

Here, Han et al. show that transcription factor Zfp335 binds DNA and drives transcription via recognition of two distinct consensus motifs by separate ZF clusters and identify the specific motif interaction disrupted by the mutation R1092W. This study presents Zfp335 as a model for understanding how C2H2-ZF TFs may use multiple recognition motifs to control gene expression.
Keywords: transcription factors, zinc fingers, protein–DNA binding, Zfp335, ZNF335
Abstract
The complexities of DNA recognition by transcription factors (TFs) with multiple Cys2–His2 zinc fingers (C2H2-ZFs) remain poorly studied. We previously reported a mutation (R1092W) in the C2H2-ZF TF Zfp335 that led to selective loss of binding at a subset of targets, although the basis for this effect was unclear. We show that Zfp335 binds DNA and drives transcription via recognition of two distinct consensus motifs by separate ZF clusters and identify the specific motif interaction disrupted by R1092W. Our work presents Zfp335 as a model for understanding how C2H2-ZF TFs may use multiple recognition motifs to control gene expression.
Cys2–His2 zinc finger (C2H2-ZF) proteins are classically recognized as transcription factors (TFs) that bind specific DNA sequences to regulate gene expression. According to the canonical model, each C2H2-ZF contacts three or more sequential bases in a manner dependent on amino acid residues at positions −1, 2, 3, and 6 on the DNA recognition α helix (Wolfe et al. 2000; Klug 2010). C2H2-ZFs are often assembled in a modular fashion to form tandem arrays that concatenate individual ZF specificities to recognize longer sequences. The human genome encodes >700 C2H2-ZF proteins (Emerson and Thomas 2009), many of which are known to play important roles in development and disease, although the biological functions and sequence specificities of most family members remain poorly characterized (Stubbs et al. 2011).
Zfp335 is a 13 C2H2-ZF protein that plays essential roles in early embryogenesis, neurogenesis, and T-cell development (Yang et al. 2012; Han et al. 2014). Zfp335 is thought to regulate transcription by recruiting H3K4 methyltransferase complexes to target gene promoters (Yang et al. 2012) and has also been reported to interact with the coactivator NCOA6 (Mahajan et al. 2002). Mutation of human ZFP335 (also called ZNF335) caused severe microcephaly, while conditional deletion in an embryonic mouse model led to brain development abnormalities related to defects in neural progenitor proliferation and differentiation (Yang et al. 2012).
We previously described an ENU-induced mouse strain bearing a mutation (R1092W) in the 12th ZF of Zfp335. Homozygous mutants were characterized by the loss of naïve T cells as the result of impaired T-cell maturation (Han et al. 2014). Zfp335 bound primarily to gene promoters, and the R1092W mutation led to altered expression of genes critical for T-cell maturation. However, reduced Zfp335R1092W occupancy was seen at only a small subset of sites, while other sites showed little to no detectable change. This suggested that Zfp335 binding to different subsets of genomic targets may be differentially regulated, raising the question of how this selectivity is achieved. In addition, we identified a 22-base-pair (bp) motif that was bound by Zfp335 in vitro. Although the predicted position of R1092 within its ZF fold led us to hypothesize that it may directly bind DNA, the R1092W mutation did not appear to impair interaction with the 22-bp motif in gel shift assays, and its impact on the biochemical function of Zfp335 remained unclear.
In this study, we use a combination of bioinformatic and biochemical approaches to investigate how Zfp335 achieves sequence-specific DNA recognition. In addition to the original consensus sequence reported in our previous work, we demonstrate here that Zfp335 recognizes a second novel motif. We map two major DNA-binding domains (DBDs) comprising distinct ZF clusters and show that each domain encodes a different sequence specificity, with R1092 being involved in binding to the second motif. Our findings highlight the complexities of DNA recognition by TFs containing several C2H2-ZF domains and offer insights into how binding to multiple sequence motifs may be functionally important in the context of specific gene regulation.
