

Abstract
Aims
Primary skull bone tumors, benign or malignant, are rare, and include a vast repertoire of lesions. These tumors are not reported systematically in the literature, with most studies being on individual entities or as single case reports.
Methods
Primary bone tumors diagnosed over a period of 12 years were retrieved, histological diagnoses reviewed, and clinical parameters noted.
Results
We identified 125 primary skull bone tumors. The mean age at diagnosis was 32 years (range: 2–65 years). Majority of patients were adults (82.4%); male preponderance was noted (72.8%). Malignant tumors were more frequent than benign tumors. Most common malignant tumor was chordoma (n = 37), while most common benign tumor was osteoma (n = 7). Tumors were most frequently located at the skull base, of which clivus was most common location. Chordomas accounted for majority of clival tumors, while chondrosarcoma predominated at other skull base locations. Benign tumors were extremely rare in skull base. Tumors of the vault bones were infrequent; with chondrosarcoma and osteoma being the most common malignant and benign tumors, respectively.
Conclusions
This is the largest series of primary skull bone tumors from India. Documentation of such a series will aid in approaching differential diagnosis of skull tumors in a systematic manner.
Keywords: bone tumors, skull, skull base, chondrosarcoma, chordoma
Introduction
Primary bony tumors of the skull are extremely rare, accounting for approximately 1% of all bone tumors. They include a vast repertoire of lesions, which may be benign or malignant. These tumors are not reported systematically in literature, as majority of published studies are on individual tumor entities or are single case reports. We, therefore, conducted this retrospective analysis of the spectrum of bony tumors involving the cranium, to document the various tumors encountered, and to analyze their clinicopathological features.
Materials and Methods
All primary bony tumors of the skull diagnosed over a period of 12 years (2002–2013) were retrieved from the archives of the Department of Pathology at our Institute (Table 1). The hematoxylin and eosin-stained slides were retrieved, and histological diagnoses were reviewed independently by three pathologists (A. K., A. N., and M. C. S.). Clinical parameters, including age, sex, and tumor location, were noted.
Table 1. Primary bone tumors of skull diagnosed over a 12-year period.
| Tumors | Number of cases (% of all tumors) | Age distribution (y): Median (range) | Male:female ratio | Most common location (number) |
|---|---|---|---|---|
| Malignant tumors | ||||
| Chordoma | 43 (34.4%) | 35 (4–60) | 36:7 | Clivus (28) |
| Chondrosarcoma | 37 (29.6%) | 32 (15–60) | 24:13 | Skull base (31) |
| Plasmacytoma | 7 (5.6%) | 54 (40–65) | 5:2 | Skull base (3) |
| Osteosarcoma | 5 (4%) | 22 (15–28) | 3:2 | Frontal bone (2) |
| ES/pPNET | 4 (3.2%) | 9.5 (2–13) | 3:1 | Parietal bone (2) |
| LCH | 2 (1.6%) | 15.5 (2–29) | 1:1 | Parietal and occipital bones |
| Tumors with intermediate malignant potential (rarely metastasizing) | ||||
| GCT | 9 (7.2%) | 32 (18–42) | 6:3 | Clivus (6) |
| Benign tumors | ||||
| Osteoma | 7 (5.6%) | 23 (14–35) | 4:3 | Frontal (3), parietal (3) |
| ABC | 4 (3.2%) | 27 (4–47) | 3:1 | Temporal (3) |
| Hemangioma | 2 (1.6%) | 17 (9–25) | Males | Frontal, temporal |
| Chondromyxoid fibroma | 1 (0.8%) | 35 | Female | Skull base |
| Osteoid osteoma | 1 (0.8%) | 16 | Male | Orbit |
| Ossifying fibroma | 1 (0.8%) | 11 | Male | Orbit |
| Cementoossifying fibroma | 1 (0.8%) | 13 | Male | Orbit |
| Fibrous dysplasia | 1 (0.8%) | 33 | Male | Orbit |
Abbreviations: ABC, aneurysmal bone cyst; ES, Ewing sarcoma; GCT, giant cell tumor; LCH, Langerhan cell histiocytosis; pPNET, peripheral primitive neuroectodermal tumor.
