1 and NA2. FEP/MD simulations were carried out for each binding site. The relative free energy determining ion selectivity is defined as
| (1) |
such that a positive number means a binding site selective for Na+ over K+. The relative free energy for Na+ over Li+ is determined in a similar manner. The FEP/MD simulations for the Li+/Na+/K+ series were carried out at three different temperatures (315K, 340K and 368K) to determine the relati ve importance of entropy and enthalpy contributions. Prior to the free energy calculations, the system was initially equilibrated for 8 ns without any restraints. The average RMS values for Cα were on the order of 1.2–1.4 Å, whereas the average RMS values for heavy atoms coordinating ions in binding sites show RMS values of 0.7–1.2 Å, depending on the binding site (values reported at T=315K). The system was further equilibrated for 1.2 ns at the higher temperatures (340K and 368K) preceding the MD/FEP simulations. Separate FEP/MD simulations were carried for each Na+ binding site in LeuT. A thermodynamic coupling parameter λ varying between zero and one was used to alchemically transform Na+ into a different ion type (K+ or Li+); i.e., 11 intermediate values of λ (“windows”) were used to go from 0 to 1 by increments of 0.1 for each of the forward and backward simulations, yielding a total aggregate simulation time of 4.4 ns for each FEP calculation. The data were collected and treated using the weighted histogram analysis method (WHAM).62; 63 The ion parameters were first tested with the FEP/MD simulations in bulk water (TIP3P potential model64; all ion parameters are included in supplementary materials. The ion parameters were modified to reproduce correctly experimental data on the free energy of Na+ and K+ hydration compared to the original CHARMM27 force-field.65 All results of the MD/FEP simulations are reported in Tables 1–3.
b) PDB Survey
To broaden our perspective of the microscopic factors governing ion selectivity in proteins, the protein database (PDB) was searched for high-resolution crystal structures containing bound Na+ and K+ ions. The search follows a strategy similar to that used in previous surveys of monovalent and divalent metal ions coordination in proteins in the PDB,24; 27; 28 The number of structures used in the present analysis is almost twice as large than any published before. A total of 483 protein structures with Na+ ions and 201 structures with K+ ion at resolution better than 2.7 Å were extracted. Multi-nuclear binding sites and sites containing other than water/amino-acids ligands, such as phosphate or sulfate groups, and entries with more than 30% of sequence homology were excluded from the analysis (e.g., there are 15 nearly identical structure of lyzosyme with a bound Na+). In such cases, the available crystal structure with the highest resolution was chosen. All structures in which Na+ ion is coordinated by more than 3 waters were removed as well. The resulting dataset contains 132 entries for Na+ and 59 entries for K+. The main results are summarized in Table 4. An extended table with statistics for each PDB entry is given in supplementary material. The ratio of structures for two ions reflects relative population of protein structures containing Na+ and K+ selected for initial analysis.
Supplementary Material
Figure 4.

Typical examples of the organization and connectivity in the Na+ binding sites found in the protein database. (A) Distorted tetrahedral binding site (methyl-coenzyme M reductase, 1HBN).70 Ligands involved in ion coordination: O—Ala544, Oγ—Thr547, O—Thr547, O—Pro548; (B) Distorted trigonal bi-pyramide binding site (30S ribosomal protein S6, 2J5A). Ligands involved in ion coordination: O—Leu86, O—Lys87, O—Asp89, O—Leu92, O—HOH3371; (C) Octahedral binding site (7,8-Diaminopelargonic Acid Synthase, 1MLY, Ligands involved in ion coordination: O—Val96, O—Thr99, Oγ—Thr99, O—Pro100, O—Leu103, O—HOH9272; (D) Seven coordinating ligands, including one negative charge (galactose oxidase, 1GOF).73 Ligands involved in ion coordination: O—Lys29, Oδ1—Asp32, O—Asn34, O—Thr37, Oγ—Thr37, O—Ala141, Oɛ2—Glu142; (E) Binding site 1 of the aspartate transporter Glt11; 74 show 4 member motif to provide coordination for Na+ ion: O—Ser349, O—Ile350, O—Gly351, O—Thr352, the further coordination is provided by O—Thr308 and probably by S—Met311 and O—Ile309.