Results and Discussion
ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) analysis reveals two enriched sequence motifs in Zfp335-bound sites
From a ChIP-seq study of Zfp335 in murine thymocytes (Han et al. 2014), we had previously identified a highly enriched 22-bp consensus sequence (“motif 1”) consisting of two nonidentical, nonpalindromic half-sites separated by a 7-bp variable spacer (GGCTGTCC-N7-TGCCTGA) (Supplemental Fig. S1A) and showed that it was specifically bound by Zfp335. Further analysis of our thymocyte ChIP-seq data revealed significant enrichment of a second DNA motif (AGGACCCC, “motif 2”) (Supplemental Fig. S1A). Both motifs were also found to be enriched in a ChIP-seq experiment performed using Flag-tagged ZNF335 expressed in the Jurkat human T-lymphoma cell line (Fig. 1A) as well as in published data from mouse embryonic brains (Yang et al. 2012; data not shown). The robust detection of these two motifs in three independent data sets across different tissues, cell types, and species argues in favor of their biological significance as Zfp335 recognition elements.
Figure 1.

ChIP-seq analysis reveals two putative binding motifs for Zfp335. (A) Motif 1 and motif 2 sequences. (B) Localization of each motif within Zfp335 ChIP-seq peaks. (C) Sequence conservation profiles for motif 1 and motif 2 sites within Zfp335 peaks versus all genomic regions within ±2 kb of the transcription start site (TSS). (D) Significance scores (−log10 P-value) for groups of Zfp335 peaks defined by the number of motif 1 or motif 2 instances found within each peak, represented as Tukey box plots. Results shown in A –D are based on Zfp335 ChIP-seq data in Jurkat cells.
Both motif 1 and motif 2 are centrally enriched within Zfp335 peaks (Fig. 1B; Supplemental Fig. S1B), consistent with the hallmarks of a direct binding motif (Bailey and Machanick 2012). Furthermore, both motifs showed higher levels of sequence conservation at Zfp335-bound sites relative to a background set of all promoter regions (Fig. 1C). Taken together, our analysis of the available genomic evidence builds on our earlier work to identify both motif 1 and motif 2 as putative consensus sequences for Zfp335 binding to target sites in vivo.
By partitioning Zfp335-binding sites based on the number of motif hits found within each peak, we observed that peaks with higher significance scores (reflecting greater Zfp335 occupancy) tended to contain more occurences of motif 1 or motif 2 (Fig. 1D), suggesting a correlation between motif occurrence and binding affinity. However, co-occurrence of both motifs was detected in just 11.1% of Zfp335-occupied regions, as compared with 12.6% containing only motif 1, 19.4% containing only motif 2, and 56.9% for which neither motif was detected (Supplemental Fig. S1C). To account for the existence of sites that lacked a canonical motif 1 match but nevertheless showed significantly enriched Zfp335 binding, we examined peaks lacking full motif 1 matches and found that the presence of motif 1 half-sites HS1 or HS2 (Supplemental Fig. S1A) correlated with higher Zfp335 occupancy (Supplemental Fig. S1C), suggesting that motif 1 half-sites may be sufficient for binding in the absence of the complete bipartite sequence. We detected several instances of linked half-sites with variant spacers occurring in Zfp335 peak regions that otherwise lacked a canonical motif 1 (Supplemental Fig. S1D), suggesting the possibility that such noncanonical motifs may account for Zfp335 binding at some locations. Similar fiindings have been made for the multi-ZF TF REST, which has a 21-bp consensus sequence comprising two well-defined half-sites and has been shown to bind to regions containing isolated half-sites or noncanonical motifs with variant spacers (Johnson et al. 2007).
Genomic evidence supporting a functional partnership between motif 1 and motif 2 at Zfp335 target sites
The discovery of two enriched motifs led us to ask whether they might cooperate in directing Zfp335 to target sites in the genome. First, we observed that Zfp335-bound regions containing both motif 1 and motif 2 exhibit a higher degree of motif sequence conservation as compared with those containing only one type of motif (Fig. 2A), suggesting that the two-motif configuration may be a hallmark of functionally important Zfp335 regulatory sequences. Furthermore, Zfp335 genomic occupancy was associated with constraints in spacing between motif 1 and motif 2. Analysis of Zfp335 peaks containing both motifs revealed two distinct clusters of pairwise spacing distances between motif 1 and motif 2: those separated by <100 bp (group A) or 100–300 bp (group B) (Fig. 2B). Both sets of spacing distances were enriched in Zfp335 peak regions relative to GC-matched control regions not bound by Zfp335 and are thus unlikely to be a sequence bias artifact arising from the high GC content of both motifs. Instead, this implies that motif spacing may be a biologically relevant property of Zfp335 binding. Taken together, our data suggest a functional relationship between motif 1 and motif 2 in determining Zfp335 occupancy at target sites.
