

Abstract
Chemical carcinogenesis studies in animals have directly contributed to a reduction of cancer burden in the human population through their ability to identify carcinogens from the workplace, diet, and environment. Reduced exposure to these carcinogens through lifestyle changes, government regulation, or change in industry practices has reduced cancer incidence in exposed populations. In addition to providing the first experimental evidence for the link between chemical and radiation exposure and cancer, animal models of environmentally induced cancer have and will continue to provide important insight into the causes, mechanisms, and conceptual frameworks of cancer. More recently, combining chemical carcinogens with genetically engineered mouse models (GEMMs) has emerged as an invaluable approach to study the complex interaction between genotype and environment that contributes to cancer development. In the future, animal models of environmentally induced cancer are likely to provide insight into areas such as the epigenetic basis of cancer, genetic modifiers of cancer susceptibility, the systems biology of cancer, inflammation and cancer, and cancer prevention.
Any model is by definition an imperfect representation of that which it seeks to emulate. Regardless of how accurate any given cancer model is, it’s true value should be measured by it’s ability to guide research and, from a practical perspective, to protect human health. Mouse, rat, and other animal models of chemically induced cancer have been remarkably faithful in revealing underlying mechanisms of carcinogenesis and pinpointing both genetic and environmental factors that influence cancer susceptibility in the human population.
There are currently two major applications of animal models of chemical carcinogenesis. One is experimental cancer research, where the objective is to learn about the causes and mechanisms of cancer, as well as translational research objectives such as chemoprevention or early detection of cancer. The second more practical application is to test chemicals with potential human exposure for carcinogenic activity, in the so-called carcinogen bioassay. This chapter focuses on mouse models of chemical and radiation carcinogenesis with an emphasis on combining such models with genetically engineered mouse models (GEMMs). Highlights of selected chemical carcinogen models, including history, applications, protocols, and future directions will be discussed. A brief description of the history and use of the carcinogen bioassay will also be presented.
Cancer is a disease caused by both genetic mutations and environmental exposures
It is widely accepted that cancer is a “genetic disease” (Vogelstein and Kinzler, 2004). Much support for this concept has come from the detection of mutated genes in tumors and the use of GEMMs which are designed to carry mutations in the orthologous genes that are mutated in human cancers. These mouse models, to varying degrees of accuracy, recapitulate the pathogenesis of human cancer and provide important experimental evidence for the genetic basis of cancer (Van Dyke and Jacks, 2002;Becher and Holland, 2006;Frese and Tuveson, 2007). However, current estimates indicate that inherited genetic predisposition makes a relatively minor contribution to most cancers (Lichtenstein et al., 2000) implying that most mutations that arise in -and contribute to- cancer are somatically acquired through spontaneous events or as a result of environmental exposure. Supporting the latter idea, human cancer has a prominent, and in some cases overwhelming environmental etiology, indicating cancer is also an “environmental disease” (IARC, 1990;Doll and Peto, 1981). Environment is defined as anything people interact with, including exposure from lifestyle choices, natural and medical radiation, sunlight, workplace exposure, drugs, and substances in the air, water, and soil (IOM, 2001;OTA, 1981). Thus, while GEM models mirror some of the genetic, biologic, and pathologic features of human cancer, on their own, i.e. under controlled environmental conditions, they do not take into account the complexity of environmental exposures that contribute to cancer. Combining chemical and other environmental exposures with genetically defined mouse models provides a useful experimental setting to study the interaction between host genotype and environmental exposures that ultimately dictate cancer risk.
Early studies, the use of animals to identify carcinogens
Over 200 years ago, the first report linking environmental exposure and cancer was published by the English physician and surgeon Percivill Pott (Pott, 1775). Dr. Pott noted that a common history provided by patients with scrotal skin cancer was employment as chimney sweeps in their youth. These young workers were chronically exposed to high levels of soot and tar and developed cancer after a long latency. The observation of tumor latency noted by Pott was highly prescient and predated the concept of multistage cancer. Over 100 years later, in 1895, Rehn reported observations of increased bladder cancers in workers expose to aniline dyes (Rehn, 1895). These and other seminal epidemiologic observations ultimately gave rise to the field of chemical carcinogenesis (Lawley, 1994). However, it wasn’t until 1915, first published in English in 1918, that a direct causal link between chemical exposure and cancer was established. Yamagiwa and Ichikawa reported that chronic application of coal tar to rabbit ears gave rise initially to benign lesions some of which eventually developed into malignant epidermal tumors (Yamagiwa and Ichikawa, 1918). This was soon followed by similar studies in mice (Tsutsui, 1918). In the 1930s, the first pure chemical compounds, the polycyclic aromatic hydrocarbons (PAHs) benzo[a]pyrene, dibenz[a,h]anthracene, and 3-methylcholanthrene were shown to induce tumors in mice, firmly establishing defined chemical entities as a cause of cancer (Cook et al., 1932;Kennaway, 1955). PAHs consist of multiple fused benzene rings and are formed during incomplete combustion of organic matter. PAHs are a major component of cigarette smoke and air pollution particulates and, to this day, are widely distributed in the environment and found in certain foods. In part, due to the widespread distribution of PAHs such as benzo[a]pyrene, there are few epidemiologic studies linking its exposure to human cancer and animal models provide the main evidence of their carcinogenicity.
Following the reports of bladder cancer in workers in the dye industry, Sasaki and Yoshida, succeeded in inducing liver cancer in rats after feeding the azo dye o-amidoazotoluene (Sasaki and Yoshida, 1935). Kinosita in 1936 reported 4-dimethyl-aminazobenzene (DAB) induced liver cancer (Kinosita, 1936), and in 1941, bladder and other cancers were found in rats exposed to 2-acetylaminofluorine (2AAF) (Weisberger and Weisburger, 1958). Studies of azo dyes also provided the first evidence that some chemicals require metabolic activation by the host to cause cancer (Miller and Miller, 1947). The epidemiology of aromatic amines and cancer is largely the epidemiology of industry related bladder cancer, and due, in no small part to these early animal studies, the exposure of workers to these compounds has been greatly reduced. Aromatic amines are still used in industry during chemical synthesis and are found in cigarette smoke. The link between cigarette smoking and bladder cancer has been attributed to aromatic amines (Vineis and Pirastu, 1997) indicating a continuing threat from these compounds.