Results
We identified 125 cases of primary bony tumors of the skull, which accounted for 1.1% of all central nervous system tumors diagnosed during the 12-year period. The mean age of patients was 32 years (range: 25–65 years). There was a male preponderance (91 males and 34 females). Majority of patients were adults (103/125; 82.4%); pediatric patients were rare (22/125; 17.6%). Malignant tumors (98/125; 78.4%) were more common than benign (18/125; 14.4%) and intermediate grade (9/125; 7.2%) tumors. Base of the skull was the most frequent location (86/125; 68.8%).
Chordoma was the most common histological type (43/125; 34.4%). It was also the most common malignant tumor (43/98; 43.9%), followed by chondrosarcoma (37/98; 37.8%), including three mesenchymal chondrosarcomas. Of the five osteosarcomas (OSs) identified, one was a radiation-induced OS. The most common benign tumor was osteoid osteoma (8/18; 44.4%). Giant cell tumor (GCT) was the only tumor with intermediate malignant potential. Malignant tumors were more prevalent than benign tumors in adults as well as children. While plasmacytomas (n = 7) were seen only in adults, Ewing sarcoma/peripheral primitive neuroectodermal tumor (ES/pPNET) (n = 4) was exclusive to the pediatric age group. GCTs were seen predominantly in adults (8/9; 88.9%). There was no other difference in histological types between children and adults. The most important differential diagnoses were between chordoma and chondrosarcoma (Fig. 1), and between ES/pPNET and mesenchymal chondrosarcoma (Fig. 2). On immunohistochemistry, chordomas were immunopositive for brachyury and cytokeratin, which helped to differentiate from chondrosarcomas, which are negative for these markers. Both, ES/pPNET and mesenchymal chondrosarcoma showed features of a malignant round cell tumor immunopositive for CD99 (MIC2); however, the former showed periodic acid-Schiff-positive, diastase-resistant intracytoplasmic glycogen which was absent in the latter. In addition, foci of well differentiated cartilage were identifiable in mesenchymal chondrosarcomas, clinching the diagnosis.
Fig. 1.
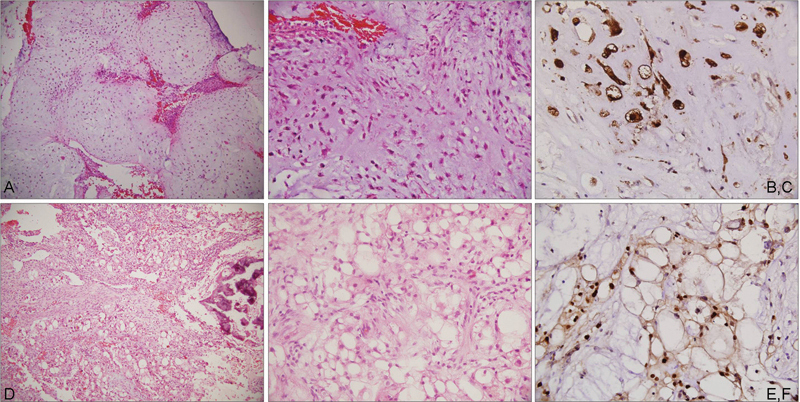
Photomicrographs showing a chondrosarcoma composed of cartilaginous lobules (a; HE, ×100) with high cellularity and pleomorphism (b; HE, ×200), immunopositive for S-100 (c; IHC, ×400); chordoma with a myxoid matrix (d; HE, ×100) small polygonal cells and large physaliphorous cells (e; HE, ×200) immunopositive for brachyury (f; IHC, ×400). HE, hematoxylin and eosin stain; IHC, immunohistochemistry.
Fig. 2.