Figure 2.
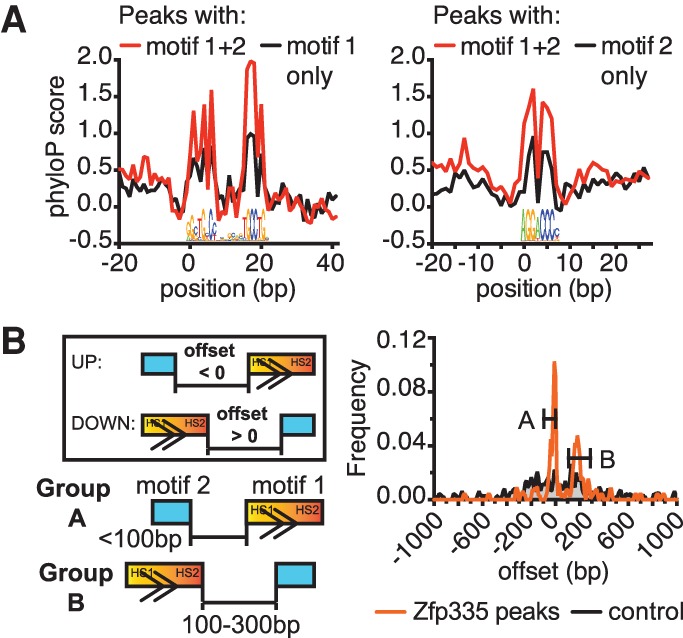
Genomic evidence for a functional partnership between motif 1 and motif 2. (A) Zfp335 peak regions containing both motifs have more highly conserved motif sequences compared with single-motif peaks. (B) Frequency distribution of spacing distances (“offset”) between motif 1 and motif 2. (Group A) Motif pairs separated by <100 bp; (group B) motif pairs separated by 100–300 bp. (Boxed inset) Negative offset values are used when motif 2 is located “upstream” of motif 1 relative to the canonical orientation of motif 1, and positive values are used when motif 2 is “downstream” from motif 1.
Zfp335 binds to motif 1 and motif 2 in vitro
We performed gel shift assays to determine whether Zfp335 bound directly to the two motifs identified from our ChIP-seq analyses. In addition, we sought to determine the effect of the R1092W mutation (shown in Fig. 3A) on DNA binding. Probes were designed based on Zfp335-bound sites upstream of two target genes: Zfp335 for motif 1 (Z1) and Ankle2 for motif 2 (A2). A negative control probe containing no Zfp335 consensus sequences was extracted from the promoter of Pdap1 (Pd) (Fig. 3B). Full-length wild-type and R1092W Zfp335 (Zfp335WT and Zfp335R1092W, respectively) was isolated from HEK293T extracts by Flag pull-down, and silver stain analysis confirmed that both preparations yielded comparable amounts of highly purified protein (Supplemental Fig. S2A). Both Zfp335WT and Zfp335R1092W formed detectable gel shift complexes with the Z1 probe, suggesting that R1092W may not affect motif 1 interaction directly. Zfp335WT binding to the A2 probe was also detected; however, the appearance of faint, diffuse A2 gel shift bands for Zfp335R1092W suggests that it interacts more weakly with motif 2 compared with Zfp335WT (Fig. 3C).
Figure 3.

Zfp335 binds to motif 1 and motif 2. (A) Schematic depicting C2H2-ZF domains of full-length Zfp335 and all mutant constructs used in this study, drawn approximately to scale. (B) Sequences of probes used in gel shift assays, containing either motif 1 (Z1), motif 2 (A2), or a negative control sequence (Pd). Consensus sequences are highlighted in bold. (C) Zfp335 binding to labeled Z1 and A2 probes. Gel shift complex formation is augmented by addition of the reciprocal unlabeled probe (lane 4 vs. 3, lane 10 vs. 9); this effect is not observed for purified Zfp335R1092W (lane 6 vs. 5, lane 12 vs. 11). Data are representative of three independent experiments.