Ethyl carbamate (urethane) was used as a sedative and large numbers of Japanese patients were exposed to this carcinogen between 1950 and 1975. Urethane was shown to induce lung cancer in mice as early as 1943 (Nettleship et al., 1943), but decades passed before its deliberate use was stopped (Miller, 1991). Urethane is found at low levels in many foods as a byproduct of fermentation. Like many other carcinogens, urethane requires metabolic activation by the p450 system, in this case to vinyl carbamate epoxide, which can covalently bind to DNA to form mutagenic DNA adducts (Forkert, 2010). The N-nitroso compounds represent another important class of chemical carcinogens. One of these, N-Nitrosodimethylamine (DMN) was first shown to be carcinogenic in rats (Magee and Barnes, 1956). Subsequently, many additional N-nitroso compounds were shown to cause cancer in animals and nearly every species tested is sensitive to cancer induced by DMN or the related N-Nitrosodiethylamine (DEN) (Magee and Barnes, 1967). Nitrosamines are present in the environment and produced during digestion and may contribute broadly to cancer (Miller and Miller, 1979). Other early animal studies establishing a causal link between environmental exposures and cancer include UV radiation induced skin cancer (Findlay, 1928) and X-ray induced lymphomas, ovarian cancer, and other tumors in mice (Furth and Furth, 1936). Collectively, these landmark studies through the early to mid 20th century established unequivocal causal connections between chemical or radiation exposure, and the subsequent development of cancer (Boyland, 1969). Importantly, these studies contributed directly to a reduction in human exposure to many of these agents, and subsequent reduction in cancer risk, through a variety of mechanisms including lifestyle changes, government regulation, and alterative industry practices.
Over the ensuing decades, additional animal studies of chemical carcinogenesis, combined with epidemiological observations linking occupational exposures to cancer risks, gave rise to two major applications of animal models in use today: the carcinogen bioassay and experimental cancer research.
The carcinogen bioassay: Mice and rats as the unsung heroes in chemical safety assessment
As a direct outcome of chemical carcinogenesis in animal models and epidemiologic evidence, combined with the burgeoning development of the chemical industry with resulting widespread environmental contamination and exposures, the National Cancer Institute (NCI) initiated animal testing programs in the 1970s to identify potential carcinogens and to establish safe levels of exposure. The NCI Carcinogenesis Bioassay Program was succeeded by the National Toxicology Program (NTP), whose mission is to coordinate federal toxicology programs and to refine the toxicology and carcinogenicity tests (National Toxicology Program, 2011). Chemicals that are selected for study are subjected to a two year bioassay using both sexes and two species, usually inbred and outbred strains of mice or rats and at several predetermined doses (NCI, 1980;Fung et al., 1995). In addition, the NTP employs GEMMs on a case by case basis in short term, more hypothesis based cancer bioassays (Pritchard et al., 2003). Laboratory tests in animals are a major source of the evidence gathered that is used to set regulatory standards for potential human exposure. This is summarized in the Handbook of Carcinogenic Potency and Genotoxicity Databases (Gold and Zeiger, 1997), which includes data from over 5,000 experiments and 1,298 chemical agents in over 1000 papers and 400 Technical Reports from the NCI/NTP.
Attesting to the value of carcinogen testing in animals, some 30 substances first shown to cause cancer in animals were subsequently linked to human cancer through epidemiologic studies, including estrogen, formaldehyde, diethylstilbestrol, DDT, vinyl chloride, 2,3,7,8-TCDD, radon gas, beryllium, asbestos, 4- aminobiphenyl, bis(chloromethyl)ether, and 1,3 butadiene (Rall, 2000;Huff, 1993). Viewed in another way, ~25% of those substances that are causally or strongly associated with human cancer were first identified as carcinogens in animals.
Accumulated evidence through the rodent bioassay permits some generalizations when comparing human to rodent cancer susceptibility. Cancer rates are low in humans and high in rodents for liver (excepting hepatitis C associated liver cancer), kidney, forestomach, and thyroid gland. Cancer rates are high in both species in lung, mammary gland, hematopoietic system, bladder, oral cavity, and skin. Cancer rates are high in humans and low in rodents for prostate, pancreas, colon/rectum, and cervix/uterus.
Extrapolating data from the rodent carcinogen bioassay to acceptable levels of human exposure involves consideration of the weight of evidence, mechanisms of action, threshold levels, and dose response patterns of the test compounds. As part of this process, it is useful to classify chemical carcinogens into two broad categories, genotoxic and nongenotoxic. These may be further divided into eight subclasses: direct acting carcinogens, procarcinogens, solid state carcinogens, hormones, immunosuppressors, co-carcinogens, and promoters (Weisburger and Williams, 1981). Much work remains to be done to determine the mechanism of action of many of these compounds and to establish safe levels of exposure.
Collectively, these findings from the NCI/NTP bioassays have been used by federal and state regulatory agencies to reduce exposure to mutagenic carcinogens. Another outcome of these studies was the development and use of the Ames test and other short term genotoxic assays widely used by regulatory agencies to screen chemicals prior to human exposure.
