

Abstract
Purpose
A hallmark of albinism is foveal hypoplasia. However, literature suggests variable foveal development. This study evaluates the association between ocular phenotype and foveal morphology to demonstrate the broad structural and functional spectrum.
Methods
Best-corrected visual acuity (BCVA), nystagmus, angle kappa, stereoacuity, iris transillumination, macular melanin presence, foveal avascular zone, and annular reflex were recorded in 14 patients with albinism. Spectral-domain optical coherence tomography provided macular images.
Results
The clinical phenotype was broad, with BCVA varying from 20/20 to 20/100. Better BCVA was associated with a preserved foveal avascular zone, annular macular reflex, stereoacuity, and macular melanin. Imaging demonstrated a continuum of foveal development correlating with BCVA. Individuals with a rudimentary pit had normal inner and outer segment lengthening and better BCVA.
Conclusions
The spectrum of ocular structure and visual function in albinism is broad, suggesting a possible diagnosis of albinism in a patient with an even more normal clinical presentation.
INTRODUCTION
Albinism, an inherited disorder of melanin biosynthesis, produces a variable phenotype, classified according to the mutation(s) on a gene known to cause albinism. Those born with white hair have oculocutaneous albinism type 1 (OCA1), with mutation(s) in the TYR (Tyrosinase) gene on chromosome 11q14-q21.1 Some never develop any melanin pigment (OCA1A; MIM #2031100), and others develop melanin pigment in their hair, skin, eyelashes, and eyes (OCA1B; MIM #606952). Mutations in the 0CA2 gene on chromosome 15ql2-ql3 cause OCA2 (MIM #203200).2 More unusual types of albinism, including OCA3 and OCA4,3 have been described and, to date, three additional types of OCA have been reported but remain incompletely described.4–6 OCA3 (MIM #203290) occurs with mutations on the TYRP1 gene (encoding tyrosinase-related protein 1) and is often characterized by a milder phenotype with nearly normal hair and skin pigmentation relative to OCA1 and OCA2,7 whereas OCA4 (MIM #606574) is caused by SLC45A2 mutations and demonstrates a variable phenotype spanning the entire spectrum of hair and iris color permutations.8 Finally, OCA is rarely associated with syndromes that necessitate systemic evaluation, including Chediak–Higashi and Hermansky–Pudlak syndromes.9,10
All types of OCA have autosomal recessive inheritance. Males with normal cutaneous and hair pigment but the ocular features of albinism have mutations in the GPR143 gene at Xp22.2; ocular albinism (OA1; MIM #300500) is as an X-linked disorder. Others with the appearance of ocular albinism have been found to have mutations in one of the genes causing OCA, indicating that skin pigmentation deficiencies in OCA can at times be subtle.10,11
The phenotypic spectrum associated with albinism continues to expand, with recent studies highlighting more atypical features.8,12–17 Creel et al.18 used visual evoked potentials and others have used functional magnetic resonance imaging19 to show excessive retinostriate decussation in albinism.20 Most individuals with albinism have a positive angle kappa.21 The ocular features in albinism vary, but some degree of foveal hypoplasia is constant. Optical coherence tomography (OCT) has shown both an absent and present foveal depression.22–26 Additionally, adaptive optics imaging has been used to look at foveal cone packing and has demonstrated that peak foveal cone density can be variable in albinism, and has also shown that normal cone packing can occur even in the absence of a foveal pit.26,27
Although arrested foveal development is considered a classic finding in albinism, both clinical and imaging studies have described some individuals with rudimentary foveal development,13,15,17 indicating that foveal hypoplasia (or fovea plana) may not be sufficient to describe macular anatomy in all individuals. In this study, we sought to examine clinical correlates that may be related to foveal development in albinism.23 The overarching goal was to qualitatively describe the phenotypic spectrum of albinism, rather than provide a formal, large cross-sectional analysis of albinism subtypes.
