

Abstract
The nanostructures of self-assembling biomaterials have been previously designed to tune the release of growth factors in order to optimize biological repair and regeneration. We report here on the discovery that weakly cohesive peptide nanostructures in terms of intermolecular hydrogen bonding, when combined with low concentrations of osteogenic growth factor, enhance both BMP-2 and Wnt mediated signaling in myoblasts and bone marrow stromal cells, respectively. Conversely, analogous nanostructures with enhanced levels of internal hydrogen bonding and cohesion lead to an overall reduction in BMP-2 signaling. We propose that the mechanism for enhanced growth factor signaling by the nanostructures is related to their ability to increase diffusion within membrane lipid rafts. The phenomenon reported here could lead to new nanomedicine strategies to mediate growth factor signaling for translational targets.
Keywords: Self-assembly, lipid raft, BMP-2, LRAP, growth factor signaling
The extracellular matrix (ECM) is a complex structural landscape that mechanically supports cells and harbors bioactive molecules that direct proliferation, differentiation, migration, and tissue morphogenesis.1,2 Growth factors in particular are proteins associated with the ECM and induce cell response by interacting with transmembrane receptors at the cell surface to initiate specific signaling cascades. Multiple external factors govern the ability of growth factors to signal cells including the ability of the growth factor to bind to ECM components, the concentration, and location of the target cell.
Direct injection of growth factors for regenerative medicine applications has had limited success in patients,3 and therefore biomaterials have been developed as reservoirs or vehicles for growth factor delivery.4 Additionally, because the local extracellular environment can dramatically influence growth factor mediated cell response, the potential of biomaterials to directly alter growth factor signaling remains an active area of research.5−8 In general, biomaterials are often designed to control spatial and temporal release of bioactive molecules; they can serve a passive role for cell signaling by encapsulating the growth factor through covalent or physical means. Alternatively, biomaterials can have a more active role by exhibiting preferential binding for growth factors of interest,9,10 directly mimicking bioactive molecules,11−14 or targeting specific cell types.15 Interest on self-assembled supramolecular nanostructures for cell signaling has been partly based on their structural versatility, allowing easy incorporation of these specific features. The typical approach for directing cell signaling has been to design the biomaterial for specific interactions at the cell-material interface, for example, with membrane bound receptors.16,17
Peptide amphiphile (PA) molecules are a class of self-assembling molecules that can self-assemble into supramolecular nanofibers and create biomimetic, synthetic components in the extracellular mileu. They have been designed to bind or mimic the activity of specific growth factors for regenerative applications ranging from ischemic disease in the heart or limbs to cartilage and bone regeneration.9−12 PA biomaterials have demonstrated an ability to interact directly with cell surface receptors to modulate downstream signaling and cell response.18,19 Molecular design can be applied to a diverse array of targets by linking covalently to an alkyl tail a highly customized peptide segment that controls the physical properties and bioactivity of the nanofiber matrix. Recently, it has been shown that intermolecular interactions, namely hydrogen bonding and electrostatic repulsion, can have significant effects on assembly morphology and cell response.20−23 In particular, tailoring hydrogen bonding within the assemblies has been used to modify the apparent stiffness of the matrix to control neuron maturation,20 or to alter drastically cell viability upon contact with cationic nanofibers.23
We have investigated here the role of intermolecular interactions within supramolecular assemblies on growth factor signaling. Two materials that differ in the degree of intermolecular hydrogen bonding were designed by choosing primary amino acid sequences with varied β-sheet propensity. The influence of these PA assemblies on growth factor mediated osteogenic differentiation was evaluated and a mechanistic evaluation of how PA supramolecular cohesion affects cell signaling was carried out by studying intracellular signaling and the mobility of cell membrane lipid rafts.
Design and Characterization of PA Assemblies
Two PAs with similar charge and molecular architecture were designed with different propensities for β-sheet hydrogen bonding. Both materials lack designed bioactivity. Altering the amino acid composition of the peptide backbone allowed for varying degrees of hydrogen bonding within the assemblies. Both molecules are similar in amino acid composition with the exception of valine, which was chosen for its strong preference to adopt a β-sheet secondary structure (strong β-sheet PA, Figure 1A) or glycine, which prefers a random coil conformation, effectively reducing the degree of intermolecular hydrogen bonding (weak β-sheet PA, Figure 1A).24 Positively charged lysine residues promote the association of PA nanostructures with the negatively charged cell surface, however, the total number of charged residues was constrained to prevent cytotoxicity, which has been observed previously when combining both high cationic charge and weak hydrogen bonding in PA assemblies.23,25
Figure 1.

Characterization of strong β-sheet and weak β-sheet PA materials. (A) Chemical structures of strong β-sheet PA (Lys-Lys-Ala-Ala-Ala-Val-Val-Val-Lys-palmitoyl, top) and weak β-sheet PA (Lys-Lys-Gly-Gly-Gly-Ala-Ala-Ala-Lys-palmitoyl, bottom) that vary the propensity for intermolecular β-sheet hydrogen bonding adjacent to the alkyl tail. (B) Wide-angle X-ray scattering from solutions of strong β-sheet and weak β-sheet assemblies (arrow: 4.7 Å, corresponds to spacing for β-sheet hydrogen bonding). (C) Cryogenic transmission electron microscopy of strong or weak β-sheet assemblies in serum-free cell media showing the presence of nanofibers. Scale bar: 200 nm.