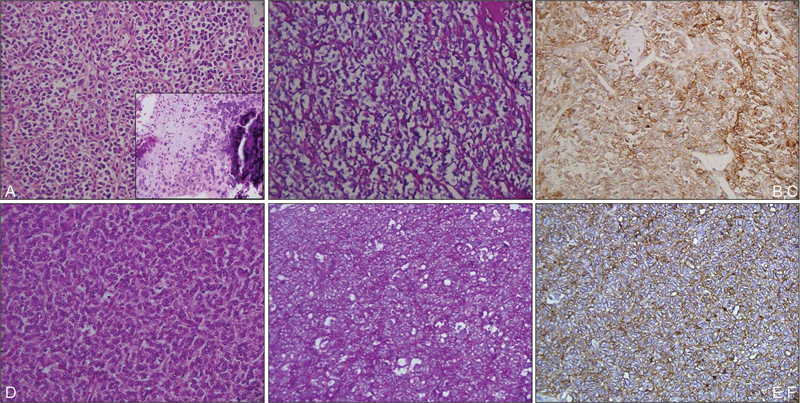
Photomicrographs showing mesenchymal chondrosarcoma with predominant malignant small round cell areas with occasional foci of well differentiated cartilage (inset) (a; HE, ×200); small round cells are negative on PAS stain (b; PAS, ×200) and are immunopositive for CD99 (c; IHC, ×200); case of Ewing sarcoma showing a malignant small round cell tumor (d; HE, ×200); with cytoplasmic PAS-positive glycogen (c; PAS, ×200) and CD99 immunopositivity (f; IHC, ×200). HE, hematoxylin and eosin stain; IHC, immunohistochemistry; PAS, periodic acid-Schiff.
Base of the skull was the most frequent location (86/125; 68.8%), and showed predominantly malignant tumors. Only one benign tumor was located at the skull base, a chondromyxoid fibroma. While midline locations in the skull base (clivus, sellar) were more frequently associated with chordomas, chondrosarcomas were seen in other parts of the skull base (sphenoid wing). The vault bones (n = 28) and orbit (n = 10) had equal distribution of benign and malignant tumors. Osteoma was the most frequent tumor of the vault (6/28; 21.4%), while fibroosseous lesions (3/10; 30%) were the commonest orbital lesions. Multiple lesions were seen in only one patient, which showed plasmacytoma on histopathology.
Discussion
Bony tumors of the skull are uncommon lesions, and therefore have not been systematically analyzed and reported in literature. They have been reported to account for around 0.8 to 1% of all bone tumors. However, one of the largest series of bone tumors, from the Mayo Clinic, found that they accounted for 4% of all bone tumors, with the majority being malignant. With the advances in neurosurgical techniques, bony tumors at previously inaccessible locations, particularly in the skull base, are now being resected more frequently. It is, therefore, imperative that pathologists acquaint themselves with the lesions that can involve the skull bones, and at which particular location they may occur. Like bone tumors at any other location, primary tumors involving the skull bones are classified based on their cell of origin, as bone-forming tumors, cartilaginous tumors, fibroosseous lesions, tumors arising from notochord remnants, etc.
Bone-Forming Tumors
Osteomas are benign, slow-growing bony tumors, seen in young adults, usually in males. They arise in the craniofacial region, particularly in the paranasal sinuses and the jaw bones, and rarely affect the skull bones.1 2 Gundewar et al reported that only 25% of all craniofacial osteomas were seen in the orbit, while 17% involved the frontal bone.1 We identified seven osteomas in our study, three in the frontal bone, three in the parietal bone, and one in the orbit. Multiple osteomas are seen in patients with Gardner syndrome, in association with intestinal polyps, desmoids, and epidermoid cysts. Haddad et al recommended the following classification of cranial osteomas: (1) skull base osteomas, (2) skull vault osteomas, (3) dural osteomas, and (4) intraparenchymal osteomas, with skull base osteomas being most common among these groups.3 Whether the latter two are true entities remains in question. On histopathology, they are characterized by compact lamellar bone with scant intervening fibrous stroma. Symptomatic tumors are managed by surgical excision. In the skull, sphenoid osteomas demand early excision, as they may grow and compress the optic apparatus, leading to blindness.2
Osteoid osteomas are tumors of the long bones, with only 1% of osteoid osteomas affecting the skull and facial bones, where they are much rarer than osteomas.4 Usually seen in young adults, around 90% occur by the third decade of life.4 A male preponderance has been noted.5 On histopathology, they have a central vascularized nidus, surrounded by dense sclerotic bone. The nidus is composed of an interlacing network of osteoid, often in microtrabecular arrays, showing variable mineralization. These osteoid trabeculae are lined by plump appositional osteoblasts. We identified one osteoid osteoma in our study, involving the orbit of a 16-year-old boy. Osteoblastomas are distinguished from osteoid osteomas only on the basis of size, with lesions greater than 1.5 cm being considered as osteoblastomas.6 These tumors frequently affect the spine, particularly the posterior elements. Osteoblastomas are infrequent in the calvaria, and only few case reports are available.7 8 Surgical excision is the treatment of choice for both these benign tumors.