Chemical carcinogenesis in experimental cancer research
In addition to their role in identifying potential human carcinogens, the judicious design and interpretation of chemical and radiation carcinogenesis studies have made seminal contributions to many cornerstone principles of experimental oncology, many predating the molecular biology era (Pitot, 1983). For example, it was noted that many chemicals did not cause cancer at the site of exposure but were often active at distal sites, often in the liver. In 1947, the Millers noticed aromatic amine liver carcinogens bound to liver proteins (Miller and Miller, 1947) and speculated that binding to such macromolecules was important for the carcinogenicity of these compounds. Following up on early skin tumor painting studies, Heidelberger showed that PAHs bound to DNA of mouse skin and Lawley established a correlation between carcinogenicity and DNA binding, implicating DNA as the relevant target (Goshman and Heidelberger, 1967;Brookes and Lawley, 1964). As chemical carcinogens were covalently bound to DNA, these and other findings led to two important concepts: that many carcinogens are inactive in their native form and require metabolic activation to become active and carcinogenic, and that mutation in DNA may be a key event in their mechanism of action as carcinogens.
Berenblum and Shubik applied DMBA and croton oil in sequence to induce skin tumors in mice and from their analysis concluded that carcinogenesis occurs through at least two discrete stages, initiation, an irreversible event, in this case mediated by DMBA and promotion, a reversible process involving chronic exposure to an irritant such as croton oil (BERENBLUM and Shubik, 1947). The phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), was later shown to be the active ingredient in croton oil (Verma and Boutwell, 1980). The stages of carcinogenesis were subsequently expanded to include malignant progression, a process where benign neoplasms covert to malignant, invasive lesions, sometimes with metastatic dissemination. It is now generally accepted that cancer arises as a multistep process, an idea that can be traced back to the writings of Dr. Pott (Pott, 1775).
Following the finding of carcinogens bound to DNA and the correlation between DNA binding and carcinogenicity (Brookes and Lawley, 1964) it wasn’t until the discovery of mutated oncogenes and tumor suppressor genes in both human tumors and chemically induced rodent tumors (Balmain and Pragnell, 1983;Zarbl et al., 1985;Burns et al., 1991;Brown et al., 1990) that completed the link which began in 1915, between environmental exposures to carcinogens, mutations, and cancer. Collectively, these findings helped to solidify the concepts of mutagens as carcinogens and the genetic basis of cancer. When viewed through the lens of Nowell’s clonal evolution model of cancer development (Nowell, 1976), it is easy to see the major impact of these concepts, even if unstated, on the design and interpretation of many oncology experiments to this day. Chemical carcinogen models have contributed to many other fundamental concepts in cancer biology and genetics and many of the first carcinogens to be identified are still widely used as experimental agents.
Combining chemical carcinogen and genetically engineered models of cancer
Over the past two decades, GEMMs have been increasing employed to model cancer. These models are the subject of other chapters in this volume, as well as recent reviews (Van Dyke and Jacks, 2002;Becher and Holland, 2006;Frese and Tuveson, 2007). Given that cancer is the product of complex interactions between the genotype and environment, the combined use of chemical carcinogen and genetically engineered models is a logical approach to unravel the complex interplay between genetic susceptibility and environmental exposure. The clearest example of this is the increased spectrum of tumors observed in some GEM models following exposure to carcinogens or radiation. The tumor suppressor p53 provides one early illustrative example. p53 knockout mice were originally reported to be susceptible to spontaneous lymphomas and sarcomas, but not epithelial tumors (Donehower et al., 1992;Jacks et al., 1994). This observation was curious as p53 is frequently mutated in both human and mouse epithelial tumors. To address this conundrum, p53 deficient mice were subjected to the DMBA/TPA multistage skin tumor protocol. Interestingly, p53 deficient mice did not develop more skin tumors, but those that did develop progressed much more rapidly to malignant, invasive, and metastatic carcinomas. This pointed to a role for p53 in suppressing malignant progression, a conclusion that has been born out in a number of subsequent studies (Lewis et al., 2005;Jackson et al., 2005). Thus, p53 plays a stage-specific role in epithelial cancer and chemical exposure was required to reveal this phenotype. As Ras mutations were found in skin tumors from both wild type and p53 deficient mice, this finding also illustrated cooperation between mutations in Ras and p53 during malignant progression.
p53 deficient mice are also susceptible to DMN induced hemangiosarcomas (Harvey et al., 1993), to ionizing radiation induced lymphomas and sarcomas (Kemp et al., 1994), and UV induced skin cancer (Ziegler et al., 1994;Jiang et al., 1999), among other chemicals and agents (Tennant et al., 1999). The susceptibility of mice deficient in Arf, a tumor suppressor that regulates p53, to DMBA/TPA induced skin cancer progression (Kelly-Spratt et al., 2004), to chemically induced hemangiosarcomas (Busch et al., 2012), and lung cancer (see below), collectively highlight the Arf/p53 signaling axis as an important barrier to chemical and radiation induced cancer.
Another notable example illustrating the value of combining chemical and radiation carcinogenesis with GEM models involved the cyclin dependent kinase inhibitor p27 (CDKN1B) While its role as a CDK inhibitor made p27 a candidate tumor suppressor, the initial paucity of mutations in CDKN1B in tumors cast doubts on its importance in human cancer (Philipp-Staheli et al., 2001). Further, p27 knockout mice displayed only modest susceptibility to spontaneous tumor formation, restricted to the pituitary gland (Fero et al., 1996;Kiyokawa et al., 1996). These findings were difficult to reconcile with the clear association of low levels of p27 protein expression with poor prognosis in breast, lung, prostate, colon, and other cancers (Chu et al., 2008). This puzzle was at least partially solved when p27 deficient mice were challenged with chemical carcinogens or ionizing radiation. Following exposure, both p27 null and heterozygous mice showed a marked susceptibility to cancer in a wide range of tissues, including lung, small intestine, colon, prostate, and hematopoietic, relative to wild type littermates (Fero et al., 1998;Philipp-Staheli et al., 2002;Di Cristofano et al., 2001;Kelly-Spratt et al., 2009). In addition to establishing a causal and pan tissue role for p27 in tumor suppression, analysis of tumors from p27+/− mice showed no mutation or loss of heterozygosity in the remaining wild type Cdkn1b allele, providing clear evidence of tumor suppressor gene haploinsufficiency. Simultaneously, Donehower reported that p53 also showed evidence of haploinsufficient tumor suppression (Venkatachalam et al., 1998). Prior to these landmark experiments, tumor suppressor genes were widely, if not universally, thought to function in a genetically recessive manner, requiring mutational hits on both alleles before cancers could develop. In contrast, these studies showed that loss of just a single allele was sufficient. Haploinsufficiency has now been described for many tumor suppressor genes (Payne and Kemp, 2005;Scuoppo et al., 2012) and may be the rule rather than the exception. Thus, combining chemical carcinogenesis and GEM models was instrumental in modifying one of the central dogmas of cancer genetics. As a postscript, recent data from cancer genome sequencing projects has found that CDKN1B is one of the most significantly mutated genes in human breast cancer (Ellis et al., 2012), underscoring the predictive value of mouse models to identify tumor suppressor genes.