The supramolecular assemblies formed by the strong β-sheet and weak β-sheet PAs were characterized using wide-angle X-ray scattering (WAXS) and cryogenic transmission electron microscopy (cryoTEM). WAXS from a solution of the strong β-sheet nanostructures revealed a Bragg reflection at 4.7 Å, indicative of regular spacing between β-strands, while the weak β-sheet PA showed only diffuse scattering, suggesting a lack of long-range order (Figure 1B). CryoTEM revealed that both PAs form one-dimensional assemblies in solution (Figure 1C). Together these results confirm that the nanostructures formed by these two molecules are similar in morphology but exhibit clear differences in the extent of hydrogen bonding.
Cell Response to PA-BMP-2 Mixtures
Next, we sought to evaluate if the differences in hydrogen bonding within the assemblies could have an effect on growth factor signaling. We selected BMP-2 as a model growth factor and C2C12 mouse myoblast cells as a model cell type, as these cells are well-known to convert from a myogenic to osteogenic lineage when treated with BMP-2.26 Cells were plated on tissue culture plastic and treated with a mixture of BMP-2 (10 nM) and PA (0.001% w/v). To exclude the possibility of any cytotoxic effect of PAs, especially from a combination of weak hydrogen bonding and cationic charge a cell viability assay was performed.23 Treatment with either strong or weak β-sheet PAs supported cell survival at the concentrations used in cell studies; however, we note that at concentrations 5-fold higher than working concentrations, some cell toxicity was observed with Weak PA and BMP-2 mixtures (Figure S1).
In addition to promoting osteogenic differentiation, BMP-2 has a potent antimitogenic effect, which can further favor the differentiation process for C2C12 cells.27 Therefore, to evaluate the effects of the assemblies on BMP-2 signaling, both cell proliferation and differentiation were studied. Cell proliferation was measured by DNA uptake of the nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU), which was added to cultures that were pretreated overnight with nanofibers alone or combinations of nanofibers and BMP-2. After 8 h of EdU exposure, a significant increase in the proportion of EdU positive cells was observed after treatment with strong β-sheet assemblies (58 ± 6% versus 40 ± 10% in untreated control), while identical treatment with weaker β-sheet assemblies did not elicit a change in proliferation (Figure 2A,B). BMP-2 treatment alone caused a 9% reduction in the EdU positive cell population and a similar 11% reduction was observed when the growth factor was combined with weak β-sheet nanostructures. Combined treatment of BMP-2 and strong β-sheet nanostructures on the other hand did not show any difference in cell proliferation relative to the strong β-sheet PA treatment alone. This result suggests that assemblies with strong hydrogen bonding not only enhance the proliferation of C2C12 cells but also block the antimitogenic effect of BMP-2.
Figure 2.

Proliferation and differentiation of C2C12 myoblast cells treated with strong or weak β-sheet PA nanofibers with and without BMP-2. (A) Representative images following an 8 h treatment with EdU to label nuclei of proliferating cells. Co-localization of EdU incorporation (red) with cell nuclei (blue) show differences in cell proliferation. Untreated control (NT). Scale bar: 100 μm. (B) Quantification of EdU positive cells (from panel A) demonstrating significant differences in cell proliferation. Kruskal–Wallis test. **p < 0.01, ***p < 0.001, compared to NT (n = 3). (C) Fast Blue staining to visualize alkaline phosphatase (ALP) activity after 3 days of culture. Scale bar: 100 μm. (D) Quantification of ALP activity following 3 days of culture using a colorimetric enzyme assay. Kruskal–Wallis test. ***p < 0.001, compared to NT (n = 4).
To evaluate the osteogenic differentiation of the C2C12 cells, alkaline phosphatase (ALP) activity was monitored. After 3 days of treatment, ALP-expressing cells were found to respond to BMP-2 in a dose-dependent manner, as expected (Figure S2). Interestingly, we observed a sharp increase for ALP expression in cells treated with both BMP-2 and weakly cohesive PA nanostructures compared to cells treated with BMP-2 alone (Figure 2C,D). Quantification confirmed this observation showing a 7.2-fold increase in ALP activity per cell when treated with a combination of the weakly cohesive supramolecular nanostructures and BMP-2 compared with cells treated with an optimal dose (10 nM) of growth factor alone. Additionally, enhanced ALP expression remained even when the dose of BMP-2 was reduced further but only in the presence of the weakly cohesive PA–nanofibers (Figure S2). Exposure to the weak β-sheet assemblies alone did not evoke measurable ALP activity, indicating that enhanced ALP activity resulted from an amplified effect of the BMP-2 signal and not due to an intrinsic osteogenic influence of the nanofibers. We also observed a noticeable reduction in ALP expression when BMP-2 was added to the media along with PA assemblies with strong β-sheet character. This observation is consistent with the proliferation result, as these data suggest an inhibitory effect of strong β-sheet PA assemblies on BMP-2 induced signaling.