PATIENTS AND METHODS
This study was approved by the Institutional Review Boards at the University of Minnesota, the Medical College of Wisconsin, and the Marshfield Clinic, and was compliant with the Health Information Portability and Accountability Act. Fourteen white patients with a diagnosis of albinism were examined by a pediatric ophthalmologist and geneticist at the University of Minnesota. The parents/patients consented to imaging at the Medical College of Wisconsin. The monocular spectral-domain OCT (SD-OCT) imaging results of six patients (JC103, JC0125, AD0063, JC0170, JC0131, and JC0140) were previously published.23 We prospectively examined the clinical features of these six patients and eight additional patients who also had SD-OCT imaging to further understand the effect of foveal morphology on visual function over a broad spectrum of albinism.
Initial diagnosis of albinism was based on several factors, but all individuals had to have evidence of hypopigmentation of the retina and underdevelopment of the fovea. History included family history of albinism, ancestry, hair color at birth, ability to suntan, photosensitivity and methods to alleviate it, and age at which prescription glasses wear was initiated. Color of hair, eyelashes, and iris were noted. We recorded features associated with albinism: nystagmus, grade of macular transparency,28 presence or absence of macular melanin pigment in the retinal pigment epithelium (RPE),13,16 an annular reflex and/or a foveal avascular zone, grade of iris transillumination,28 stereopsis, strabismus, and angle kappa. Procedures are described below.
Blood was drawn for molecular DNA analysis on all patients. Based on phenotype, patients were evaluated for mutations in TYR, OCA2, TYRP1, SLC45A2 (OCA1, 2, 3, and 4, respectively), and/or GPR143 (OA1) genes by Sanger sequencing, as previously described.29–32 Patients of possible African descent were screened for the 2.7 kb P gene deletion.33 DNA sequences were aligned and analyzed for mutations using DNASTAR Lasergene software (DNASTAR Inc., Madison, WI). Identified mutations were compared to the Albinism database (http://www.ifpcs.org/albinism/index) to determine novelty.
Monocular and binocular best-corrected visual acuity (BCVA) was measured with linear letters at 20 feet (M & S Technologies, Niles, IL). Preferred head posture was allowed. Monocular testing used a fogging technique. Stereoacuity was assessed with the Titmus vectograph (Stereo Optical Co., Inc., Chicago, IL). A positive angle kappa (measured in millimeters of nasal displacement of the light reflex) was noted with the subject fixing monocularly on a penlight. Prism and alternate cover test determined binocular alignment at 20 feet. Type of nystagmus was recorded; when absent, reassessment with slit-lamp biomicroscopy and direct ophthalmoscopy ensured its absence. Iris transillumination was graded with slit-lamp biomicroscopy (grade 1 represents punctuate transillumination; grade 4 is complete iris transillumination [ie, no iris pigment]).28 Dilation and cycloplegia were obtained with phenylephrine 2.5% and tropicamide 1%, with streak retinoscopy and refinement 20 minutes later. Macular transparency was graded (choroidal vessels easily visible in grade 1; no choroidal vessels visible in grade 3).28 Presence or absence of granular melanin pigment in the macula was determined with direct ophthalmoscopy. The macular vascular pattern was evaluated for normal vascular wreathing and a foveal avascular zone with 30° fundus photographs (Zeiss FF4 fundus camera; Carl Zeiss, Jena, Germany). A rudimentary annular reflex in the macula was assessed with binocular indirect ophthalmoscopy and a 20-diopter (D) condensing lens, facilitated by slightly rocking the lens. Patients with a milder phenotype underwent visual evoked potential testing to detect retinostriate misrouting, using a technique described by Creel et al.34 The presence of retinostriate misrouting is useful in clinching the diagnosis of albinism when it is uncertain by clinical examination.