OSs are malignant tumors composed of osteoblastic cells that produce osteoid. To date, approximately 150 cases of OS have been reported to involve the skull, accounting for around 2% of all OS.9 10 OS of skull are usually seen in the vault, and rarely occur in the skull base.4 Hadley et al analyzed all pediatric skull OS reported in literature and found that 61% of de novo OS were calvarial, while the remainder arose in the skull base.9 Of the five OS in our study, three (60%) involved the vault bones, while two (40%) were located at the skull base. Histopathologically, OSs can be classified into osteoblastic, chondroblastic, and fibroblastic subtypes, based on the predominant matrix produced. In our series, we identified three osteoblastic, one chondroblastic, and one telangiectatic OS. However, unlike OS of the jaw, histological type has no prognostic connotations.11 Cranial OS have been found to have poorer survival than OS of the mandible and maxilla, as well as those of long bones.12 13 This may be related to the difficulty in achieving complete excision of OS in the former location. Adjuvant or neoadjuvant chemotherapy along with surgery has been reported to improve the outcome of cranial OS.14 15
Chondrogenic Tumors
Cartilaginous tumors including chondrosarcoma are more common in the skull base than in the vault, as the bones of the former are formed by enchondral ossification while those of the latter are formed by intramembranous ossification. Chondrosarcoma accounts for 0.15% of all intracranial tumors and 6% of skull base neoplasms.16 In the skull base, the petrous temporal bone, clivus, and temporooccipital junction are the most commonly reported sites for chondrosarcoma.4 17 In our series, we found 16 tumors in the petrous temporal bone, 10 in the clivus, and 5 in the sphenoid bone. Apart from the skull base, we also found two each in the parietal, occipital, and orbital bones. On histopathology, chondrosarcomas are classified as conventional, mesenchymal, clear cell, dedifferentiated, of which conventional chondrosarcoma is the most common.18 Mesenchymal chondrosarcoma more commonly arises from the meninges than the skull bone, and is associated with a better prognosis than conventional chondrosarcoma. More importantly, chondrosarcomas are accorded one of three grades, I to III, for well-, moderately-, and poorly differentiated tumors, which has a significant impact on the patient outcome.17 Other prognostic factors include postoperative radiotherapy, and extent of surgical excision. Due to their deep location, chondrosarcomas are difficult to resect completely; hence, surgery along with adjuvant radiotherapy is the treatment of choice. Mortality rates with surgery alone are higher than for surgery followed by radiotherapy.17
Chondromyxoid fibroma (CMF) is a benign chondroid tumor that occasionally affects the skull base, particularly the petrous temporal bone, leading to deafness.4 19 20 It has also been documented at various other cranial locations, including frontal bone, orbit, etc.19 21 22 These tumors usually occur in the second and third decades of life.19 21 Histologically, CMF are lobulated tumors with stellate to spindled cells embedded in a myxoid matrix. The lobules are hypocellular at their center and more cellular at their periphery, with many spindle cells and interspersed osteoclastic giant cells. The principal differential diagnosis is with chondrosarcoma, which can also have a lobulated appearance. However, in chondrosarcoma, even the center of the lobules would be relatively hypercellular, unlike in CMF. Surgical excision is the mainstay of therapy for these tumors; adjuvant radiotherapy may be considered in incompletely resected tumors.19 In our study, we had one CMF located in the anterior skull base in a 35-year-old woman.
Other rare chondroid tumors, such as chondroma and chondroblastoma, have also been reported to involve the skull. However, these were not identified in our series.