Intracellular Signaling with PA-BMP-2 Mixtures
To begin interrogating signaling events at the cell surface, we first studied the known downstream signaling cascades associated with BMP receptor activation. We performed Western blot analysis to determine if the PA assembly/BMP-2 combinations influence the canonical BMP-2 signaling cascade. There are two distinct modes of intracellular BMP-2 signaling that eventually modulate gene expression. These two modes are Smad-dependent (Smad) and Smad-independent (non-Smad) signaling pathways. The former pathway involves sequential activation of a number of Smad proteins, while the latter involves the MAPK (mitogen-activated protein kinase) pathway leading to the activation of p38 MAPK. Both pathways increase ALP expression.
The activation of Smad was probed by assaying for the expression of phosphorylated Smad 1/5/8 (p-Smad). However, enhanced accumulation of p-Smad protein was not observed following the addition of strong or weak β-sheet PA nanofibers alone (Figure S3). This finding further supports the observation that the BMP-2 ligand was required to initiate BMP-2 specific signaling (Figure 3A,B). When treated with weakly cohesive PA nanostructures and BMP-2 for 30 min, the mean values of p-Smad protein accumulation measured by densitometry (3.05 ± 0.41) was found to be slightly higher relative to cells treated with BMP-2 alone (2.45 ± 0.25); however, the increased detection of p-Smad protein was absent when the cells were similarly treated with strong β-sheet PA assemblies and BMP-2 (1.24 ± 0.29). A similar trend was observed with the phosphorylated p38 protein (Figure S3). The crosstalk and fine-tuning of the Smad and non-Smad signaling pathways remain complex, but both Smad and non-Smad signaling contribute to osteoblastic differentiation and bone formation.28−30 These results demonstrate that the combination of weak β-sheet PA and BMP-2 successfully activates intracellular events associated with BMP-2 induced signaling.
Figure 3.

Probing cell-material interactions at the cell surface and downstream BMP-2 mediated signaling. (A) Whole cell lysates were extracted from C2C12 cells following treatment with strong or weak β-sheet PA assemblies and/or BMP-2. Western blot analysis was performed for phospho-Smad 1/5/8 (p-Smad 1/5/8). (B) Densitometry measurements from Western blot bands in the p-Smad experiment that appears in (A) normalized to β-actin protein content. (C) RT-PCR experiments evaluating gene expression of alkaline phosphatase (ALP) and osteocalcin (OCN) mRNAs following 3 days of culture. Data is normalized to GAPDH and the values from untreated controls (NT) serve as the baseline. Statistical analysis was performed using a Kruskal–Wallis test with Dunn’s post-test to compare between groups. ***p < 0.001 compared to untreated controls (NT), †††p < 0.001 comparing between designated groups (n = 3).
The upregulation of BMP-mediated Smad and non-Smad signaling pathways in the presence of weak β-sheet PA nanofibers prompted us to investigate whether such changes are reflected in the expression of genes involved in osteogenic differentiation. Gene expression was evaluated using quantitative RT-PCR measurement of mRNAs encoding for ALP and the protein osteocalcin (OCN), which are early and late markers of osteogenic differentiation, respectively (Figure 3C). Exposing the cells to exogenous BMP-2 for 3 days of culture resulted in a significant enhancement in expression of both ALP and OCN compared to untreated controls.31,32 When cells were treated with both weak β-sheet PA assemblies and BMP-2, expression of ALP and OCN mRNAs increased by 340 ± 148 and 157 ± 30 fold, respectively (Figure 3C). In contrast, a combined treatment of strong β-sheet nanofibers with BMP-2 reduced the expression of these two genes relative to treatments with BMP-2 alone (Figure 3C). The gene expression findings corroborate the results from ALP staining experiments and confirm that the weak assemblies are capable of enhancing the effect of BMP-2 in C2C12 cells, resulting in upregulation of osteogenic markers and genes associated with the BMP-2 signaling pathway.
Additionally, because the strong and weak PAs were not designed to mimic any specific biological signal and did not promote osteogenic differentiation when used alone, we hypothesized that the observed PA effect is due to their influence on BMP-2 signaling. One possible mechanism for this effect could be due to differences in sequestration of the growth factor to the nanofiber surface. Similar to charged polymers such as chitosan, alginate, and hyaluronan that have been used previously to sequester and deliver BMP-2,33 PA materials have demonstrated their ability to increase the local concentration of growth factors (such as BMP-2) to improve bioactivity9−11 To assess if there were differences in affinity for BMP-2 between the strong and weak β-sheet PA nanofibers, we used an enzyme-linked immunosorbent assay (ELISA) to measure BMP-2 levels. ELISA results suggest that both strong and weak β-sheet PA assemblies bound similar amounts of BMP-2, as over 94% of the growth factor was retained by the PA nanostructures (Figure S4). Because binding of BMP-2 was similar for both PAs, we concluded that the observed differences in signaling involved interactions occurring at the cell-PA interface.