Acknowledgments
We are grateful to E. Gouaux and S. Singh for providing us with the access to experimental data on Na+/K+ selectivity in LeuT before the actual publication and stimulating discussions. We would also like to thank E. Di Cera, O.Boudker, L. DeFelice, A. Ribeiro, and P. Tieleman for constructive criticism of the paper. This work would not be possible without computer-time allocation at the West-Grid Canada and National Computer Scientific Alliance (NCSA) USA. This work was supported by the National Sciences and Engineering Research Council (NSERC) Discovery grant to S.N. and the National Institute of Health grant number GM-62342 to B.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hille B. Ion Channels of Excitable Membranes. 3rd. Sinauer; Sunderland, MA: 2001. [Google Scholar]
- 2.DeFelice L. Transporter structure and mechanism. Trends Neurosci. 2004;27:352–359. doi: 10.1016/j.tins.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Rudnick G. In: Neurotransmitter transporters: Structure, Functions and Regulation. Reith RA, editor. Humana Press; Totowa, NJ, USA: 2002. [Google Scholar]
- 4.Iversen LL. Role of transmitter uptake mechanism in synaptic transmission. Brit J Pharmacology. 1971;41:571–577. doi: 10.1111/j.1476-5381.1971.tb07066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blakely RD, Robinson MB, Amara SG. Expression of neurotransmitter transport from rat brain mRNA in Xenopus laevis oocytes. Proc Natl Acad Sci. 1988;85:9846–9850. doi: 10.1073/pnas.85.24.9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 7.Russ WP, Engelman DM. The GxxxG motif: a framework for transmemberna helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 8.Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol. 2006;70:1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 9.Noskov SY. Mechanism of substrate specificity in the leucine transporter LeuT. Proteins:Struct Funct Bioinf. 2008 Submitted. [Google Scholar]
- 10.Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar GxxxG-like and proline motifs. Curr Opin Struct Biol. 2004;14:465–479. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 12.Singh S, Piscitelli C, Yamashita A, Gouaux E. Manuscript in preparation 2007 [Google Scholar]
- 13.Di Cera E, Guinto ER, Vindogni A, Dang QD, Ayala YM, Wyui M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 14.Prasad S, Wright K, Roy DB, Bush LA, Cantwell AM, Di Cera E. Redesgning the monovalent cation specificity of an enzyme. Proc Natl Acad Sci. 2003;100:13785–13890. doi: 10.1073/pnas.2333109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Cahit BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 17.Noskov SY, Berneche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 18.Noskov SY, Roux B. Ion Selectivity in Potassium Channel. Biophys Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 19.Noskov SY, Roux B. Importance of Hydration and Dynamics on the Selectivity of the KcsA and NaK Channels. J Gen Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi N, Ye S, Alam A, Chen L, Jiang Y. Atomic structure of a Na+- and K+-conducting channel. Nature. 2006;440:570–574. doi: 10.1038/nature04508. [DOI] [PubMed] [Google Scholar]
- 21.Bernèche S, Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 22.MacKerell ADJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 23.Cornell W, Cieplak P, Bayly C, Gould I, Jr, K M, Ferguson D, Spellmeyer D, Fox T, Caldwell J, Kollman P. A second generation force field for the simulation of proteins and nucleic acids. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 24.Page MJ, Di Cera E. Role of Na+ and K+ in Enzyme function. Physiol Reviews. 2006;86:1049–1092. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 25.Eisenman G. Cation selective electrodes and their mode of operation. Biophys J. 1962;2:259–323. doi: 10.1016/s0006-3495(62)86959-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallographica Section D-Biological Crystallography. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]
- 27.Dudev T, Lim C. Effect of Carboxylate-Binding Mode on Metal Binding/Selectivity and Function in Proteins. Acc Chem Res. 2007;40:85–93. doi: 10.1021/ar068181i. [DOI] [PubMed] [Google Scholar]