Interestingly, we observed the appearance of a stronger electrophoretic band—potentially signifying a more stable protein–DNA complex—upon inclusion of an unlabeled A2 probe in binding reactions containing a labeled Z1 probe and Zfp335WT protein; this effect was absent with Zfp335R1092W (Fig. 3C). A similar effect was observed upon inclusion of an unlabeled Z1 probe in binding reactions containing a labeled A1 probe (Fig. 3C). In assays using total nuclear extracts instead of purified protein, the relative increase in gel shift band intensities was even more striking (Supplemental Fig. S2B,C), possibly due to competition by other DNA-binding proteins raising the affinity threshold for Zfp335 to form detectable complexes with its cognate probes. Control probes lacking motif 1 or motif 2 consensus sequences failed to elicit the same outcome, suggesting that this phenomenon is dependent on sequence-specific interactions (Fig. 3C; Supplemental Fig. S2D–E). These data suggest that Zfp335 engagement with both of its motifs is associated with increased protein–DNA complex stability over binding to either motif in isolation, although further studies will be needed to investigate this phenomenon in more quantitative detail as well as to uncover its mechanistic basis. We suggest a hypothetical model in which Zfp335 switches between an inactive conformational state with low DNA-binding affinity and a higher-affinity active state and further speculate that the low-affinity state is enforced by an autoinhibitory mechanism that is released when both motifs are present, which may promote more stable binding to target sites. Autoinhibition of DNA binding has been characterized in several TFs, most notably in Ets-1 and other Ets family members (Jonsen et al. 1996; Greenall et al. 2001; Hollenhorst et al. 2011; Regan et al. 2013), and is recognized as a widespread control mechanism to modulate TF activity (Graves et al. 1998). It would be of interest to investigate the possibility of an autoinhibitory mechanism in the context of Zfp335 or other multi-ZF TFs in future studies.
Each motif is recognized by a different ZF cluster
The ability of Zfp335 to recognize both motifs strongly suggests the possibility that it contains multiple DBDs with distinct sequence specificities. Zfp335 has 13 ZFs segregated into two distinct clusters: an N-terminal cluster consisting of two neighboring ZF arrays (ZF2–4 and ZF5–9) and a C-terminal cluster of four ZFs (ZF10–13). To map the domains responsible for interaction with each motif, we made a series of deletion mutants (shown in Fig. 3A). The N-terminal constructs N1 (ZF1–9), N3 (ZF2–9), and N4 (ZF5–9) formed detectable and specific gel shift complexes with the Z1 probe (Fig. 4A). N2, which contained the only ZF not associated with a larger cluster, failed to bind Z1 (Fig. 4A). None of the N-terminal constructs tested showed specific binding activity with the A2 probe (Supplemental Fig. S3A). In contrast, electrophoretic mobility shift assay (EMSA) analysis of the C-terminal ZF10–13 cluster showed that it was sufficient for binding to the A2 probe (further described below and shown in Supplemental Fig. S3B). These results indicate that the N-terminal ZF cluster mediates sequence-specific binding to motif 1, whereas recognition of the motif 2 consensus sequence is achieved via the C-terminal ZFs. Thus, the N-terminal and C-terminal ZF clusters function as separate DBDs, each of which encodes different sequence specificities and is able to bind independently to its corresponding motif.
Figure 4.

N-terminal and C-terminal ZF clusters function as DBDs for motif 1 and motif 2, respectively. (A) EMSA analysis of N1, N3, and N4 protein fragments binding to motif 1 in the presence of excess unlabeled competitor (comp.) probe. Black arrowheads indicate sequence-specific gel shift complexes. (B) Probes were designed with mutations in either HS1 (MH1), HS2 (MH2), or both half-sites (MH12), as indicated. ZS is identical to Z1 except that the original 7-bp spacer sequence was substituted (bold red) to eliminate residual binding activity due to a weak match to HS1 (not shown). (C) N3 (ZF2–9) binds to both half-sites; N4 (ZF5–9) binding requires HS1 but not HS2. (D) Model for motif 1 binding by Zfp335: ZF5–9 constitute the major recognition module for HS1, and ZF2–4 contribute to HS2 binding. Data in A and C are representative of three independent experiments. (E) C1R1092W [Kd(app) = 1407 nM ± 117 nM] binds motif 2 with lower affinity compared with C1WT [Kd(app) = 73.4 nM ± 7.1 nM]. Data are pooled from three experiments.