PA Nanofiber Association with the Cell Membrane and Its Influence on Membrane Dynamics
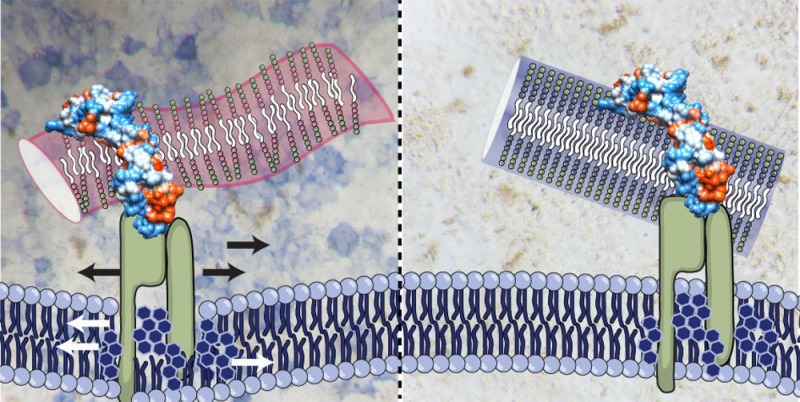
Because the initial step in most growth factor signal cascades involve receptor–ligand binding at the cell surface, factors in the local environment such as membrane fluidity can play a crucial role.34 Therefore, we next chose to evaluate changes in lipid raft mobility in response to strong and weak β-sheet PA nanofibers. Visualization of PA nanofiber treated cells by confocal microscopy and scanning electron microscopy confirmed the association of the PA nanofibers with cell surfaces for both strong and weak β-sheet PA nanofibers (Figure 4A,B, Figure S5). Our previous study demonstrated that highly cationic PA assemblies with weak hydrogen bonding can lead to cytotoxicity due to their ability to disrupt cell membranes.23 Although the PA nanostructures used here did not elicit cytotoxicity at the concentrations used, the combination of positive charge and weak hydrogen bonding within the supramolecular nanostructure could result in direct interactions between PA nanofibers and cell membranes. Thus, we chose to investigate the dynamics of the lipid rafts, as modification of lipid raft mobility by the presence of the PA nanofibers could affect growth factor accessibility and signaling.
Figure 4.

PA nanofiber association with the cell membrane and their effect on lipid raft fluidity. (A) Confocal microscopy images of C2C12 cells following treatment with fluorescently labeled strong or weak β-sheet PA nanofibers (red). Cells are stained with phalloidin to visualize actin (green) and DAPI to visualize cell nuclei (blue). Scale bar: 10 μm. (B) Scanning electron micrographs of cells treated with PA-BMP-2 mixtures. PA nanofibers associate with the cell surface (black arrows). The surface on which the cells are cultured have been pseudocolored yellow for clarity. Scale bar: 2 μm. (C,D) Confocal images from fluorescence recovery after photobleaching experiments showing recovery of CTxB-Alexa 488 fluorescence in the photobleached area; cells were treated with (C) strong β-sheet or (D) weak β-sheet PA nanofibers. Scale bars: 10 μm. (E) Representative recovery curves have been normalized to prebleach intensities and corrected for fluorescence decay during imaging for each treatment condition: untreated (black), strong β-sheet PA nanofibers (red), or weak β-sheet PA nanofibers (blue). (F) Measured values for the diffusion coefficient D of CTxB-Alexa 488 treated C2C12 cells with the following conditions: untreated (black), strong β-sheet PA nanofibers (red), or weak β-sheet PA nanofibers (blue). Statistical analysis was performed using a one-way ANOVA with a Dunnett’s post-test comparing to untreated controls (NT). **p < 0.01 compared to NT, (n = 9–10 cells for each condition).
Lipid rafts are tightly packed and ordered nanoscale assemblies of proteins and lipids that float freely in the cell membrane. It has been proposed that these rafts, which are sterol-sphingolipid-enriched, play a critical role in subcellular processes such as membrane sorting, formation of signaling complexes, and endocytic trafficking.35−37 Signaling events initiated by a number of growth factors, such as glial cell-derived neurotrophic factor (GDNF), epidermal growth factor (EGF), and BMP, are thought to involve lipid rafts.38 The exact mechanism by which lipid rafts enable or augment signaling is unknown, however, current research suggests that metastable rafts with receptors bound to their ligand can coalesce into larger, more stable raft domains to serve as “concentrating platforms” for individual receptors so that large signaling complexes can be formed. In fact, integrin-binding PA assemblies have been proposed to affect lipid raft-mediated signaling by binding surface-bound integrins and impairing their ability to traffic into a “concentrated lipid raft platform”.18 These results suggest that supramolecular nanofibers could potentially alter raft organization and dynamics by binding surface receptors. However, because the present study uses PA assemblies that lack designed bioactivity for receptors, we instead hypothesized that the combination of weak cohesion, cationic charge, and amphiphilic nature of the nanofibers could directly affect the mobility of lipid rafts.