Several SD-OCT images were acquired for each patient due to nystagmus. Volumetric scans were repeated for determination of preferred retinal locus of fixation, and until a scan with relatively low motion distortion at the expected fovea was obtained. Line scans were registered and averaged to reduce speckle noise in the image prior to analysis. SD-OCT imaging parameters included presence or absence of a foveal pit, doming of the fovea (elevation around the intended area of a fovea with visible central depression), determination of preferred retinal locus with replicate SD-OCT imaging, retinal thickness (from the central subfield, as defined by Early Treatment of Diabetic Retinopathy Study [ETDRS], at the expected fovea), and photoreceptor inner and outer segment lengthening, measured as the relative inner or outer segment length at the fovea compared to that at 1.75 mm from the foveal center. Several points were considered to assign the preferred retinal locus compared to individuals without albinism.23,35 Normal inner segment/outer segment length was defined as a ratio within two standard deviations of the normal mean.23
RESULTS
Details of the history and clinical examination are in Table A (available in the online version of the article), with patients listed from better binocular BCVA to relatively worse BCVA. Figure 1 shows the spectrum of foveal morphology in this study. Binocular BCVA ranged from 20/20- to 20/80 and BCVA of the imaged eye from 20/20- to 20/100. Five patients had granular melanin pigment in the macula and all of these had BVCA of 20/40 or better. Additionally, four patients with macular melanin pigmentation had a foveal avascular zone and three possessed a rudimentary annular reflex. More iris pigment was found in these individuals and four of the five had stereoacuity. Three patients had granular macular pigmentation and normal inner and outer segment lengths and an indistinct foveal pit, suggesting that these findings are associated with less arrest in foveal development. Additional imaging results23 showed that neither retinal thickness nor axial length correlated with BCVA. As expected, longer axial length was associated with myopia, whereas shorter axial length was associated with hyperopia. All 14 patients had a positive angle kappa value.21 Eight had a temporal preferred retinal locus for fixation (based on imaging), consistent with their positive angle kappa value. Patients with the worst BCVA had a larger positive angle kappa value than those with better acuity, but a temporal preferred retinal locus alone was not consistently associated with reduced BCVA. The preferred retinal locus had some variability during SD-OCT imaging. Four of the six patients with BCVA of 20/40 or better lacked nystagmus; the 10 remaining patients all demonstrated some type of nystagmus.
Figure 1.
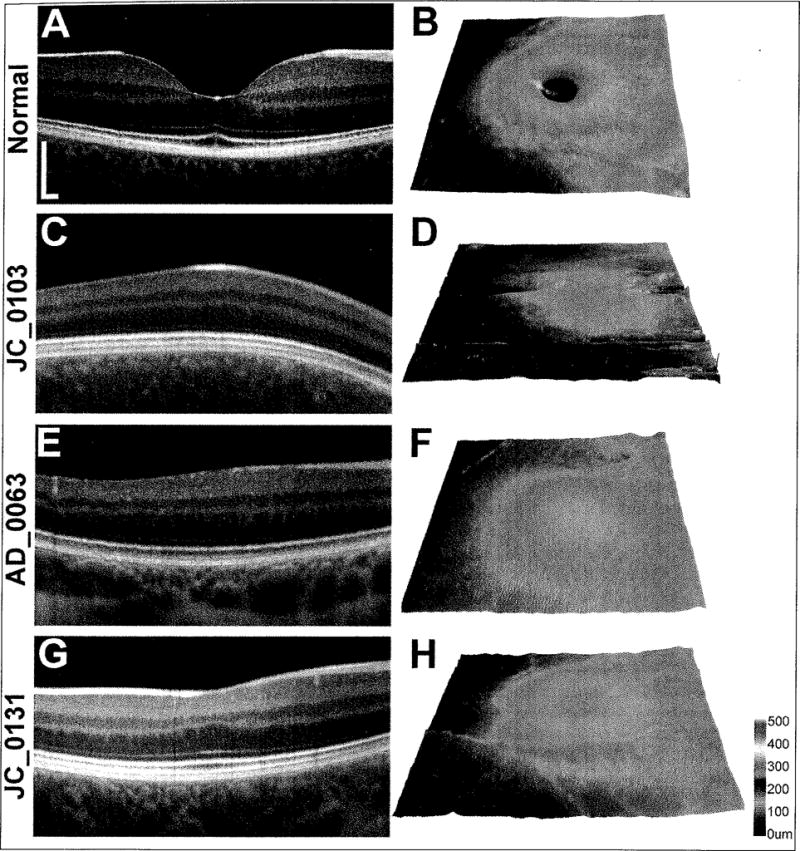
(A,C,E,G) Linear spectral-domain optical coherence tomography scans at the expected fovea (Bioptigen, Research Triangle Park, NC). (B,D,F,H) Retinal thickness maps (Cirrus HD-OCT; Carl Zeiss Meditec, Dublin, CA). (A,B) Normal foveal pit morphology includes doming of the retina with a deep depression at the point of cone outer segment elongation, indicating cone packing. (C,D) JC0103 lacks both the doming and depression of the retina at the expected fovea. (E,F) AD0063 is a representative subject with doming but no depression. (G,H) JC0131 demonstrates foveal morphology that includes both doming and a slight depression at the expected fovea. (Panels C–H were reprinted from McAllister JT, Dubis AM,Tait DM, et al. Arrested development: high-resolution imaging of foveal morphology in albinism. Vis Res. 2010;50:810–817, with permission from Elsevier.)