An important lesson from these and numerous other examples, is that the spontaneous tumor spectra of GEM models only tells part of the story, and appropriate environmental or dietary exposures should be routinely considered in the phenotypic analysis of GEMMs. Furthermore, the different spectra of tumors observed between mice and humans has been attributed to inherent differences between the species and has been used to argue against the value of mice as a model of human cancer (Anisimov et al., 2005). However, at least some of these differences are almost certainly due to very different environmental exposures between laboratory mice and humans. Another potentially confounding difference when comparing mouse to human cancer susceptibility is that mouse model experiments are frequently performed on one or two inbred strain backgrounds, while the human population is genetically diverse. It is well established that genetic background affects cancer risk in both humans and mice (Balmain, 2002;Dragani, 2003).
Chemical carcinogen models to identify genetic modifiers of cancer risk
Inbred strains differ widely in their susceptibility to spontaneous or chemically induced neoplasia in most if not all tissues, including lung, liver, (Drinkwater and Ginsler, 1986;Dragani et al., 1995a)(Drinkwater and Ginsler, 1986;Dragani et al., 1995a)(Drinkwater and Ginsler, 1986;Dragani et al., 1995a) skin, and colon (Demant, 2003). In most cases, treatment with tissue specific chemical carcinogens to induce tumors is an essential strategy for the discovery, mapping, and eventual identification of genetic modifiers of cancer risk. For example, topical application of the carcinogen DMBA was used to develop the highly susceptible SENCAR mouse strain (Slaga, 1986) and to map and characterize multiple skin cancer susceptibility loci (Skts) using both inbred strains and interspecies mus musculus x mus spretus crosses (Angel and DiGiovanni, 1999;Nagase et al., 1995;Quigley et al., 2009). Urethane or N-ethyl-N-nitrosourea (ENU) treatment of A/J and other strains was used to identify lung cancer susceptibility loci including the Pas, Par, and Sluc alleles (Lynch, 1926;Dragani et al., 1995b;Liu et al., 2006;Tripodis et al., 2001). Treatment of inbred strains with DEN, ENU, or urethane to induce liver tumors revealed dramatic differences in susceptibility to hepatocarcinogenesis, leading to the identification of Hcs and Hcr modifier alleles (Drinkwater and Ginsler, 1986;Dragani et al., 1995a;Poole et al., 1996). The carcinogens 1,2-dimethylhydrazine (DMH) and azoxymethane (AOM) as well as use of the germline susceptible strain ApcMin was used to identify small intestine and colon cancer susceptibility alleles Mom and Scc (Dietrich et al., 1993;Bissahoyo et al., 2005;van et al., 1999). Going forward, in depth analysis of these genetic modifiers will provide insight into gene x gene or epistatic interactions that predict cancer risk, novel mechanisms in cancer progression, and by extrapolation, human cancer susceptibility.
In addition to revealing mechanisms of genotype x environment interaction and identifying genes that modulate environmentally induced cancer, the enhanced susceptibility of some GEMMs to chemical agents is valuable to regulatory agencies. For example, transgenic Ras and p53 heterozygous knockout mice are routinely used by the NTP in the carcinogen bioassay, thus reducing time and saving money, allowing for increasing throughput to test additional potential human carcinogens (Ashby, 2001;Tennant et al., 1996;Tennant et al., 1999). Due to the broad susceptibility of p27 deficient mice to epithelial cancer, it has been argued that p27 deficient mice would also be useful for the carcinogen bioassay (Payne and Kemp, 2003).
Common models of chemical carcinogenesis
Skin cancer
Probably the most extensively researched animal model of cancer is multistage chemical carcinogenesis of mouse skin (see (Abel et al., 2009) for detailed protocols). In addition to having an unbroken experimental history that goes back for the better part of a century, this model has a number of experimental advantages, including simplicity, the ability to observe and quantify the number and growth rate of tumors, and the ability to study initiation, promotion, progression, and metastasis in sequence and the genes associated with these stages. One of the more useful attributes of the DMBA/TPA protocol is that the induced tumors harbor mutations in the oncogene Ras and the tumor suppressor p53 with remarkable consistency. Since Quintanilla and Balmain discovered mutations in Hras in chemically induced skin tumors 30 years ago (Balmain and Pragnell, 1983;Quintanilla et al., 1986), studies in many laboratories have confirmed that DMBA/TPA induced tumors almost uniformly contain the identical A>T mutation in codon 61 of Hras. This remarkable consistency lends itself to the progressive accumulation of knowledge about genetic, biologic, and environmental modifiers of oncogenic Ras (Kemp, 2005). Dozens if not hundreds of different inbred mouse strains and GEM models have been subjected to the DMBA/TPA protocol providing a rich repository of knowledge on the genetics and biology of multistage cancer. Virtually any general feature of cancer biology can be and/or has been explored with this model including inflammation, angiogenesis, epigenetics, chemoprevention, to name just a few. Recent examples include dissecting the role of the Arf, p53 and EMT in metastasis (Ruddell et al., 2008;Tsai et al., 2012) and lineage tracing to determine the clonal origin of cancer (Driessens et al., 2012).