To test our hypothesis, we probed cell membrane lipid raft mobility by performing fluorescence recovery after photobleaching (FRAP) experiments.39,40 Live C2C12 cells were stained with fluorescently labeled cholera toxin subtype B (CTxB), a protein marker that is commonly used to visualize lipid rafts. To evaluate the effect of PA nanostructures on lipid raft dynamics, cells were treated with either strong β-sheet PA or weak β-sheet PA nanofibers and the recovery of CTxB fluorescence was monitored over time (Figure 4C,D). Additionally, to minimize variability in FRAP measurements the bleaching area was restricted to the leading edge of the cell, where the cell membrane and cytosol assume a flat and spread morphology to create a two-dimensional object. Upon quantification, our experiments revealed that CTxB in naïve C2C12 cell membranes have a diffusion coefficient (D) of 0.11 ± 0.3 μm2/s (Figure 4E,F), similar to values reported for other cell lines.41 The diffusion of CTxB remained similar after adding strong β-sheet nanofibers (D, 0.09 ± 0.4 μm2/s). However, after treatment with weak nanofibers a significant increase in CTxB diffusion was observed (D, 0.17 ± 0.04 μm2/s), suggesting that the lipid raft domains have enhanced mobility in the presence of assemblies with weak hydrogen bonding.
Raft assembly is both dynamic and reversible; moreover, palmitoylated and myristoylated proteins such as flotillins and caveolins preferentially associate with lipid rafts.35,38,42 Previous research has demonstrated that palmitoylated PA assemblies with weak hydrogen bonding can have high affinities for the cell membrane.23 We postulate that the increased raft mobility in samples treated with weak β-sheet PA nanofibers is attributed to PA molecules intercalating within the cell membrane and lipid-rich microdomains. Initiation of BMP-2 signaling occurs at the cell surface through receptors that reside in specific membrane domains that enrich in specific membrane subdomains, such as caveolae.43,44 Enhanced signaling is observed when cells are treated with a mixture of both growth factor and weakly cohesive nanofibers, therefore we postulate that the weak PA assemblies promote preferential association with lipid rafts through two mechanisms. First, the electrostatic interaction between the PA and growth factor can localize the ligand at the cell surface in proximity to the signaling receptors that reside in lipid-rich microdomains and second, the increased mobility of the lipid rafts caused by treatment with weakly cohesive PA nanofibers directly increases the statistical probability of a ligand–receptor interaction.
Exploring Additional Signaling Mechanisms When Varying Nanostructure Cohesion
To determine if the relationship between nanostructure cohesion and enhanced cell signaling could be generalized to other signaling molecules and pathways, we chose to explore cell differentiation of a second widely used cell model: biopotential bone marrow stromal ST2 cells. The potent signaling molecule leucine-rich amelogenin peptide (LRAP) was used in combination with the PA assemblies, as it is known to both stimulate osteogenesis and inhibit adipogenesis of mesenchymal stem cells by activating the canonical Wnt/ β-catenin signaling pathway.45−47 We treated ST2 cells with combinations of LRAP and either strong or weak β-sheet PA nanostructures to determine if weak β-sheet PA assemblies could enhance osteogenesis by potentiating the Wnt signaling cascade. First, cells were treated with varied concentrations of Wnt activators (LRAP, 0.15 nM, 0.25 nM, 1.5 nM) in the absence or presence of the weak β-sheet PA nanofibers (0.001% w/v) and RT-PCR was performed to determine differences in mRNA expression levels of bone marker genes. As previously reported, LRAP exhibited a dose-dependent bioactivity, as shown by increased expression of osteogenic markers (Figure S6).45−47 Similar to the C2C12 cells treated with weak β-sheet PA assemblies and BMP-2 (Figure S2), cotreatment of ST2 cells with LRAP and weak β-sheet PA assemblies significantly enhanced the bioactivity at low doses, achieving osteogenic gene expression levels comparable to those observed in treatments with higher doses of LRAP (Figure 5A). The cotreatment with the weak β-sheet PA assemblies did not enhance the efficacy of LRAP at higher doses, potentially due to the previously reported effect of saturation of the Wnt signaling cascade by LRAP over 1.5 nM.47
Figure 5.

Differentiation of ST2 bone marrow stromal cells treated with the LRAP signaling molecule to activate the Wnt pathway and weak β-sheet PA assemblies. Real time quantitative RT-PCR analysis of osteoblast cell marker gene mRNA expression and mineral deposition by ST2 cells. (A) Two days after osteo-induction, RNA was isolated for qRT-PCR analysis of osteoblast marker genes: Runx2, Osx, Dlx5, and type I collagen (coll. I). Data is expressed as a relative fold increase in gene expression of conditions containing weak β-sheet PA assemblies normalized to conditions with LRAP but lacking PA assemblies. ***p < 0.001, **p < 0.01, nonparametric t-test comparing to conditions with the same LRAP concentration but lacking PA nanofibers (n = 3). (B) Two weeks after bone induction, mineral deposition was assayed with Alizarin Red staining and (C) quantified. *p = 0.018, nonparametric t-test comparing conditions with the same concentration of LRAP but lacking PA nanofibers (n = 3). Data is represented as a relative increase in mineral deposition for cells treated with the weak β-sheet PA nanofibers normalized to the LRAP only treatment baseline. OM: Osteogenic media without LRAP.
The enhancement of gene expression observed in bone marrow stromal ST2 cells was similar to that observed for the C2C12 myoblasts with LRAP and BMP-2, respectively. Next, we sought to confirm cell differentiation and function by evaluating mineralization potential of the cells using an Alizarin Red S stain. Co-treatment of ST2 cells with the weakly cohesive nanostructures again demonstrated enhanced bioactivity at low doses of LRAP, demonstrating increased mineralization similar to that of the high dose LRAP (Figure 5B, C). These results suggest that the weak β-sheet nanostructures in combination with LRAP are capable of enhancing osteogenic differentiation in bone marrow stromal cells through activation of the canonical Wnt/β-catenin signaling pathway.