Chordoma
Chordoma is a rare tumor that arises from the remnants of the notochord, an embryonal structure. It most commonly arises from the sacrum, followed by the skull base in approximately 30% of cases, where clivus is the predominant site of origin.4 23 Skull base chordomas occur in patients a decade younger than those with sacral chordomas.4 Chordoma is considered a low-grade malignant tumor due to its propensity for locally invasive growth. These are lobulated tumors, which on microscopy show the pathognomonic physaliphorous cells with vacuolated cytoplasm, surrounded by spindled to epithelioid cells embedded in a variably myxoid matrix. They show nuclear immunopositivity for brachyury, a transcription factor, accompanied by immunopositivity for cytokeratin, epithelial membrane antigen (EMA), and S-100 protein. Histological subtypes of chordoma include chondroid chordoma, with areas resembling hyaline cartilage; sarcomatoid chordoma with malignant spindle cell areas immunopositive for brachyury; and dedifferentiated chordoma, with malignant spindle cell areas that are negative for brachyury.23 Although initial studies denoted a better prognosis to the chondroid variant, more recent studies have found similar prognosis as conventional chordoma.24 Sarcomatoid and dedifferentiated variants have worse prognosis. The most important differential diagnosis is with chondrosarcoma, as these tumors may be indistinguishable on imaging. In our study, we found that chordomas were more common than chondrosarcomas in midline locations in the skull base, while chondrosarcomas were laterally located unlike chordomas. Immunohistochemically, while chondrosarcomas also show positivity for S-100 protein, they are negative for cytokeratin, EMA, and brachyury, which thus help in this differential diagnosis. D2–40 has been suggested as a true chondroid marker expressed in chondrosarcoma, aiding in differential diagnosis with chordoma and other chondroid lesions.25 Recent studies have also revealed presence of IDH1 and IDH2 mutations in chondrosarcomas, implicating their role in differentiating chondrosarcomas from chordomas.26 Loss of INI-1 immunoexpression has also been identified in a subset of aggressive pediatric chordomas.27
Outcome of patients with chordoma is dependent on the extent of resection. Maximal resection, with minimal loss of neurological function, and preservation of quality of life should be the goal of treatment. The American Joint Committee on Cancer/Union for International Cancer Control recommends the following classification for extent of resection: R0 for negative microscopic margin of ≥ 1 mm or greater; R1 for microscopic margin of ≤ 1 mm, but no evidence of residual macroscopic tumor; R2 for residual macroscopic tumor or tumor spillage in the intraoperative field, which has been adopted for application to chordomas.23 28 In spite of aggressive surgery, chordomas frequently recur locally; therefore, patients should be given adjuvant high-dose radiotherapy following resection.29 For inoperable cases, a biopsy confirmation followed by radiotherapy is the recommended protocol. A recent phase II study has hinted at the efficacy of imatinib mesylate in chordomas that express platelet-derived growth factor β, laying the groundwork for future-targeted therapy in this aggressive tumor.30
Ewing Sarcoma/Peripheral Primitive Neuroectodermal Tumor
ES is a malignant tumor that shows varying degree of neuroectodermal differentiation, and is characterized by recurrent balanced translocations involving the EWSR1 gene on chromosome 22. It usually occurs in patients younger than 20 years of age, and involves the diaphysis of long bones.31 The skull and vertebrae are involved only occasionally. It may also arise primarily from the dura, where it can mimic a meningioma on radiology. Occasional reports of primary diffuse leptomeningeal ES/pPNET are also available.32