N-terminal ZF sequence specificities mapped to individual motif 1 half-sites
C2H2-ZFs typically function in arrays of three or more units that enable recognition of multinucleotide sequences (Stubbs et al. 2011). The bipartite structure of motif 1, with its two well-defined half-sites separated by a spacer of constrained length, raised the possibility that these half-sites may be bound separately by distinct yet spatially linked ZF arrays. As previously noted, the N-terminal ZFs cluster into two arrays, ZF2–4 and ZF5–9, separated by a short stretch of 16 amino acids. To map the ZF arrays necessary for binding to each half-site, we competed a panel of mutant probes (Fig. 4B) against the Z1 consensus probe for binding to N3 (ZF2–9) or N4 (ZF5–9). We found that HS1 is both necessary and sufficient for binding to N4. Eliminating HS1 strongly affected binding of the MH1 probe to N4, similar to that of mutating both half-sites; conversely, the MH2 probe, with an intact HS1 and a mutated HS2, competed for binding to N4 just as efficiently as control probes with two intact half-sites (Fig. 4C). In binding reactions with N3 protein, the MH2 probe competed less well relative to wild-type probes but more efficiently compared with a probe lacking both half-sites (Fig. 4C), indicating that HS2 is partially required for motif 1 binding by N3, most likely through ZF2–4. These results suggest that full motif 1 binding by Zfp335 involves modular recognition of HS1 by ZF5–9 and HS2 by ZF2–4 (Fig. 4D).
The R1092W mutation impairs motif 2 binding by C-terminal ZFs
A major goal of this study was to understand the molecular basis for the partial loss of function in Zfp335R1092W. Our identification of the C-terminal ZF cluster as the motif 2-binding domain led us to hypothesize that the R1092W mutation specifically disrupts binding to motif 2. We tested this hypothesis using a quantitative EMSA approach with purified protein containing the C-terminal ZF10–13 cluster (C1 construct in Fig. 3A). Although the R1092W mutation did not completely abolish DNA binding (Supplemental Fig. S3B), it resulted in an almost 20-fold reduction in binding affinity, with an apparent Kd of 1407 nM ± 117 nM for C1R1092W compared with 73.4 nM ± 7.1 nM for C1WT (Fig. 4E). These data indicate that the R1092W mutation significantly lowers affinity of Zfp335 binding to motif 2, likely by disrupting a key amino acid–DNA base contact. Nonetheless, some binding activity is preserved, as ZF–DNA interactions at other motif positions remain intact, resulting in a hypomorphic mutant protein whose phenotypic effects have been shown previously to be partially compensated by overexpression (Han et al. 2014).
Zfp335 activates transcription in a sequence-specific manner dependent on motif 1 -binding and motif 2-binding sites
Prior studies have presented evidence suggesting that Zfp335 functions primarily to activate expression of target genes, possibly via the recruitment of H3K4 methyltransferases (Yang et al. 2012), although it has also been suggested to act as a repressor in certain cases (Han et al. 2014). However, the presumed function of Zfp335 as a positive regulator of gene expression has not been directly demonstrated by biochemical evidence. Furthermore, the cis-acting DNA sequence determinants necessary for transcriptional regulation by Zfp335 have not been tested functionally. Having established that both motif 1 and motif 2 were bound by Zfp335 in vitro, we next investigated whether these motifs functioned as cis-regulatory elements for Zfp335-dependent transcriptional activity in a reporter gene assay.
We created luciferase reporter constructs containing single copies of both motif sequences in the same relative orientation cloned upstream of a minimal promoter element as well as mutant variants that lacked either motif 2 or motif 1. We tested the ability of Zfp335WT or Zfp335R1092W to activate these reporter constructs in transiently transfected HEK293T cells (Fig. 5A). Expression of Zfp335WT resulted in a 10-fold increase in activation of the dual-motif reporter relative to endogenous background but had little effect on parental reporter plasmid activity, indicating that a DNA element combining both motifs supported Zfp335-induced transcriptional activation. We also observed robust activation of the dual-motif reporter by Zfp335R1092W. Loss of motif 2 alone led to decreased activation by Zfp335WT but did not significantly affect Zfp335R1092W-induced activity. In contrast, removal of motif 1 resulted in significantly decreased activation driven by Zfp335R1092W but not Zfp335WT. These observations are in line with the expectation that selective disruption of motif 2 binding makes Zfp335R1092W disproportionately dependent on motif 1, whereas Zfp335WT is able to use both motifs as binding sites and retains the ability to activate reporter gene expression when only one motif is present.