We hypothesize that the increased fluidity of the lipid rafts has similar effects on both Wnt/ β-catenin and BMP-2 signaling cascades. The environment of the cell membrane has the potential to affect receptor activation, receptor deactivation, and/or signal propagation. Membrane and lipid raft fluidity can potentially alter the physical state of the membrane, and alter the viscosity of membrane microdomains. Recent studies have demonstrated that the lateral mobility of membrane ligands affects both receptor clustering and activity.48,49 Additionally, in experiments with induced pluripotent stem (iPS) cells and cell feeder layers, positive correlations between membrane mobility and biological activity were observed,50 while cancer cells demonstrated reduced growth factor signaling and apoptosis with conditions that reduced membrane fluidity.51 The cell membrane can be subject to fusion with PA assemblies to cause a slightly “leaky” membrane and this may be the mechanism through which membrane fluidity is enhanced. Although toxicity is not observed, antimicrobial surfactant-like peptides and cell penetrating peptides that exhibit both cationic and amphiphilic properties similar to strong and weak PA assemblies have demonstrated this effect on lipid membranes.52−54 Our observations suggest that weakly cohesive nanostructures that interact with cell membranes to modify lipid raft mobility may offer a strategy to improve growth factor signaling.
We have demonstrated here that reducing the internal cohesion of supramolecular peptide nanofibers can significantly enhance growth factor signaling in two separate pathways each mediated by unique receptors and subcellular signaling pathways. We specifically found that nanostructures with weaker β-sheet hyrogen bonding promoted cell differentiation in two established cell model systems for osteogenesis. In contrast, assemblies with stronger hydrogen bonding inhibited differentiation while promoting cell proliferation. We postulate that intercalation of weak, positively charged PA nanostructures or individual molecules into the cell membrane cause the observed increase in lipid raft mobility, which in turn enhances cell signaling. Our study has identified a new mechanism to potentiate signaling by a growth factor using supramolecular nanostructures that could ultimately reduce the necessary therapeutic doses of growth factors, thereby reducing both growth factor induced complications and health care costs.
Acknowledgments
We are grateful to the following core facilities at Northwestern University: Cell Imaging Facility (generously supported by CCSG P30 CA 060553 awarded to the Robert H. Lurie Comprehensive Cancer Center), Biological Imaging Facility (graciously supported by the NU Office of Research), and Northwestern University’s Atomic and Nanoscale Characterization Experimental Center (NUANCE). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We acknowledge Guy Macha and Vukica Srajer for assistance with XRD. We are grateful to the following core facilities at Northwestern University: the Peptide Synthesis Core at the Simpson Querrey Institute for BioNanotechnology, the Biological Imaging Facility (BIF), the Cell Imaging Facility (CIF), and Keck Biophysics Facility for instrument use.
Glossary
Abbreviations
- BMP-2
bone morphogenetic protein 2
- FRAP
fluorescence recovery after photobleaching
- CTxB
cholera toxin subtype B
- WAXS
wide-angle X-ray scattering
- cryoTEM
cryogenic transmission electron microscopy
- OCN
osteocalcin
- ALP
alkaline phosphatase
- ECM
extracellular matrix
- EdU
5-ethynyl-2′-deoxyuridine
- MAPK
mitogen activated protein kinase
- ELISA
enzyme-linked immunosorbent assay
- LRAP
leucine rich amelogenin peptide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.6b00054.
Detailed description of experimental methods, supplementary figures showing cell viability response to PA treatment, C2C12 cell differentiation after exposure to PAs and varying BMP-2 concentration, activation of non-Smad BMP-2 signaling, BMP-2 affinity to PAs, and cellular distribution of PAs. (PDF)
Author Present Address
C.J.N.: Pacific Northwest National Laboratory, Physical Sciences Division, Richland, WA, 99306.
Author Present Address
S.S.: Clarkson University, Department of Biology, Potsdam, NY, 13699.
Author Contributions
C.J.N. and S.S. contributed equally to this work. C.J.N, S.S, M.L.S., and Y.Z. designed and performed the research and wrote the manuscript. S.S.L. and J.Y. performed experiments and S.I.S. supervised the research and wrote the manuscript. All authors have given approval to the final version of the manuscript.
This work was supported by the NIH, the National Institute for Dental and Craniofacial Research Grant 2R01 DE015920-06, and the National Institute for Biomedical Imaging and Bioengineering Grant 2R01EB003806–06A2.
The authors declare no competing financial interest.