Histologically, ES are composed of sheets of malignant small round cells with scant cytoplasm containing glycogen, which is responsible for the cytoplasmic clearing of the tumor cells. Homer-Wright rosettes may be present, indicating neuroectodermal differentiation. Diffuse strong membranous positivity for CD99, the MIC2 gene product, is typical of this tumor; however, this marker has low specificity as it is positive in several other round cell tumors that enter the differential diagnosis. In the skull, the closest differential diagnosis is mesenchymal chondrosarcoma, as the round cell component of this tumor closely resembles ES/pPNET morphologically and immunohistochemically, being immunopositive for MIC2, as well as neural markers such as neuron-specific enolase and CD57. Therefore, testing for the EWSR1 translocation may be necessary, especially in small biopsies where the cartilaginous component of mesenchymal chondrosarcoma may be easily missed. This may be performed by fluorescence in situ hybridization or reverse transcriptase-polymerase chain reaction. FLI1 is a new immunohistochemical marker that detects the EWSR1-FLI1 fusion protein, and may be helpful in such situations when genetic testing is unavailable. In our study, we found that all ES/pPNET were located in the vault/orbit, while all mesenchymal chondrosarcomas involved the skull base. Thus, the location of the tumor should be taken into consideration when arriving at a diagnosis. ES/pPNET may extend intra-axially and cause confusion with central PNET, which primarily arises from the brain parenchyma. However, the latter tumor does not show cytoplasmic glycogen, CD99 positivity, or EWSR1 rearrangements.31 Unlike ES/pPNET, central PNET have a poor outcome, making it important to differentiate between the two.33
Other Rare Tumors
In our study, we identified few rarer tumors such as GCT, aneurysmal bone cyst (ABC), hemangioma, plasmacytoma, Langerhan cell histiocytosis, and fibroosseous lesions including ossifying fibroma, cementoossifying fibroma, and fibrous dysplasia (Fig. 3). Approximately 1% of GCTs affect the bones of the skull and face, with the sphenoid being the most common location.4 We identified six GCTs in the skull base and three involving the vault bones. ABC is a differential diagnosis of GCT, and can be distinguished from the former by distribution of the giant cells predominantly along the walls of vascular channels. Skull hemangiomas usually affect the vault, particularly the frontal bone.4 In our series, one was located in the frontal, and the second was in the temporal bone. One case each of ossifying fibroma, cementoossifying fibroma, and fibrous dysplasia was identified in our series, all of which involved the orbit. Skull involvement by fibrous dysplasia may be isolated or a part of generalized disease associated with GNAS1 somatic mutations.
Fig. 3.
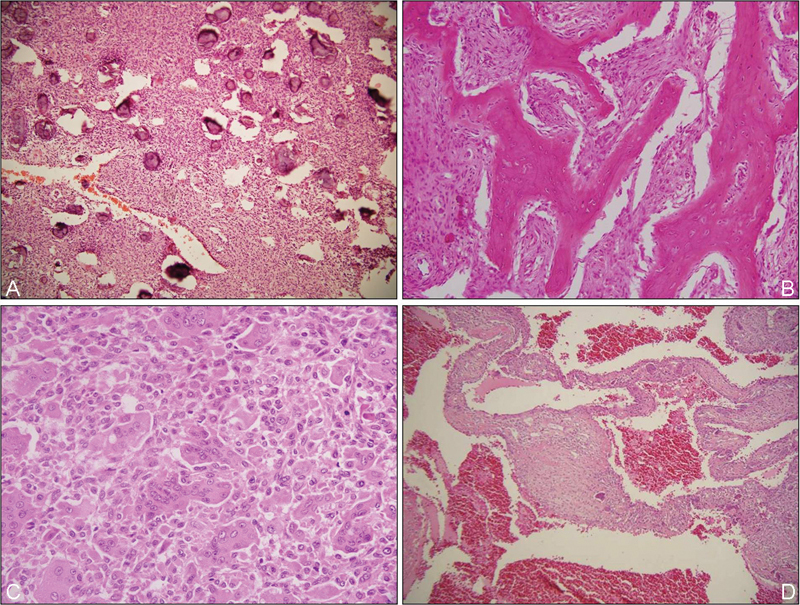
Rare skull bone tumors: cementoossifying fibroma (a; HE, ×200); fibrous dysplasia (b; HE, ×200); giant cell tumor (c; HE, ×200) and aneurysmal bone cyst (d; HE, ×200). HE, hematoxylin and eosin stain.
Conclusion
This is one of the largest series of primary bone tumors of the skull. Documentation of such a series is important as it will aid clinicians in approaching the differential diagnosis of skull bone tumors in a systematic manner based on parameters, such as patient age and tumor location, and thus provide an objective protocol for patient management. As the morphology of these tumors in the skull is similar to those at other locations, it should be familiar to all pathologists. Judicial use of immunohistochemistry and few molecular studies can help in diagnosis of most of these lesions.
Footnotes
Conflict of Interest None. Financial Support None.