Molecular analysis was completed for all 14 patients. Five patients with OCA1A or OCA1B had TYR mutations, three had GPR143 mutations (consistent with their diagnosis of OA1), two had 0CA2 c.2228C mutations, one had a different variant OCA2 mutation, and one had a mutation of the SLC45A2 transporter protein known to cause OCA4. No mutations were found in the remaining two patients. In this study, we identified four novel mutations (GPR143 c.797, GPR143 c.933, SLC45A2 c.264, and SLC45A2 c.1417) in three different patients.
DISCUSSION
Foveal hypoplasia (ie, absence of a foveal pit) is commonly thought to be an inevitable finding in albinism. However, the imaging results reported here, in addition to the clinical findings of the 14 patients included in this report, indicate that some individuals with albinism have rudimentary foveal development and that this is associated with relatively better BCVA than those without any foveal development, consistent with previous findings.25 The ophthalmoscopic appearance of the fovea may include a macular annular reflex even though a foveal pit is missing. Other associated clinical findings that seem to be associated with better BCVA include the presence of stereoacuity, a mildly positive angle kappa value, a moderate amount of iris pigment, and macular melanin pigment in the RPE. Therefore, the spectrum of structural and visual findings in albinism appears to be greater than commonly thought. We have shown that precise documentation of ocular structure allows correlation with visual function. The results of this study alert the clinician to consider a diagnosis of albinism in an individual with relatively good BCVA, measurable stereoacuity, macular melanin pigment, no nystagmus, and rudimentary foveal development who might not have been previously diagnosed as having albinism.
Determining the type of albinism with only history and examination is possible for some types of albinism (eg, an individual with longstanding white hair and eyelashes, absence of suntanning, and full iris transillumination has OCA1A), whereas a male with the ocular findings of albinism and pigmentary mosaicism in his mother has OA1. Determining the type of albinism in other individuals often requires molecular testing. We found mutations in 12 of 14 patients tested. Interestingly, patient JC0131 reported white hair at birth, suggesting OCA1, but actually had OCA2 with genetic evaluation. When molecular analysis is negative, the diagnosis can be made clinically, based on ocular (and often skin and hair) hypopigmentation, an underdeveloped fovea, and misrouting of the retinostriate fibers detected with a visual evoked potential, as was done in two of our patients who had relatively better BCVA. Undetectable mutations or detection of only one mutation in albinism has been reported by others.3,36–38 Although a limited sample size, the patients described herein exhibited the phenotypic breadth of clinical and imaging variability in albinism. Ten of the 14 patients had some degree of horizontal nystagmus, consistent with data that nystagmus is present in most patients with albinism, regardless of type.3 The four patients lacking nystagmus all had BCVA of 20/40 better, demonstrating a possible marker of a more subtle phenotype. All patients in this study had a positive angle kappa value, as previously reported.21 In this study, we show that it appears that a more positive angle kappa value is present in those with more reduced BCVA, recognizing that the assessment of a positive angle kappa value is not precise. Stereoacuity, present in five patients, correlated positively with better BCVA and reduced iris transillumination, confirming an earlier study.13
Emmetropization, the developmental balance of anterior segment refraction with axial length to produce an ocular system that perfectly focuses light rays on the retina, has been described to be impaired in animals with albinism.39,40 This was classically assumed to be primarily due to foveal hypoplasia; however, these studies pointed to the importance of the other ocular features (eg, RPE functional abnormalities) during emmetropization. Additionally, a higher amount of astigmatism occurs in albinism, associated with decreased emmetropization during the years of ocular development.41 The current study was unable to correlate the size of refractive error with BCVA. In our study, seven of the patients had hyperopia, generally associated with shorter axial lengths, and the others had myopia and longer axial lengths. With-the-rule astigmatism was common, and was even present in many of the individuals with almost normal vision. Corrective lenses during childhood results in improved BCVA in those with albinism.42 Our patients began wearing glasses in childhood, although earlier age of beginning wear did not correlate with better visual acuity outcomes or decreased refractive errors.