Figure 5.

Motif 1-binding and motif 2-binding sites are sufficient for Zfp335-mediated transcriptional activation. (A) Zfp335WT or Zfp335R1092W expression constructs were transfected into HEK293T cells along with a luciferase reporter vector containing no motifs, both motifs, motif 1 only, or motif 2 only. Luciferase activity was normalized to Renilla cotransfection control and is expressed as mean ± SE of duplicate transfections. Data shown are from one representative experiment out of three. (B) Model showing how Zfp335 binds to its two consensus motifs.
In summary, we demonstrated that Zfp335 associates with two distinct motifs both in vitro and in vivo and that these interactions are mediated by specific ZF clusters separated into N-terminal and C-terminal DBDs that can bind their respective motifs independently (Fig. 5B). This work represents a significant advance in elucidating the biochemical and genomic aspects of how Zfp335 functions as a TF, informing efforts to understand its role in development and disease. Furthermore, we believe that these findings may offer insights into other C2H2-ZF proteins, which collectively represent the largest class of eukaryotic TFs, of which a vast majority remains poorly characterized (Vaquerizas et al. 2009). Although recent high-throughput efforts have greatly increased the number of known binding motifs (Badis et al. 2009; Jolma et al. 2013; Najafabadi et al. 2015), the structural complexity widespread in C2H2-ZF proteins continues to present unique challenges. On average, human C2H2-ZF proteins contain approximately nine ZFs, and some have 30 or more (Emerson and Thomas 2009), suggesting a potential for recognizing motifs longer and more complex than that of other TF families. Furthermore, the modular nature of C2H2-ZF proteins allows for the differential use of specific ZF subsets to achieve different sequence specificities in various biological contexts (Stubbs et al. 2011).
Our discovery of multiple binding specificities for Zfp335 underscores the notion that multi-C2H2-domain TFs have the potential to recognize more than one sequence motif using discrete ZF clusters that function as independent DBDs. Prior understanding of this phenomenon has been limited to a small number of cases. One example is EVI-1, which contains two autonomous DBDs (an N-terminal cluster of seven ZFs and a C-terminal cluster of three ZFs), each recognizing a different motif (Delwel et al. 1993; Morishita et al. 1995). Others include OAZ (30 ZFs), which uses different subsets of fingers to bind two distinct DNA sequences (Hata et al. 2000), and CTCF (11 ZFs) (Nakahashi et al. 2013). Recent large-scale studies have suggested that DNA binding via multiple distinct high-affinity motifs may be more prevalent amongst TFs than previously thought (Badis et al. 2009). As most TF motifs are short and degenerate (Spitz and Furlong 2012), usage of multiple motifs and diversification of the DNA-binding repertoire may allow for more efficient targeting of TFs to functional binding sites and contribute to context-dependent regulation of TF occupancy and function. However, attempts to determine binding specificities of C2H2-ZF proteins using high-throughput approaches such as PBMs and HT-SELEX have met with limited success (Jolma et al. 2013). As a result, the multiple motif-binding behavior that we described for Zfp335 still remains relatively undocumented within the C2H2-ZF family aside from the few examples mentioned above. Hence, our study positions Zfp335 as a model for investigating the complexities of sequence recognition by C2H2-ZF proteins to further our understanding of this important class of transcriptional regulators.