- 28.Dudev T, Lim C. A DFT/CDM Study of Metal-Carboxylate Interactions in Metalloproteins: Factors Governing the Maximum Number of Metal-Bound Carboxylates. J Am Chem Soc. 2006;128:1553–1561. doi: 10.1021/ja055797e. [DOI] [PubMed] [Google Scholar]
- 29.Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Crystallographica Section D-Biological Crystallography. 2006;62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- 30.Neilson GW, Enderby JE. The coordination of metal aquaions. Adv Inorg Chem. 1989;34:195–218. [Google Scholar]
- 31.Rudnick G. Serotonin transporters - Structure and function. J Membr Biol. 2006;213:101–110. doi: 10.1007/s00232-006-0878-4. [DOI] [PubMed] [Google Scholar]
- 32.Quick M. Regulating the conducting states of a mammalian serotonin transporter. Neuron. 2003;40:537–549. doi: 10.1016/s0896-6273(03)00605-6. [DOI] [PubMed] [Google Scholar]
- 33.Keynan S, Kanner BI. GABA transport in reconstituted preparations from rat-brain-coupled sodium and chloride. Biochemistry and Cell Biology-Biochimie Et Biologie Cellulaire. 1988;27:12–17. doi: 10.1021/bi00401a003. [DOI] [PubMed] [Google Scholar]
- 34.Roux MJ, Supplisson S. Neuronal and glial glycine transporters have different stoichiometries. Neuron. 2000;25:373–383. doi: 10.1016/s0896-6273(00)80901-0. [DOI] [PubMed] [Google Scholar]
- 35.Zhang YW, Rudnick G. Serotonin transporter mutations associated with obsessive-compulsive disorder and phosphorylation alter binding affinity for inhibitors. Neuropharmacology. 2005;49:791–797. doi: 10.1016/j.neuropharm.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 36.Ryan RM, Mindell JA. The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat Struct Mol Biol. 2007;14:365–371. doi: 10.1038/nsmb1230. [DOI] [PubMed] [Google Scholar]
- 37.Adams SV, DeFelice LJ. Ionic currents in the human serotonin transporter reveal inconsistencies in the alternating access hypothesis. Biophys J. 2003;85:1548–1559. doi: 10.1016/S0006-3495(03)74587-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dixon JF, Hokin LE. Lithium acutely inhibits and chronically up-regulates and stabilizes glutamate uptake by presynaptic nerve endings in mouse cerebral cortex. Proc Natl Acad Sci. 1998;95:8363–8368. doi: 10.1073/pnas.95.14.8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pifl C, Singer EA. Ion dependence of carrier-mediated release in dopamine or norepinephrine transporter-transfected cells questions the hypothesis of facilitated exchange diffusion. Molecular Pharmacology. 1999;56:1047–1054. doi: 10.1124/mol.56.5.1047. [DOI] [PubMed] [Google Scholar]
- 40.Collier DA, Arranz MJ, Sham P, Battersby S, Vallada H, Gill P, Aitchison KJ, Sodhi M, Li T, Roberts GW, Smith B, Morton J, Murray RM, Smith D, Kirov G. The serotonin transporter is a potential susceptibility factor for bipolar affective disorder. Neuroreport. 1996;7:1675–1679. doi: 10.1097/00001756-199607080-00030. [DOI] [PubMed] [Google Scholar]
- 41.Menaker D, Bendahan A, Kanner BI. The substrate specificity of a neuronal glutamate transporter is determined by the nature of the coupling ion. J Neurochem. 2006;99:20–28. doi: 10.1111/j.1471-4159.2006.04003.x. [DOI] [PubMed] [Google Scholar]
- 42.Bostick DL, Brooks CL., III Selectivity in K+ channels is due to topological control of the permeant ion’s coordinated state. Proc Natl Acad Sci. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lockless SW, Zhou M, MacKinnon R. Structural and thermodynami properties of selective ion binding in K+ channel. PLOS Biology. 2007;5:e21. doi: 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gouaux E, MacKinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 45.Allen TW, Andersen OS, Roux B. On the Importance of Atomic Fluctuations, Protein Flexibility and Solvent in Ion Permeation. J Gen Physiol. 2004;124:679–690. doi: 10.1085/jgp.200409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aqvist J, Alvarez O, Eisenman G. Ion-Selective Properties of a Small Ionophore in Methanol Studied by Free Energy Perturbation Simulations. J Phys Chem. 1992;96:10019–10025. [Google Scholar]
- 47.Marrone TJ, Merz KM., Jr Molecular Recognition of K+ and Na+ by Valinomycin in Methanol. J Am Chem Soc. 1995;117:779–791. [Google Scholar]