A spectrum of BCVA and clinical and imaging features in individuals with albinism is reported here, but a larger sample size would help to confirm the trends suggested by our 14 patients. Our results showed that retinal thickness does not correlate with BCVA, similar to normal individuals,43 and that gender did not appear to have a meaningful association with retinal thickness or pit depth, in contrast to normal individuals.44 One final limitation of our study is that it lacked ethnic variability, because all patients were white with Western European ancestry. Moving forward, a larger sample size will be essential to elicit the interplay among shared familial genes, specific mutations, age, gender, and other factors in determining the morphological and functional outcomes in individuals with albinism. Such could have a strong impact on our ability to predict visual outcomes and viable treatment options for patients.
The clinical and imaging features noted in this study define the spectrum of albinism. A large cross-sectional study to demonstrate genotype-phenotype correlations would be of great utility in further defining this spectrum. Identifying the clinical and imaging findings determinant of visual outcomes holds important implications in the future for patients and their families dealing with a new diagnosis of albinism. As we continue to improve our ability to predict ocular developmental outcomes in this population and to clinically identify those less severely affected with albinism, patients may be able to be diagnosed earlier and counseled more thoroughly on what they can expect in the years to come.
Acknowledgments
Supported by the Albinism and Related Eye Disorders Research Fund, University of Minnesota Foundation; unrestricted grants to the Department of Ophthalmology & Visual Neurosciences, University of Minnesota, and the Department of Ophthalmology, Medical College of Wisconsin from Research to Prevent Blindness, Inc., New York, NY; NIH Grants P30 EY001931, T32 EY014537, C06 RR016511, R01 EY017607; RD & Linda Peters Foundation; Vision for Tomorrow; and The Gene & Ruth Posner Foundation. Partially supported by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR000427.
The authors thank Phyllis Summerfelt for making the travel arrangements and the patients and their families for their time and assistance.
Footnotes
The authors have no financial or proprietary interest in the materials presented herein.
References
- 1.Oetting W. The tyrosinase gene and oculocutaneous albinism type 1 (OCA1): a model for understanding the molecular biology of melanin formation. Pigment Cell Res. 2000;13:320–325. doi: 10.1034/j.1600-0749.2000.130503.x. [DOI] [PubMed] [Google Scholar]
- 2.Abadi R, Pascal E. The recognition and management of albinism. Ophthalmic Physiol Opt. 1989;9:3–15. doi: 10.1111/j.1475-1313.1989.tb00797.x. [DOI] [PubMed] [Google Scholar]
- 3.Gargiulo A, Testa F, Rossi S, et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest Ophthalmol Vis Sci. 2011;52:1281–1289. doi: 10.1167/iovs.10-6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kausar T, Bhatti MA, Ali M, Shaik RS, Ahmed ZM. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet. 2013;84:91–93. doi: 10.1111/cge.12019. [DOI] [PubMed] [Google Scholar]
- 5.Wei AH, Zang DJ, Zhang Z, et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J Invest Dermatol. 2013;133:1834–1840. doi: 10.1038/jid.2013.49. [DOI] [PubMed] [Google Scholar]
- 6.Gronskov K, Dooley CM, Ostergaard E, et al. Mutations in c10orf11, a melanocyte-diffetentiation gene, cause autosomal-recessive albinism. Am J Hum Genet. 2013;92:415–421. doi: 10.1016/j.ajhg.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang K, Li Z, Lei J, et al. Oculocutaneous albinism type 3 (OCA3): Analysis of two novel mutations in TYRP1 gene in two Chinese patients. Cell Biochem Biophys. 2011;61:523–529. doi: 10.1007/s12013-011-9234-0. [DOI] [PubMed] [Google Scholar]