Supplementary Material
References
- Hynes R. O. Science 2009, 326 (5957), 1216–1219. 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross M.; Dexter T. Cell 1991, 64 (2), 271–280. 10.1016/0092-8674(91)90638-F. [DOI] [PubMed] [Google Scholar]
- Henry T. D.; Annex B. H.; McKendall G. R.; Azrin M. A.; Lopez J. J.; Giordano F. J.; Shah P.; Willerson J. T.; Benza R. L.; Berman D. S. Circulation 2003, 107 (10), 1359–1365. 10.1161/01.CIR.0000061911.47710.8A. [DOI] [PubMed] [Google Scholar]
- Lee K.; Silva E. A.; Mooney D. J. J. R. Soc., Interface 2011, 8 (55), 153–170. 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez C. M. R.; Stephanopoulos N.; Sur S.; Lee S. S.; Newcomb C.; Stupp S. I. Ann. Biomed. Eng. 2015, 43 (3), 501–514. 10.1007/s10439-014-1166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F.; Rifkin D. B. Curr. Opin. Cell Biol. 2009, 21 (5), 616–622. 10.1016/j.ceb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher D. E.; Mooney D. J.; Zandstra P. Science (Washington, DC, U. S.) 2009, 324, 1673–1677. 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leight J. L. J.; Wozniak M. A. M.; Chen S. S.; Lynch M. L. M.; Chen C. S. C. Mol. Biol. Cell 2012, 23 (5), 781–791. 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R. N.; Shah N. A.; Del Rosario Lim M. M.; Hsieh C.; Nuber G.; Stupp S. I. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (8), 3293–3298. 10.1073/pnas.0906501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. S.; Hsu E. L.; Mendoza M.; Ghodasra J.; Nickoli M. S.; Ashtekar A.; Polavarapu M.; Babu J.; Riaz R. M.; Nicolas J. D.; Nelson D.; Hashmi S. Z.; Kaltz S. R.; Earhart J. S.; Merk B. R.; McKee J. S.; Bairstow S. F.; Shah R. N.; Hsu W. K.; Stupp S. I. Adv. Healthcare Mater. 2015, 4 (1), 131–141. 10.1002/adhm.201400129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. S.; Huang B. J.; Kaltz S. R.; Sur S.; Newcomb C. J.; Stock S. R.; Shah R. N.; Stupp S. I. Biomaterials 2013, 34 (2), 452–459. 10.1016/j.biomaterials.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber M. J.; Tongers J.; Newcomb C. J.; Marquardt K.-T.; Bauersachs J.; Losordo D. W.; Stupp S. I. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (33), 13438–13443. 10.1073/pnas.1016546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha R. H.; Sur S.; Boekhoven J.; Shi H. Y.; Zhang M.; Stupp S. I. Acta Biomater. 2015, 12, 1–10. 10.1016/j.actbio.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X.; Zhou J.; Guvench O.; Sangiorgi F. O.; Li X.; Zhou N.; Xu B. Bioconjugate Chem. 2014, 25 (6), 1031–1035. 10.1021/bc500187m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoven J.; Zha R. H.; Tantakitti F.; Zhuang E.; Zandi R.; Newcomb C. J.; Stupp S. I. RSC Adv. 2015, 5 (12), 8753–8756. 10.1039/C4RA16593D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M. M.; George J. H. Science (Washington, DC, U. S.) 2005, 310 (5751), 1135–1138. 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- Shin H.; Jo S.; Mikos A. G. Biomaterials 2003, 24 (24), 4353–4364. 10.1016/S0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- Srikanth M.; Das S.; Berns E. J.; Kim J.; Stupp S. I.; Kessler J. A. Neuro-Oncology 2013, 15 (3), 319–329. 10.1093/neuonc/nos316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Newcomb C. J.; Zhou Y.; Lei Y. P.; Bringas P.; Stupp S. I.; Snead M. L. Biomaterials 2013, 34 (13), 3303–14. 10.1016/j.biomaterials.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur S.; Newcomb C. J.; Webber M. J.; Stupp S. I. Biomaterials 2013, 34, 4749. 10.1016/j.biomaterials.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger J. E.; Berns E. J.; Bitton R.; Newcomb C. J.; Stupp S. I. Angew. Chem., Int. Ed. 2011, 50 (28), 6292–6295. 10.1002/anie.201100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramonov S. E.; Jun H.-W.; Hartgerink J. D. J. Am. Chem. Soc. 2006, 128 (22), 7291–7298. 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- Newcomb C. J.; Sur S.; Ortony J. H.; Lee O.-S.; Matson J. B.; Boekhoven J.; Yu J. M.; Schatz G. C.; Stupp S. I. Nat. Commun. 2014, 5, 3321. 10.1038/ncomms4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M. Biochemistry 1978, 17 (20), 4277–4285. 10.1021/bi00613a026. [DOI] [PubMed] [Google Scholar]