Table 1 highlights selected examples of tissue specific (liver, lung, breast, colon) or pan-tissue models of chemical carcinogenesis that have been widely used in both regulatory settings and experimental cancer research. Further details are provided in the accompanying “Protocols” chapters. A schematic outlining some critical concepts of chemical carcinogenesis, in this case urethane induced lung cancer, is shown in Figure 1.
Table 1.
Common models of chemical carcinogenesis.
| Tissue | Agent | Route |
|---|---|---|
| Pan tissue | Ionizing radiation ENU |
Whole body exposure i.p. injection |
| Skin | DMBA B [a]P UV |
Topical application Topical application Topical exposure |
| Liver | DEN ENU Phenobarbital |
i.p. injection i.p. injection Drinking water |
| Lung | Urethane NNK |
i.p. injection i.p. injection |
| Breast | DMBA | Oral gavage |
| Colon | AOM, DMH | i.p injection |
Figure 1. Model of urethane induced lung cancer.
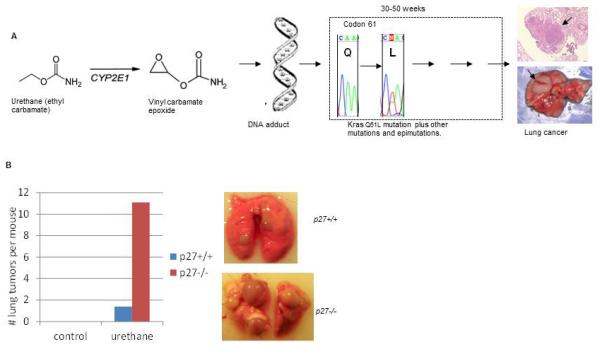
A. Following intraperitoneal injection, urethane is metabolized by the p450 system to highly reactive vinyl carbamate epoxide which covalently binds to DNA, and can result in base misincorporation during DNA replication and point mutations. ~80% of urethane induced lung tumors contain KrasQ61L mutations and additional genetic and epigenetic events that contribute to tumor progression. These additional events include mutation of p53 (Horio et al., 1996), mislocalization of p27 (Kelly-Spratt et al., 2009), altered DNA methylation (Alyaqoub et al., 2007), and loss of p19/Arf expression (S. Bush and C. Kemp, unpublished data). B: Combined urethane with GEM models to reveal genotype x environment interaction. Shown are the mean number of urethane induced lung tumors >1 mm in diameter in 30 week old 129 × C57BL/6J F1 p27+/+ (wild type) and p27−/− (nullizygous) littermate mice. Mice were either untreated (control) or injected with urethane at 12 days of age (1mg/g body wt). Neither wild type nor p27 nullizygous mice spontaneously developed lung tumors. Urethane treated wild type mice developed an average of 1.5 tumors per mouse while p27−/− mice averaged 11 tumors per mouse. On the right are representative lungs from urethane treated 50 week old mice of the indicated genotype. This reveals that urethane exposure and germline deletion of p27 interact synergistically to accelerate lung tumor development and demonstrates that p27 is a potent barrier to chemically induced cancer.
In summary, the ease of applying chemical carcinogens to any strain background or GEM model facilitates the study gene x gene and gene x environment interactions and is valuable to reveal latent tumor predisposition of GEMMs. The versatility and widespread use of animal models of chemical carcinogenesis, which began in the mid part of the last century, engenders progressive accumulative of insight into the genetics and biology of cancer.
The future of chemical carcinogenesis
Animal models of chemical carcinogenesis have made a major contribution to the precept that the most effective way to prevent cancer is to reduce exposure to mutagenic carcinogens. However, many important chemicals and agents are carcinogenic but are not obviously mutagenic, for example hormones, phenobarbital, asbestos, some metals, and chlorinated hydrocarbons such as DDT and PCBs. Application of these agents to selected GEM models which carry mutations in genes that influence the response to these agents may help unravel additional mechanisms of carcinogenesis. It can reasonably be anticipated that some of these mechanisms will involve epigenetic events. Abundant epidemiologic and molecular data point to epigenetic mechanisms as an important component in the link between early environmental exposure and later risk of diseases such as cancer (Jirtle and Skinner, 2007). For example, the carcinogens nickel, arsenic, and diethystilbestrol induce epigenetic changes (Anderson, 2004;Herceg, 2007), but the mechanistic role of these alterations in the carcinogenic process remain to be elucidated. Novel approaches that combine environmental exposures with GEM models are needed to determine both the environmental causes of epigenetic alterations and the mechanistic role of these alterations in cancer development. Clonal analysis of cancer has confirmed that tumors are genetically and phenotypically heterogeneous (Greaves and Maley, 2012;Driessens et al., 2012). This heterogeneity likely contributes to treatment failure and modeling this heterogeneity in mice is a significant challenge that lies ahead. As models of chemical carcinogenesis recapitulate important aspects of the etiology of human cancer and chemically induced tumors are often highly heterogeneous, these models may serve as a useful venue to study tumor heterogeneity and the implications for tumor response to therapy. Animal models of cancer that combine both genetic and environmental elements will continue to provide a critical role in the quest to reduce the burden of cancer on the human population.
Acknowledgements
The author would like to thank Norman Drinkwater, Jef French, and Russell Moser for helpful comments on the manuscript. Work in author’s laboratory is funded by grants from the NCI Mouse Models of Human Cancer Consortium U01 CA141550, NIEHS R01 ES020116, and DOD CA093616.