Materials and methods
EMSAs
For qualitative EMSA studies, double-stranded probes were prepared by annealing 5′-biotinylated oligonucleotides (Integrated DNA Technologies). All probe sequences are listed in Supplemental Table S1. Protein extracts and probes were incubated in binding buffer consisting of 8 mM HEPES (pH 7.9), 80 μM EDTA, 50 mM KCl, 8% (v/v) glycerol, 40 μg/mL BSA, 10 μM ZnCl2, 20–40 μg/mL poly(dI-dC), and 0.2 mM DTT for 20 min at room temperature. Samples were loaded on nondenaturing gels (4.5% or 6% polyacrylamide, 3.5% glycerol, 0.25× TBE), run in 0.25× TBE buffer for 1–1.5 h under constant voltage (160 V) at 4°C, and transferred to Hybond N+ nylon membranes (Amersham Pharmacia Biotech) in 0.5× TBE at 380 mA for 1 h. Biotin-labeled DNA was detected using the LightShift chemiluminescent nucleic acid detection module kit (Thermo Scientific). Blots were imaged using the ChemiDoc MP system (Bio-Rad), and chemiluminescent signals were quantified using Image Lab version 5.1 (Bio-Rad). For quantitative EMSA analyses, duplex probes were prepared from 3′-Alexa fluor 488-labeled oligonucleotides (Integrated DNA Technologies). Purified recombinant protein was incubated with 5 nM labeled probe for 20 min at room temperature in reaction mixtures consisting of 30 mM NaCl, 60 μM EDTA, 8% (v/v) glycerol, 100 μg/mL BSA, 10 μM ZnCl2, 20 μg/mL poly(dI-dC), and 0.2 mM DTT. Samples were run on pre-equilibrated 6% polyacrylamide gels. Gels were imaged on the ChemiDoc MP using default instrument settings for Alexa fluor 488 detection. The ratio of free probe signal at each protein concentration relative to the total probe signal in a control lane containing no protein ([DNAfree]/[DNAtotal]) was determined using Image Lab, and the fraction of DNA bound to protein was calculated as 1 − [DNAfree]/[DNAtotal]. Data points from three independent experiments were fitted using nonlinear least-squares analysis and plotted using Prism 6 (GraphPad). Apparent Kd values were measured as the protein concentration required for 50% of the probe to be bound.
Luciferase reporter assays
Synthetic oligonucleotides (Supplemental Table S2) were annealed to form double-stranded inserts with KpnI and NheI restriction site overhangs and ligated into the pGL4.23 [luc2/minP] reporter vector (Promega). pGL4.23 reporter constructs were cotransfected into HEK293T cells with the Renilla luciferase plasmid pGL4.74 [hRluc/TK] as a normalization control and Zfp335 pcDNA3 expression constructs. Transfections were done in duplicate using 24-well plates. Cells were harvested after 48 h, and luciferase activity was assayed using the dual-luciferase reporter assay kit (Promega) according to the manufacturer's protocols.
ChIP-seq
Formaldehyde cross-linked Jurkat cells were resuspended in lysis buffer (50 mM HEPES-KOH at pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) followed by nucleus wash buffer (10 mM Tris-HCl at pH 8.0, 200 mM NacCl, 1 mM EDTA, 0.5 mM EGTA). Chromatin was fragmented in buffer containing 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM CaCl2, 3 mM MgCl2, 0.5% IGEPAL CA-630, and 1% Triton X-100 using a combination of probe sonication and digestion with 0.6 U/µL micrococcal nuclease (Worthington). ChIP was performed using anti-Flag M2 antibody (Sigma) and Protein G Dynabeads (Life Technologies). Beads were washed in low-salt buffer (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA), high-salt buffer (10 mM Tris-HCl at pH 8.0, 500 mM NaCl, 1% Triton X-100, 1 mM EDTA), LiCl buffer (50 mM HEPES-KOH at pH 7.5, 250 mM LiCl, 1 mM EDTA, 1% IGEPAL CA-630, 0.7% sodium deoxycholate), and TE buffer. Elution and DNA purification were performed as described (Han et al. 2014). Approximately 150 ng of DNA was used for library construction according to Illumina's TruSeq protocol. Libraries were size-selected (200–500 bp) by agarose gel purification and sequenced on the Illumina HiSeq 2000 platform as 50-bp single-end reads.
ChIP-seq data analysis
Sequence reads from Jurkat ChIP-seq libraries were adapter-trimmed and aligned to the hg19 reference genome. Data processing and peak calling were carried out as described (Han et al. 2014).
Data availability
Zfp335 ChIP-seq data obtained from mouse thymocytes were previously deposited to NCBI's Gene Expression Omnibus (GEO) under SuperSeries GSE58293. Jurkat T-cell ChIP-seq data are available from GEO under accession number GSE83116.