- 8.Mauri L, Barone L, Al Oum M, et al. SLC45A2 mutation frequency in oculocutaneous albinism Italian patients doesn’t differ from other European studies. Gene. 2014;533:398–402. doi: 10.1016/j.gene.2013.09.053. [DOI] [PubMed] [Google Scholar]
- 9.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Introne W, Boissy R, Gahl W. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet and Metab. 1999;68:283–303. doi: 10.1006/mgme.1999.2927. [DOI] [PubMed] [Google Scholar]
- 11.Summers CG, Oetting WS, King RA. Diagnosis of oculocutaneous albinism with molecular analysis. Am J Ophthalmol. 1996;121:724–726. doi: 10.1016/s0002-9394(14)70647-6. [DOI] [PubMed] [Google Scholar]
- 12.Kasmann-Kellner B, Seitz B. Phenotype of the visual system in oculocutaneous and ocular albinism [article in German] Ophthalmology. 2007;104:648–661. doi: 10.1007/s00347-007-1571-4. [DOI] [PubMed] [Google Scholar]
- 13.Lee KA, King RA, Summers CG. Stereopsis in patients with albinism: clinical correlates. J AAPOS. 2001;5:98–104. doi: 10.1067/mpa.2001.112441. [DOI] [PubMed] [Google Scholar]
- 14.Whang SJ, King RA, Summers CG. Grating acuity in albinism in the first three years of life. J AAPOS. 2002;6:393–396. doi: 10.1067/mpa.2002.129047. [DOI] [PubMed] [Google Scholar]
- 15.Harvey PS, King RA, Summers CG. Spectrum of foveal development in albinism detected with optical coherence tomography. J AAPOS. 2006;10:237–242. doi: 10.1016/j.jaapos.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Summers CG. Vision in albinism. Trans Am Ophthal Soc. 1996;94:1095–155. [PMC free article] [PubMed] [Google Scholar]
- 17.Chong GT, Farsiu S, Freedman SF, et al. Abnormal foveal morphology in ocular albinism imaged with spectral-domain optical coherence tomography. Arch Ophthalmol. 2009;127:37–44. doi: 10.1001/archophthalmol.2008.550. [DOI] [PubMed] [Google Scholar]
- 18.Creel DJ, Summers CG, King RA. Visual anomalies associated with albinism. Ophthalmic Paediatric Genet. 1990;11:193–200. doi: 10.3109/13816819009020979. [DOI] [PubMed] [Google Scholar]
- 19.Schmitz B, Krick C, Kasmann-Kellner B. Morphology of the optic chiasm in albinism [article in German] Ophthalmologe. 2007;104:662–665. doi: 10.1007/s00347-007-1572-3. [DOI] [PubMed] [Google Scholar]
- 20.Neveu M, von dem Hagen E, Morland A, Jeffery G. The fovea regulates symmetrical development of the visual cortex. J Comp Neurol. 2008;506:791–800. doi: 10.1002/cne.21574. [DOI] [PubMed] [Google Scholar]
- 21.Merrill KS, Lavoie JD, King RA, Summers CG. Positive angle kappa in albinism. J AAPOS. 2004;8:237–239. doi: 10.1016/j.jaapos.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Kubal A, Dagnelie G, Goldberg M. Ocular albinism with absent foveal pits but without nystagmus, photophobia, or severely reduced vision. J AAPOS. 2009;13:610–612. doi: 10.1016/j.jaapos.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 23.McAllister JT, Dubis AM, Tait DM, et al. Arrested development: high-resolution imaging of foveal morphology in albinism. Vis Res. 2010;50:810–817. doi: 10.1016/j.visres.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas MG, Kumar A, Mohammad S, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011;118:1653–1660. doi: 10.1016/j.ophtha.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammad S, Gottlob I, Kumar A, et al. The functional significance of foveal abnormalities in albinism measured using spectral-domain optical coherence tomography. Ophthalmology. 2011;118:1645–1652. doi: 10.1016/j.ophtha.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 26.Wilk M, McAllister J, Dubis A, Cooper RF. Relationship between fovea] cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014;13:5923. doi: 10.1167/iovs.13-13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marmor MF, Choi SS, Zawadzki RJ, Werner JS. Visual insignificance of the foveal pit. Arch Ophthalmol. 2008;126:907–913. doi: 10.1001/archopht.126.7.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers CG, Knobloch WH, King RA, Witkop C. Herman-sky-Pudlak syndrome: ophthalmic findings. Ophthalmology. 1988;95:545–554. doi: 10.1016/s0161-6420(88)33152-0. [DOI] [PubMed] [Google Scholar]