References
- 1.Gundewar S, Kothari D S, Mokal N J, Ghalme A. Osteomas of the craniofacial region: A case series and review of literature. Indian J Plast Surg. 2013;46(3):479–485. doi: 10.4103/0970-0358.121982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boffano P, Roccia F, Campisi P, Gallesio C. Review of 43 osteomas of the craniomaxillofacial region. J Oral Maxillofac Surg. 2012;70(5):1093–1095. doi: 10.1016/j.joms.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Haddad F S, Haddad G F, Zaatari G. Cranial osteomas: their classification and management. Report on a giant osteoma and review of the literature. Surg Neurol. 1997;48(2):143–147. doi: 10.1016/s0090-3019(96)00485-5. [DOI] [PubMed] [Google Scholar]
- 4.Ellison D W, Perry A, Rosenblum M, Asa S, Reid R, Louis D N. London, United Kingdom: Edward Arnold; 2008. Tumors: non-neuroepithelial tumours and secondary effects; pp. 2002–2182. [Google Scholar]
- 5.Dugert E, Lagleyre S, Brouchet A, Deguine O, Cognard C, Bonneville F. Osteoid osteoma invading the posterior labyrinth of the petrous bone. AJNR Am J Neuroradiol. 2010;31(9):1764–1766. doi: 10.3174/ajnr.A1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inwards C Y, Oliveira A M. Philadelphia, PA: Saunders Elsevier; 2013. Tumors of the osteoarticular system; pp. 1871–1932. [Google Scholar]
- 7.Lin Y C, Commins D L, Fedenko A N, Pinsky G S. A rare case of periosteal osteoblastoma located in the frontal cranial bone. Arch Pathol Lab Med. 2005;129(6):787–789. doi: 10.5858/2005-129-787-ARCOPO. [DOI] [PubMed] [Google Scholar]
- 8.Moon K S, Jung S, Lee J H. et al. Benign osteoblastoma of the occipital bone: case report and literature review. Neuropathology. 2006;26(2):141–146. doi: 10.1111/j.1440-1789.2006.00643.x. [DOI] [PubMed] [Google Scholar]
- 9.Hadley C, Gressot L V, Patel A J. et al. Osteosarcoma of the cranial vault and skull base in pediatric patients. J Neurosurg Pediatr. 2014;13(4):380–387. doi: 10.3171/2013.12.PEDS13359. [DOI] [PubMed] [Google Scholar]
- 10.Nora F E, Unni K K, Pritchard D J, Dahlin D C. Osteosarcoma of extragnathic craniofacial bones. Mayo Clin Proc. 1983;58(4):268–272. [PubMed] [Google Scholar]
- 11.Clark J L, Unni K K, Dahlin D C, Devine K D. Osteosarcoma of the jaw. Cancer. 1983;51(12):2311–2316. doi: 10.1002/1097-0142(19830615)51:12<2311::aid-cncr2820511224>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 12.Kassir R R, Rassekh C H, Kinsella J B, Segas J, Carrau R L, Hokanson J A. Osteosarcoma of the head and neck: meta-analysis of nonrandomized studies. Laryngoscope. 1997;107(1):56–61. doi: 10.1097/00005537-199701000-00013. [DOI] [PubMed] [Google Scholar]
- 13.Haque F, Fazal S T, Ahmad S A, Abbas S Z, Naseem S. Primary osteosarcoma of the skull. Australas Radiol. 2006;50(1):63–65. doi: 10.1111/j.1440-1673.2005.01537.x. [DOI] [PubMed] [Google Scholar]
- 14.Ashkan K, Pollock J, D'Arrigo C, Kitchen N D. Intracranial osteosarcomas: report of four cases and review of the literature. J Neurooncol. 1998;40(1):87–96. doi: 10.1023/a:1006007411312. [DOI] [PubMed] [Google Scholar]
- 15.Salvati M, Ciappetta P, Raco A. Osteosarcomas of the skull. Clinical remarks on 19 cases. Cancer. 1993;71(7):2210–2216. doi: 10.1002/1097-0142(19930401)71:7<2210::aid-cncr2820710708>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 16.Cianfriglia F, Pompili A, Occhipinti E. Intracranial malignant cartilaginous tumours. Report of two cases and review of literature. Acta Neurochir (Wien) 1978;45(1–2):163–175. doi: 10.1007/BF01774391. [DOI] [PubMed] [Google Scholar]