Reference List
- Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat. Protoc. 2009;4:1350–1362. doi: 10.1038/nprot.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alyaqoub FS, Tao L, Kramer PM, Steele VE, Lubet RA, Gunning WT, Pereira MA. Prevention of mouse lung tumors and modulation of DNA methylation by combined treatment with budesonide and R115777 (Zarnestra MT) Carcinogenesis. 2007;28:124–129. doi: 10.1093/carcin/bgl136. [DOI] [PubMed] [Google Scholar]
- Anderson LM. Introduction and overview. Perinatal carcinogenesis: growing a node for epidemiology, risk management, and animal studies. Toxicol. Appl. Pharmacol. 2004;199:85–90. doi: 10.1016/j.taap.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Angel JM, DiGiovanni J. Genetics of skin tumor promotion. Prog. Exp. Tumor. Res. 1999;35:143–57. doi: 10.1159/000062010. 143-157. [DOI] [PubMed] [Google Scholar]
- Anisimov VN, Ukraintseva SV, Yashin AI. Cancer in rodents: does it tell us about cancer in humans? Nat. Rev. Cancer. 2005;5:807–819. doi: 10.1038/nrc1715. [DOI] [PubMed] [Google Scholar]
- Ashby J. Expectations for transgenic rodent cancer bioassay models. Toxicol. Pathol. 2001;29(Suppl):177–182. doi: 10.1080/019262301753178591. [DOI] [PubMed] [Google Scholar]
- Balmain A. Cancer as a complex genetic trait: tumor susceptibility in humans and mouse models. Cell. 2002;108:145–152. doi: 10.1016/s0092-8674(02)00622-0. [DOI] [PubMed] [Google Scholar]
- Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature. 1983;303:72–74. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006;66:3355–8. doi: 10.1158/0008-5472.CAN-05-3827. discussion. [DOI] [PubMed] [Google Scholar]
- BERENBLUM I, Shubik P. A new, quantitative, approach to the study of the stages of chemical carcinogenesis in the mouse's skin. Br. J. Cancer. 1947;1:383–391. doi: 10.1038/bjc.1947.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissahoyo A, Pearsall RS, Hanlon K, Amann V, Hicks D, Godfrey VL, Threadgill DW. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol. Sci. 2005;88:340–345. doi: 10.1093/toxsci/kfi313. [DOI] [PubMed] [Google Scholar]
- Boyland F. The correlation of experimental carcinogenesis and cancer in man. Progress in Experimental Tumor Research. 1969;11:222–234. doi: 10.1159/000391396. [DOI] [PubMed] [Google Scholar]
- Brookes P, Lawley PD. Evidence for the binding of polynuclear aromatic hydrocarbons to the nucleic acids of mouse skin: relation between carcinogenic power of hydrocarbons and their binding to deoxynucleic acid. Nature. 1964;202:781–784. doi: 10.1038/202781a0. [DOI] [PubMed] [Google Scholar]
- Brown K, Buchmann A, Balmain A. Carcinogen-induced mutations in the mouse c-Ha-ras gene provide evidence of multiple pathways for tumour progression. Proc. Natl. Acad. Sci. USA. 1990;87:538–542. doi: 10.1073/pnas.87.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns PA, Kemp CJ, Gannon JV, Lane DP, Bremner R, Balmain A. Loss of heterozygosity and mutational alterations of the p53 gene in skin tumors of interspecific hybrid mice. Oncogene. 1991;6:2363–2369. [PubMed] [Google Scholar]
- Busch SE, Gurley KE, Moser RD, Kemp CJ. ARF suppresses hepatic vascular neoplasia in a carcinogen-exposed murine model. J. Pathol. 2012;227:298–305. doi: 10.1002/path.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Cook JW, Hieger I, Kennaway EL, Mayneord WV. The production of cancer by pure hydrocarbons. Royal Society Proceedings. 1932;111:455–484. [Google Scholar]
- Demant P. Cancer susceptibility in the mouse: genetics, biology and implications for human cancer. Nat. Rev. Genet. 2003;4:721–734. doi: 10.1038/nrg1157. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dragani TA. 10 years of mouse cancer modifier loci: human relevance. Cancer Res. 2003;63:3011–3018. [PubMed] [Google Scholar]
- Dragani TA, Manenti G, Gariboldi M, De Gregorio L, Pierotti MA. Genetics of liver tumor susceptibility in mice. Toxicology Letters. 1995a;82:613–619. doi: 10.1016/0378-4274(95)03505-2. [DOI] [PubMed] [Google Scholar]
- Dragani TA, Manenti G, Pierotti MA. Genetics of murine lung tumors. Adv. Cancer Res. 1995b;67:83–112. doi: 10.1016/s0065-230x(08)60711-3. [DOI] [PubMed] [Google Scholar]
- Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–530. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinkwater NR, Ginsler JJ. Genetic control of hepatocarcinogenesis in C57BL/6J and C3H/HeJ inbred mice. Carcinogenesis. 1986;7:1701–1707. doi: 10.1093/carcin/7.10.1701. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Findlay GM. Ultra-violet light and skin cancer. The Lancet. 1928;ii:1070–1073. [Google Scholar]
- Forkert PG. Mechanisms of lung tumorigenesis by ethyl carbamate and vinyl carbamate. Drug Metab Rev. 2010;42:355–378. doi: 10.3109/03602531003611915. [DOI] [PubMed] [Google Scholar]
- Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat. Rev. Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- Fung VA, Barrett JC, Huff J. The carcinogenesis bioassay in perspective: application in identifying human cancer hazards. Environ. Health Perspect. 1995;103:680–683. doi: 10.1289/ehp.95103680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth J, Furth OB. Neoplastic diseases produced in mice by general irradiation with X-rays. American Journal of Cancer. 1936;28:54–65. [Google Scholar]
- Gold LS, Zeiger E. Handbook of Carcinogenic Potency and Genotoxicity Databases. CRC Press; Boca Raton: [Google Scholar]
- Goshman LM, Heidelberger C. Binding of tritium-labeled polycyclic hydrocarbons to DNA of mouse skin. Cancer Res. 1967;27:1678–1688. [PubMed] [Google Scholar]
- Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53- deficient mice. Nature Genet. 1993;5:225–229. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- Herceg Z. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis. 2007;22:91–103. doi: 10.1093/mutage/gel068. [DOI] [PubMed] [Google Scholar]
- Horio Y, Chen A, Rice P, Roth JA, Malkinson AM, Schrump DS. Ki-ras and p53 mutations are early and late events, respectively, in urethane-induced pulmonary carcinogenesis in A/J mice. Mol. Carcinog. 1996;17:217–223. doi: 10.1002/(SICI)1098-2744(199612)17:4<217::AID-MC5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Huff J. Chemicals and cancer in humans: first evidence in experimental animals. Environ. Health Perspect. 1993;100:201–210. doi: 10.1289/ehp.93100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC Cancer: causes, occurrence, and control. 1990.