Supplementary Material
Acknowledgments
We thank Hilde Schjerven, Matthew Kneusel, and Khanh Ngo for helpful discussion; Ying Xu for technical assistance; and Ernesto Guccione for critical reading of the manuscript. This work was supported by the Howard Hughes Medical Institute and the National Institutes of Health (grant R01AI74847).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.279406.116.
References
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. 2009. Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Machanick P. 2012. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40: e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwel R, Funabiki T, Kreider BL, Morishita K, Ihle JN. 1993. Four of the seven zinc fingers of the Evi-1 myeloid-transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol 13: 4291–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson RO, Thomas JH. 2009. Adaptive evolution in zinc finger transcription factors. PLoS Genet 5: e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves BJ, Cowley DO, Goetz TL, Petersen JM, Jonsen MD, Gillespie ME. 1998. Autoinhibition as a transcriptional regulatory mechanism. Cold Spring Harb Symp Quant Biol 63: 621–629. [DOI] [PubMed] [Google Scholar]
- Greenall A, Willingham N, Cheung E, Boam DS, Sharrocks AD. 2001. DNA binding by the ETS-domain transcription factor PEA3 is regulated by intramolecular and intermolecular protein-protein interactions. J Biol Chem 276: 16207–16215. [DOI] [PubMed] [Google Scholar]
- Han BY, Wu S, Foo C-S, Horton RM, Jenne CN, Watson SR, Whittle B, Goodnow CC, Cyster JG. 2014. Zinc finger protein Zfp335 is required for the formation of the naïve T cell compartment. eLife 3: e03549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Seoane J, Lagna G, Montalvo E, Hemmati-Brivanlou A, Massagué J. 2000. OAZ uses distinct DNA- and protein-binding zinc fingers in separate BMP-Smad and Olf signaling pathways. Cell 100: 229–240. [DOI] [PubMed] [Google Scholar]
- Hollenhorst PC, McIntosh LP, Graves BJ. 2011. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 80: 437–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. 2013. DNA-binding specificities of human transcription factors. Cell 152: 327–339. [DOI] [PubMed] [Google Scholar]
- Jonsen MD, Petersen JM, Xu QP, Graves BJ. 1996. Characterization of the cooperative function of inhibitory sequences in Ets-1. Mol Cell Biol 16: 2065–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug A. 2010. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Q Rev Biophys 43: 1–21. [DOI] [PubMed] [Google Scholar]
- Mahajan MA, Murray A, Samuels HH. 2002. NRC-interacting factor 1 is a novel cotransducer that interacts with and regulates the activity of the nuclear hormone receptor coactivator NRC. Mol Cell Biol 22: 6883–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita K, Suzukawa K, Taki T, Ihle JN, Yokota J. 1995. EVI-1 zinc finger protein works as a transcriptional activator via binding to a consensus sequence of GACAAGATAAGATAAN1-28 CTCATCTTC. Oncogene 10: 1961–1967. [PubMed] [Google Scholar]
- Najafabadi HS, Mnaimneh S, Schmitges FW, Garton M, Lam KN, Yang A, Albu M, Weirauch MT, Radovani E, Kim PM, et al. 2015. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol 33: 555–562. [DOI] [PubMed] [Google Scholar]
- Nakahashi H, Kwon K-RK, Resch W, Vian L, Dose M, Stavreva D, Hakim O, Pruett N, Nelson S, Yamane A, et al. 2013. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep 3: 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MC, Horanyi PS, Pryor EE, Sarver JL, Cafiso DS, Bushweller JH. 2013. Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc Natl Acad Sci 110: 13374–13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626. [DOI] [PubMed] [Google Scholar]
- Stubbs L, Sun Y, Caetano-Anolles D. 2011. Function and evolution of C2H2 zinc finger arrays. Subcell Biochem 52: 75–94. [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. 2009. A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10: 252–263. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO. 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 29: 183–212. [DOI] [PubMed] [Google Scholar]
- Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, et al. 2012. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151: 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Zfp335 ChIP-seq data obtained from mouse thymocytes were previously deposited to NCBI's Gene Expression Omnibus (GEO) under SuperSeries GSE58293. Jurkat T-cell ChIP-seq data are available from GEO under accession number GSE83116.