- 17.Bloch O G, Jian B J, Yang I. et al. A systematic review of intracranial chondrosarcoma and survival. J Clin Neurosci. 2009;16(12):1547–1551. doi: 10.1016/j.jocn.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogendoorn P CW, Bovee J VMG, Nielsen G P. Lyon, France: IARC; 2013. Chondrosarcoma (grades I–III), including primary and secondary variants and periosteal chondrosarcoma; pp. 264–268. [Google Scholar]
- 19.Hakan T, Vardar Aker F. Chondromyxoid fibroma of frontal bone: a case report and review of the literature. Turk Neurosurg. 2008;18(3):249–253. [PubMed] [Google Scholar]
- 20.Xu H, Qin Z, Shi Z. Chondromyxoid fibroma in the sella turcica region. J Clin Neurosci. 2011;18(10):1419–1421. doi: 10.1016/j.jocn.2011.01.035. [DOI] [PubMed] [Google Scholar]
- 21.Miyamoto E, Kuriyama T, Iwamoto M, Tsuji N, Shizuki H. Cranial chondromyxoid fibroma. Case report. J Neurosurg. 1981;55(6):1001–1003. doi: 10.3171/jns.1981.55.6.1001. [DOI] [PubMed] [Google Scholar]
- 22.Khalatbari M R, Hamidi M, Moharamzad Y. Chondromyxoid fibroma of the anterior skull base invading the orbit in a pediatric patient: case report and review of the literature. Neuropediatrics. 2012;43(3):140–145. doi: 10.1055/s-0032-1307460. [DOI] [PubMed] [Google Scholar]
- 23.Stacchiotti S Sommer J; Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community Lancet Oncol 2015162e71–e83. [DOI] [PubMed] [Google Scholar]
- 24.Jian B J, Bloch O G, Yang I, Han S J, Aranda D, Parsa A T. A comprehensive analysis of intracranial chordoma and survival: a systematic review. Br J Neurosurg. 2011;25(4):446–453. doi: 10.3109/02688697.2010.546896. [DOI] [PubMed] [Google Scholar]
- 25.Cho H Y, Lee M, Takei H, Dancer J, Ro J Y, Zhai Q J. Immunohistochemical comparison of chordoma with chondrosarcoma, myxopapillary ependymoma, and chordoid meningioma. Appl Immunohistochem Mol Morphol. 2009;17(2):131–138. doi: 10.1097/PAI.0b013e3181866a13. [DOI] [PubMed] [Google Scholar]
- 26.Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y. Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol. 2012;29(4):201–206. doi: 10.1007/s10014-012-0085-1. [DOI] [PubMed] [Google Scholar]
- 27.Yadav R, Sharma M C, Malgulwar P B. et al. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro-oncol. 2014;16(3):372–381. doi: 10.1093/neuonc/not228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Gompel J J, Janus J R. Chordoma and chondrosarcoma. Otolaryngol Clin North Am. 2015;48(3):501–514. doi: 10.1016/j.otc.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Wittekind C, Compton C, Quirke P. et al. A uniform residual tumor (R) classification: integration of the R classification and the circumferential margin status. Cancer. 2009;115(15):3483–3488. doi: 10.1002/cncr.24320. [DOI] [PubMed] [Google Scholar]
- 30.Stacchiotti S, Longhi A, Ferraresi V. et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30(9):914–920. doi: 10.1200/JCO.2011.35.3656. [DOI] [PubMed] [Google Scholar]
- 31.Mobley B C, Roulston D, Shah G V, Bijwaard K E, McKeever P E. Peripheral primitive neuroectodermal tumor/Ewing's sarcoma of the craniospinal vault: case reports and review. Hum Pathol. 2006;37(7):845–853. doi: 10.1016/j.humpath.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Nambirajan A, Suri V, Sharma M C. et al. A 7-year-old girl with recurrent episodes of abdominal pain, seizures, and loss of vision: Primary diffuse leptomeningeal primitive neuroectodermal tumor masquerading as chronic meningitis. Neurol India. 2015;63(5):736–742. doi: 10.4103/0028-3886.166552. [DOI] [PubMed] [Google Scholar]
- 33.Furuno Y, Nishimura S, Kamiyama H. et al. Intracranial peripheral-type primitive neuroectodermal tumor. Neurol Med Chir (Tokyo) 2008;48(2):72–76. doi: 10.2176/nmc.48.72. [DOI] [PubMed] [Google Scholar]