- IOM Rebuiliding the Unity of Health and the Environment: a New Vision of Environmental Health for the 21st Century. In: Institute of Medicine, editor. Workshop Summary; Washington, D.C.. National Academy of Sciences; 2001. [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- Jiang W, Ananthaswamy HN, Muller HK, Kripke ML. p53 protects against skin cancer induction by UV-B radiation. Oncogene. 1999;18:4247–4253. doi: 10.1038/sj.onc.1202789. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS Biology. 2004;2:1138–1149. doi: 10.1371/journal.pbio.0020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Spratt KS, Philipp-Staheli J, Gurley KE, Hoon-Kim K, Knoblaugh S, Kemp CJ. Inhibition of PI-3K restores nuclear p27(Kip1) expression in a mouse model of Kras-driven lung cancer. Oncogene. 2009;38:3652–3662. doi: 10.1038/onc.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ. Multistep skin cancer in mice as a model to study the evolvability of cancer cells. Sem. Cancer Biol. 2005;15:460–473. doi: 10.1016/j.semcancer.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Kemp CJ, Wheldon T, Balmain A. p53 deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nature Genet. 1994;8:66–69. doi: 10.1038/ng0994-66. [DOI] [PubMed] [Google Scholar]
- Kennaway EL. The identification of a carcinogenic compound in coal tar. Brit. Med. J. 1955;ii:749–752. doi: 10.1136/bmj.2.4942.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinosita R. Researches on the carcinogenesis of the various chemical substances. Gann. 1936;30:423–426. [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Lawley PD. Historical origins of current concepts of carcinogenesis. Adv. Cancer Res. 1994;65:17–111. doi: 10.1016/s0065-230x(08)60065-2. [DOI] [PubMed] [Google Scholar]
- Lewis BC, Klimstra DS, Socci ND, Xu S, Koutcher JA, Varmus HE. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol. Cell Biol. 2005;25:1228–1237. doi: 10.1128/MCB.25.4.1228-1237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- Liu P, Wang Y, Vikis H, Maciag A, Wang D, Lu Y, Liu Y, You M. Candidate lung tumor susceptibility genes identified through whole-genome association analyses in inbred mice. Nat. Genet. 2006;38:888–895. doi: 10.1038/ng1849. [DOI] [PubMed] [Google Scholar]
- Lynch CJ. Studies on the relation between tumor susceptibility and heredity. III Spontaneous tumors of the lung in mice. J. Exp. Med. 1926;43:339–355. doi: 10.1084/jem.43.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee PN, Barnes JM. The production of malignant primary hepatic tumours in the rat by feeding dimethylnitrosamine. Br. J. Cancer. 1956;10:114–122. doi: 10.1038/bjc.1956.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee PN, Barnes JM. Carcinogenic nitroso compounds. Adv. Cancer Res. 1967;10:163–246. doi: 10.1016/s0065-230x(08)60079-2. [DOI] [PubMed] [Google Scholar]
- Miller EC, Miller JA. Milestones in chemical carcinogenesis. Semin. Oncol. 1979;6:445–460. [PubMed] [Google Scholar]
- Miller JA. The need for epidemiological studies of the medical exposures of Japanese patients to the carcinogenic ethyl carbamate (urethane) from 1950 to 1975. Jpn. J Cancer Res. 1991;82:1323–1324. doi: 10.1111/j.1349-7006.1991.tb01799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Miller EC. The metabolism and carcinogenicity of p-dimethylaminoazobenzene and related compounds in the rat. Cancer Res. 1947;7:39–41. [PubMed] [Google Scholar]
- Nagase H, Bryson S, Cordell H, Kemp CJ, Fee F, Balmain A. Distinct genetic loci control development of benign and malignant skin tumours in mice. Nature Genet. 1995;10:424–429. doi: 10.1038/ng0895-424. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program . 12th Report on Carcinogens. U.S. Department of Health and Human Services Public Health Service National Toxicoloy Program; 2011. [Google Scholar]
- NCI NCI bioassay of chemical for possible carcinogenicity. 1980.