- 48.Mullins LJ. In: Molecular Structure and functional activity of nerve cells. Grenel RG, Mullins LJ, editors. AIBS; Washington: 1956. [Google Scholar]
- 49.Mullins LJ. The penetration of some cation into muscle. J Gen Physiol. 1959;42:817–829. doi: 10.1085/jgp.42.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eisenman G, Horn R. Ion selectivity revisited:the role of kinetic and equilibrium processes in ion permeation through channels. J Membr Biol. 1983;76:197–225. doi: 10.1007/BF01870364. [DOI] [PubMed] [Google Scholar]
- 51.Bezanilla F, Armstrong CM. Negative conductance caused by entry of sodium and cesium ions into the K channels of squid axon. J Gen Physiol. 1972;53:342–347. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hille B. Potassium channels in myelinated nerve-selective permeability to small cations. J Gen Physiol. 1973;61:599. doi: 10.1085/jgp.61.6.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Åqvist J, Luzhkov V. Ion permeation mechanism of the potassium channel. Nature. 2000;404:881–884. doi: 10.1038/35009114. [DOI] [PubMed] [Google Scholar]
- 54.Luzhkov VB, Åqvist J. K+/Na+ selectivity of the KcsA potassium channel from microscopic free energy perturbation calculations. Biochim Biophys Acta. 2001;1548:194–202. doi: 10.1016/s0167-4838(01)00213-8. [DOI] [PubMed] [Google Scholar]
- 55.Asthagiri D, Pratt LR, Paulaitis ME. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J Chem Phys. 2006;125 doi: 10.1063/1.2205853. Artn 024701. [DOI] [PubMed] [Google Scholar]
- 56.Heinemann SH, Terlau H, Stuhmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441–443. doi: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- 57.Brooks BR, et al. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 58.Woolf TB, Roux B. Molecular dynamics simulations of proteins in lipid membranes: the first steps. Biophys J. 1993;A354 [Google Scholar]
- 59.Allen TW, Andersen OS, Roux B. Energetics of Ion Conduction through the Gramicidin Channel. Proc Nat Acad Sci. 2004;101:117–122. doi: 10.1073/pnas.2635314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feller S, Zhang Y, Pastor R, Brooks B. Constant pressure molecular dynamics simulation - the Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 61.Essman U, Perera M, Berkowitz M, Darden T, Lee H, Pedersen L. A smooth particle mesh ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 62.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for the free-energy calculations on biomolecules. J Comp Chem. 1992;13:1011–1021. [Google Scholar]
- 63.Souaille M, Roux B. Extension to the Weighted Histogram Analysis Method: Combining Umbrella Sampling with Free Energy Calculations. Comp Phys Comm. 2001;135:40–57. [Google Scholar]
- 64.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 65.Nina M, Beglov D, Roux B. Atomic Radii for Continuum Electrostatics Calculations based on Molecular Dynamics Free Energy Simulations. J Phys Chem B. 1997;101:5239–5248. [Google Scholar]
- 66.Roux B. The calculation of the potential of mean force using computer simulations. Comp Phys Comm. 1995;91:275–282. [Google Scholar]
- 67.DeLano WL. The PyMol Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA, USA: [Google Scholar]
- 68.Grabarse W, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U. On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J Mol Biol. 2001;309:315–330. doi: 10.1006/jmbi.2001.4647. [DOI] [PubMed] [Google Scholar]
- 69.Olofsson M, Hansson S, Hedberg L, Logan DT, Oliveberg M. Folding of S6 Structures with Divergent Amino Acid Composition: Pathway Flexibility within Partly Overlapping Foldons. J Mol Biol. 2007;365:237. doi: 10.1016/j.jmb.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 70.Sandmark J, Mann S, Marquet A, Schneider G. Structural basis for the inhibition of the biosynthesis of biotin by the antibiotic amiclenomycin. J Biol Chem. 2002;277:43352–43358. doi: 10.1074/jbc.M207239200. [DOI] [PubMed] [Google Scholar]
- 71.Ito N, Phillips SE, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KD, Knowles PF. Novel thioether bond revealed by a 1.7 A crystal structure of galactose oxidase. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 72.Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.