- 29.Giebel LB, Strunk KM, Spritz RA. Organization and nucleotide sequences of the human tyrosinase gene and a truncated tyrosinase-related segment. Genomics. 1991;9:435–445. doi: 10.1016/0888-7543(91)90409-8. [DOI] [PubMed] [Google Scholar]
- 30.Lee ST, Nicholls RD, Jong MT, Fukai K, Spritz RA. Organization and sequence of the human P gene and identification of a new family of transport proteins. Genomics. 1995;26:354–363. doi: 10.1016/0888-7543(95)80220-g. [DOI] [PubMed] [Google Scholar]
- 31.Newton JM, Cohen-Barak O, Hagiwara N, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet. 2001;69:981–988. doi: 10.1086/324340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schiaffino MV, Bassi MT, Galli L, et al. Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hum Mol Genet. 1995;4:2319–2325. doi: 10.1093/hmg/4.12.2319. [DOI] [PubMed] [Google Scholar]
- 33.Durham-Pierre D, Gardner JM, Nakatsu Y, et al. African origin of an intragenic deletion of the human P gene in tyrosinase positive oculocutaneous albinism. Nat Genet. 1994;7:176–179. doi: 10.1038/ng0694-176. [DOI] [PubMed] [Google Scholar]
- 34.Creel D, O’Donnell FE, Jr, Witkop CJ., Jr Visual system anomalies in human ocular albinos. Science. 1978;201:931–933. doi: 10.1126/science.684419. [DOI] [PubMed] [Google Scholar]
- 35.Rohrschneider K. Determination of the location of the fovea on the fundus. Invest Ophthalmol Vis Sci. 2004;45:3257–3258. doi: 10.1167/iovs.03-1157. [DOI] [PubMed] [Google Scholar]
- 36.Hutton SM, Spritz RA. A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Invest Ophthalmol Vis Sci. 2008;49:868–872. doi: 10.1167/iovs.07-0791. [DOI] [PubMed] [Google Scholar]
- 37.Preising MN, Forster H, Gonser M, Lorenz B. Screening of TYR, OCA2, GPR143, and MC1R in patients with congenital nystagmus, macular hypoplasia, and fundus hypopigmentation indicating albinism. Mol Vis. 2011;17:939–945. [PMC free article] [PubMed] [Google Scholar]
- 38.Gronskov K, Ek J, Sand A, et al. Birth prevalence and mutation spectrum in Danish patients with autosomal recessive albinism. Invest Ophthalmol Vis Sci. 2009;50:1058–1064. doi: 10.1167/iovs.08-2639. [DOI] [PubMed] [Google Scholar]
- 39.Jiang L, Long K, Schaeffel F, et al. Disruption of emmetropization and high susceptibility to deprivation myopia in albino guinea pigs. Invest Ophthalmol Vis Sci. 2011;52:6124–6132. doi: 10.1167/iovs.10-7088. [DOI] [PubMed] [Google Scholar]
- 40.Rymer J, Choh V, Bharadwaj S, et al. The albino chick as a model for studying ocular developmental anomalies, including refractive errors, associated with albinism. Exp Eye Res. 2007;85:431–442. doi: 10.1016/j.exer.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Wyatt LM, Felius J, et al. Onset and progression of with-the-rule astigmatism in children with infantile nystagmus syndrome. Invest Ophthalmol Vis Sci. 2010;51:594–601. doi: 10.1167/iovs.09-3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson J, Lavoie J, Merrill K, King RA, Summers CG. Efficacy of spectacles in persons with albinism. J AAPOS. 2004;8:515–520. doi: 10.1016/j.jaapos.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Wong AC, Chan CW, Hui SP. Relationship of gender, body mass index, and axial length with central retinal thickness using optical coherence tomography. Eye (Lond) 2005;19:292–297. doi: 10.1038/sj.eye.6701466. [DOI] [PubMed] [Google Scholar]
- 44.Wagner-Schuman M, Dubis AM, Nordgren RN, et al. Race- and sex-related differences in retinal thickness and foveal pit morphology. Invest Ophthalmol Vis Sci. 2011;52:625–663. doi: 10.1167/iovs.10-5886. [DOI] [PMC free article] [PubMed] [Google Scholar]