- Nettleship A, Henshaw PS, Meyer HL. Induction of pulmonary tumors in mice with ethyl carbamate (urethane) Journal of the National Cancer Institute. 1943;4:309–319. [Google Scholar]
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- OTA . In: Assesment of Technologies for Determining Cancer Risks from the Environment. Ofice of Technology Assesment, editor. U.S. Government Printing Office; Washington, D.C.: 1981. [Google Scholar]
- Payne SR, Kemp CJ. p27(Kip1) (Cdkn1b)-deficient mice are susceptible to chemical carcinogenesis and may be a useful model for carcinogen screening. Toxicologic Pathology. 2003;31:355–363. doi: 10.1080/01926230390201273. [DOI] [PubMed] [Google Scholar]
- Payne SR, Kemp CJ. Tumor suppressor genetics. Carcinogenesis. 2005;26:2031–2045. doi: 10.1093/carcin/bgi223. [DOI] [PubMed] [Google Scholar]
- Philipp-Staheli J, Kim KH, Payne SR, Gurley KE, Liggitt D, Longton G, Kemp CJ. Pathway-specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell. 2002;1:355–368. doi: 10.1016/s1535-6108(02)00054-5. [DOI] [PubMed] [Google Scholar]
- Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp. Cell Res. 2001;264:148–168. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
- Pitot HC. Contributions to our understanding of the natural history of neoplastic development in lower animals to the cause and control of human cancer. Cancer Surveys. 1983;2:519–538. [Google Scholar]
- Poole TM, Chiaverotti TA, Carabeo RA, Drinkwater NR. Genetic analysis of multistage hepatocarcinogenesis. Prog. Clin. Biol. Res. 1996;395:33–45. [PubMed] [Google Scholar]
- Pott P. Chirugical Observations Relative to the Cataract, the Polypus of the Nose, the Cancer of the Scrotum, the Different Kinds of Ruptures and the Mortifications of the Toes and Feet. Hawkes, Clarke, and Collins; London: 1775. [Google Scholar]
- Pritchard JB, French JE, Davis BJ, Haseman JK. The role of transgenic mouse models in carcinogen identification. Environ. Health Perspect. 2003;111:444–454. doi: 10.1289/ehp.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley DA, To MD, Perez-Losada J, Pelorosso FG, Mao JH, Nagase H, Ginzinger DG, Balmain A. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature. 2009;458:505–508. doi: 10.1038/nature07683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- Rall DP. Laboratory animal tests and human cancer. Drug Metab Rev. 2000;32:119–128. doi: 10.1081/dmr-100100565. [DOI] [PubMed] [Google Scholar]
- Rehn L. Uberblasentumoren bei fuchsinarbeiten. Arch Klin Chir. 1895;50:588. [Google Scholar]
- Ruddell A, Kelly-Spratt KS, Furuya M, Parghi SS, Kemp CJ. p19/Arf and p53 suppress sentinel lymph node lymphangiogenesis and carcinoma metastasis. Oncogene. 2008;27:3145–3155. doi: 10.1038/sj.onc.1210973. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Yoshida T. Liver carcinoma induced by feeding O-Amidoazotoluene. Archiv fur Pathologische Anatomie. 1935;295:175–220. [Google Scholar]
- Scuoppo C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I, Yoon S, Krasnitz A, Teruya-Feldstein J, Pappin D, Pelletier J, Lowe SW. A tumour suppressor network relying on the polyamine-hypusine axis. Nature. 2012;487:244–248. doi: 10.1038/nature11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaga TJ. SENCAR mouse skin tumorigenesis model versus other strains and stocks of mice. Environ. Health Perspect. 1986;68:27–32. doi: 10.1289/ehp.866827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant RW, Spalding J, French JE. Evaluation of transgenic mouse bioassays for identifying carcinogens and noncarcinogens. Mutat. Res. 1996;365:119–127. doi: 10.1016/s0165-1110(96)90016-0. [DOI] [PubMed] [Google Scholar]
- Tennant RW, Stasiewicz S, Mennear J, French JE, Spalding JW. Genetically altered mouse models for identifying carcinogens. IARC Sci. Publ. 1999:123–150. [PubMed] [Google Scholar]
- Tripodis N, Hart AA, Fijneman RJ, Demant P. Complexity of lung cancer modifiers: mapping of thirty genes and twenty-five interactions in half of the mouse genome. J. Natl. Cancer Inst. 2001;93:1484–1491. doi: 10.1093/jnci/93.19.1484. [DOI] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H. Uber das kustlich erzeugte cancroid bei der maus. Gann. 1918;12:17–21. [Google Scholar]
- Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell. 2002;108:135–144. doi: 10.1016/s0092-8674(02)00621-9. [DOI] [PubMed] [Google Scholar]
- van WT, Ruivenkamp CA, Stassen AP, Moen CJ, Demant P. Four new colon cancer susceptibility loci, Scc6 to Scc9 in the mouse. Cancer Res. 1999;59:4216–4218. [PubMed] [Google Scholar]
- Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. Embo Journal. 1998;17:4657–4667. doi: 10.1093/emboj/17.16.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AK, Boutwell RK. Effects of dose and duration of treatment with the tumour promoting agent, 12-O-tetradecanoylphorbol-13-acetate. Carcinogenesis. 1980;1:271–276. doi: 10.1093/carcin/1.3.271. [DOI] [PubMed] [Google Scholar]
- Vineis P, Pirastu R. Aromatic amines and cancer. Cancer Causes Control. 1997;8:346–355. doi: 10.1023/a:1018453104303. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Weisberger EK, Weisburger JH. Chemistry, carcinogenicity, and metabolism of 2-fluorenamine and related compounds. Adv. Cancer Res. 1958;5:331–431. doi: 10.1016/s0065-230x(08)60415-7. [DOI] [PubMed] [Google Scholar]
- Weisburger JH, Williams GM. Carcinogen testing: current problems and new approaches. Science. 1981;214:401–407. doi: 10.1126/science.7291981. [DOI] [PubMed] [Google Scholar]
- Yamagiwa K, Ichikawa K. Experimental study of the pathogenesis of carcinoma. Journal of Cancer Research. 1918;3:1–29. doi: 10.3322/canjclin.27.3.174. [DOI] [PubMed] [Google Scholar]
- Zarbl H, Sukumar S, Arthur AV, Martin-Zanca D, Barbacid M. Direct mutagenesis of Ha-ras-1 oncogenes by N-nitroso- N- methylurea during initiation of mammary carcinogenesis in rats. Nature. 1985;315:382–385. doi: 10.1038/315382a0. [DOI] [PubMed] [Google Scholar]
- Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]