- Standley S. M.; Toft D. J.; Cheng H.; Soukasene S.; Chen J.; Raja S. M.; Band V.; Band H.; Cryns V. L.; Stupp S. I. Cancer Res. 2010, 70 (8), 3020–3026. 10.1158/0008-5472.CAN-09-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T. T.; Yamaguchi A. A.; Komaki M. M.; Abe E. E.; Takahashi N. N.; Ikeda T. T.; Rosen V. V.; Wozney J. M. J.; Fujisawa-Sehara A. A.; Suda T. T. J. Cell Biol. 1994, 127 (6), 1755–1766. 10.1083/jcb.127.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. Nat. Rev. Mol. Cell Biol. 2000, 1 (3), 169–178. 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Chen G.; Deng C.; Li Y.-P. Int. J. Biol. Sci. 2012, 8 (2), 272–288. 10.7150/ijbs.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen A.; Verschueren K.; Huylebroeck D. FEBS Lett. 2003, 546 (1), 133–139. 10.1016/S0014-5793(03)00566-0. [DOI] [PubMed] [Google Scholar]
- Derynck R.; Zhang Y. E. Nature 2003, 425 (6958), 577–584. 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Luu H. H.; Song W.-X.; Luo X.; Manning D.; Luo J.; Deng Z.-L.; Sharff K. A.; Montag A. G.; Haydon R. C.; He T.-C. J. Orthop. Res. 2007, 25 (5), 665–677. 10.1002/jor.20359. [DOI] [PubMed] [Google Scholar]
- Nishimura R. R.; Kato Y. Y.; Chen D. D.; Harris S. E. S.; Mundy G. R. G.; Yoneda T. T. J. Biol. Chem. 1998, 273 (4), 1872–1879. 10.1074/jbc.273.4.1872. [DOI] [PubMed] [Google Scholar]
- Bessa P. C.; Casal M.; Reis R. L. J. Tissue Eng. Regener. Med. 2008, 2 (2–3), 81–96. 10.1002/term.74. [DOI] [PubMed] [Google Scholar]
- Helmreich E. J. Biophys. Chem. 2002, 100 (1), 519–534. 10.1016/S0301-4622(02)00303-4. [DOI] [PubMed] [Google Scholar]
- Rajendran L.; Simons K. J. Cell Sci. 2005, 118, 1099–1102. 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- Lai E. C. J. Cell Biol. 2003, 162 (3), 365–370. 10.1083/jcb.200307087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Gerl M. J. Nat. Rev. Mol. Cell Biol. 2010, 11 (10), 688–699. 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- Simons K.; Toomre D. Nat. Rev. Mol. Cell Biol. 2000, 1 (1), 31–39. 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Blank N.; Schiller M.; Krienke S.; Wabnitz G.; Ho A. D.; Lorenz H.-M. Immunol. Cell Biol. 2007, 85 (5), 378–382. 10.1038/sj.icb.7100045. [DOI] [PubMed] [Google Scholar]
- Viola A. A.; Schroeder S. S.; Sakakibara Y. Y.; Lanzavecchia A. A. Science 1999, 283 (5402), 680–682. 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- Day C. A.; Kenworthy A. K. PLoS One 2012, 7 (4), e34923–e34923. 10.1371/journal.pone.0034923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Science 2010, 327 (5961), 46–50. 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Ramos M.; Lamé M. W.; Segall H. J.; Wilson D. W. Vasc. Pharmacol. 2006, 44 (1), 50–59. 10.1016/j.vph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Wertz J. W.; Bauer P. M. Biochem. Biophys. Res. Commun. 2008, 375 (4), 557–561. 10.1016/j.bbrc.2008.08.066. [DOI] [PubMed] [Google Scholar]
- Wen X.; Cawthorn W. P.; MacDougald O. A.; Stupp S. I.; Snead M. L.; Zhou Y. Biomaterials 2011, 32 (27), 6478–86. 10.1016/j.biomaterials.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Newcomb C. J.; Warotayanont R.; Snead M. L.; Stupp S. I.. Signalling effect of leucine-rich amelogenin peotide (LRAP) on bone regeneration. In Amelogenins: Multifaceted protein for dental and bone formation and repair; Goldberg M., Ed.; Bentham Science Publisher: Saif Zone, Sharjah, United Arab Emirates, 2010; pp 219–225. [Google Scholar]
- Warotayanont R.; Frenkel B.; Snead M. L.; Zhou Y. Biochem. Biophys. Res. Commun. 2009, 387, 558–563. 10.1016/j.bbrc.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabley D.; Retterer S.; Marshall S.; Salaita K. Integr. Biol. 2013, 5 (4), 659–668. 10.1039/c3ib20239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaita K.; Nair P. M.; Petit R. S.; Neve R. M.; Das D.; Gray J. W.; Groves J. T. Science 2010, 327 (5971), 1380–1385. 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Mao H.; Joddar B.; Umeki N.; Sako Y.; Wada K.-I.; Nishioka C.; Takahashi E.; Wang Y.; Ito Y. Sci. Rep. 2015, 5, 11386. 10.1038/srep11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavolari S.; Munarini A.; Storci G.; Laufer S.; Chieco P.; Guarnieri T. Cancer Lett. 2012, 321 (2), 187–194. 10.1016/j.canlet.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Yang S.-T.; Zaitseva E.; Chernomordik L. V.; Melikov K. Biophys. J. 2010, 99 (8), 2525–2533. 10.1016/j.bpj.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehsorkhi A.; Castelletto V.; Hamley I. W.; Seitsonen J.; Ruokolainen J. Langmuir 2013, 29 (46), 14246–14253. 10.1021/la403447u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Jang H.; Harlen K.; Puri A.; Nussinov R.; Schneider J. P.; Blumenthal R. Biophys. J. 2013, 105 (9), 2093–2103. 10.1016/j.bpj.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.